Telómeros, cáncer y envejecimiento
Yuliia Fatych, Patricia Tato Moreno. 3º Biología Sanitaria. Universidad de Alcalá
Introducción histórica
En 1938, Hermann J. Müller observó que en los extremos de cromosomas expuestos a rayos X de Drosophila melanogaster no se producían mutaciones como deleciones o inversiones, mientras que esto sí ocurría en el resto del genoma. Esto se debía a la presencia de un “casquete protector” al que denominó “gen terminal” y, más tarde, “telómero”.1
Dos años después, Barbara McClintock, que realizaba estudios genéticos con maíz, describió la formación de cromosomas dicéntricos mediante la ruptura de los cromosomas y su posterior adhesión y fusión de sus extremos. Gracias a estas investigaciones, McClintock demostró que los extremos de los cromosomas se podían restaurar gracias a la obtención de un nuevo telómero.1
Sin embargo, la investigación sobre los telómeros no se volvió a retomar hasta treinta años más tarde, cuando James Watson identificó el problema de la replicación del DNA en los extremos de los cromosomas. Debido a que las DNA-polimerasas sólo pueden sintetizar DNA en sentido 5’ → 3’, la cadena 5’ → 3’ se forma mediante la síntesis de fragmentos de Okazaki, cada uno de los cuales necesita un cebador. Esto hace que el extremo 3’ del cromosoma no se pueda replicar por completo y, por tanto, en cada replicación, éste se acorta. Esto limita la capacidad de replicación de la célula. Olovnikov descubrió que la senescencia celular se producía como consecuencia del sobrepaso de ese límite, lo que provoca la alteración de la célula.1

Más tarde, tras muchos años de trabajo e investigación sobre los telómeros del protozoo Tetrahymena thermophila y de la levadura Saccharomyces cerevisiae, Elizabeth Blackburn, Jack Szostak y Carol Greider, descubrieron la existencia de una actividad enzimática que denominaron “transferasa telómero terminal”. Esta actividad estaba presente en la enzima telomerasa, a la que le atribuyeron el papel de la replicación del DNA telomérico, impidiendo el acortamiento progresivo de los telómeros en cada división celular.1 Finalmente, en el año 2009, Blackburn, Szostak y Greider recibieron el Premio Nobel de Medicina y Fisiología por sus estudios sobre los telómeros y el descubrimiento de la telomerasa.2
Estructura de los telómeros
Los telómeros son unas estructuras que se ubican en los extremos de los cromosomas lineales eucarióticos y que están compuestos por proteínas y secuencias de DNA no codificante repetidas en tándem. La secuencia repetida es la secuencia (TTAGGG)n, lo que hace que en los telómeros exista una hebra rica en nucleótidos de guanina, conocida como hebra G, y otra hebra rica en nucleótidos de citosina. La hebra G es la que se encuentra orientada en dirección 5’ → 3’ y, en su extremo, sobresale de la cadena de DNA, por lo que no se aparea con la hebra antiparalela, debido a que ésta es más corta. Este fragmento de DNA simple de la hebra G se conoce como overhang-3’ y tiene una longitud que varía según la especie. Además, la longitud de los telómeros también es variable y en cada cromosoma la cantidad de DNA telomérico puede ser diferente.3,4
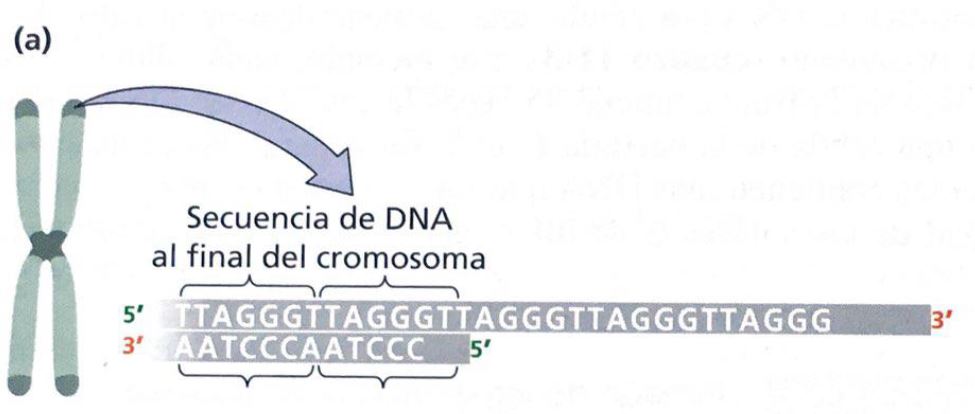
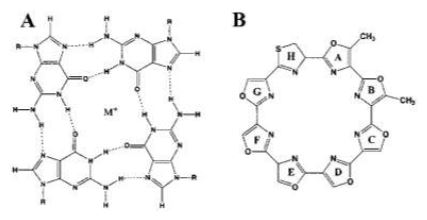
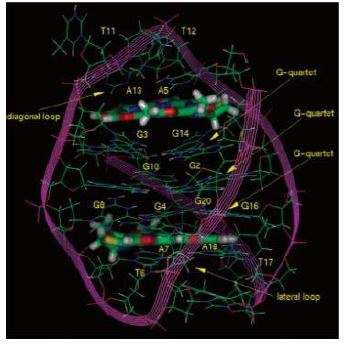
Es importante señalar la existencia de estructuras complejas en el extremo 3’ rico en G, denominadas G-cuadruplexos. Dichas estructuras se pueden encontrar en diferentes conformaciones, unidas por planos cuadrados que contienen 4 guaninas (que a su vez interaccionan por puentes de hidrógeno de Hoogsteen). Esta estructura ordenada impide la acción de las nucleasas, las cuales trabajan sobre las hebras sueltas de DNA.5
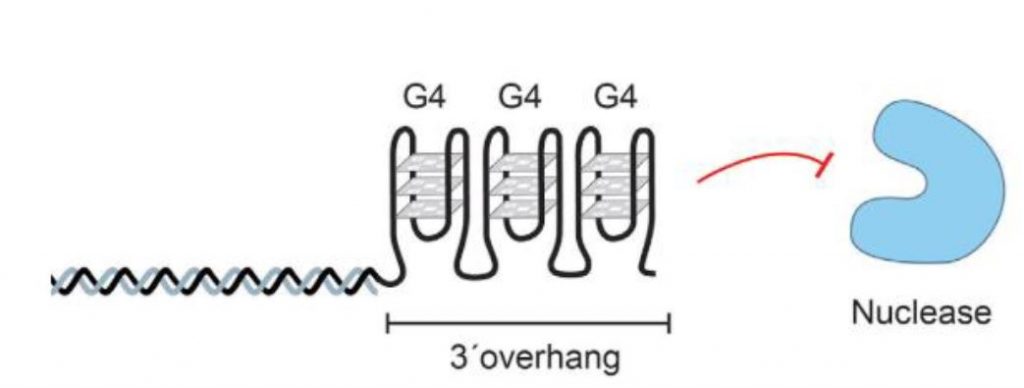
Por otro lado, en los telómeros también podemos encontrar unas secuencias repetidas, conocidas como secuencias asociadas a los telómeros. Estas estructuras varían según la especie en cuanto a su longitud, secuencia y complejidad. Además, parece ser que no tienen un papel importante en la estabilidad del cromosoma y todavía no se conoce cuál es exactamente su función.4
Esta estructura especial de los telómeros, hace que éstos tengan la función de evitar que se fusionen con los extremos de otros cromosomas, lo que se conoce como fusión telomérica. Además, también van a tener otras funciones como preservar la región codificante del DNA de la acción enzimática, permitir la interacción entre los cromosomas y la matriz nuclear, e intervenir en la transcripción de genes subteloméricos que regulan el ciclo celular. 6
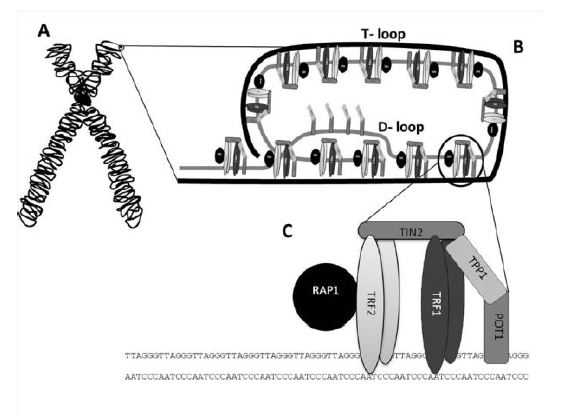
Los telómeros de mamíferos se encuentran asociados a un complejo multiproteico formado por seis proteínas que recibe el nombre de shelterina, cuya función es favorecer la formación de un lazo (“loop T”) que permite que el telómero se doble, secuestrando el extremo terminal de los cromosomas. Esto evita que el DNA telomérico sea dañado por nucleasas. Además, dicho complejo multiproteico impide que se lleve a cabo un mecanismo de reparación de DNA (MRA) en los telómeros, y regula la actividad de la telomerasa.5
Las proteínas que forman el complejo de la shelterina son TRF1, TIN2, TRF2, RAP1, POT1 y TPP1. La shelterina es reclutada por los telómeros a través de TRF1 Y TRF2. La proteína TRF1 (factor 1 de unión a las repeticiones teloméricas) se une a las secuencias repetidas TTAGGG de doble cadena e interacciona con TIN2 (factor 2 nuclear de interacción con TRF2). TRF2 también se une a las repeticiones teloméricas de doble cadena y, a su vez, interacciona con RAP1 (proteína 1 represora/activadora). Además, TIN2 también se encuentra asociada a TRF2. Sin embargo, a diferencia de TRF1 y TRF2, POT1 (protección del telómero 1) se puede unir a las secuencias TTAGGG formadas por una hebra simple y se conecta a TRF1 y TRF2 a través de la proteína TPP1 (proteína homóloga de la displasia adrenocortical). Al mismo tiempo, TPP1 también se asocia a TIN2.7,8
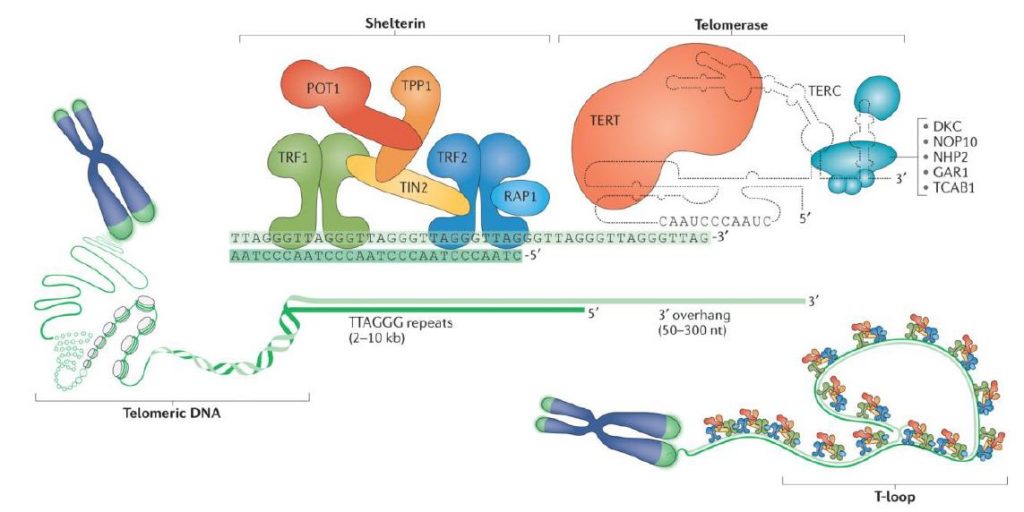
Además del “loop T”, en el cual el DNA monocatenario se enrosca alrededor de un círculo estabilizado por shelterina, existe al final de éste otro bucle, el “loop D”. Este último loop consiste en una estructura de triple hebra denominado como bucle de desplazamiento, en el cual el DNA telomérico monocatenario se entrelaza con una región de DNA bicatenario.5
Aspectos moleculares de la telomerasa: estructura, función, regulación.
Arquitectura funcional de la telomerasa
La telomerasa es una enzima de tipo ribonucleoproteína que participa en la síntesis de las secuencias repetitivas de DNA de los telómeros, estabilizando su longitud.6
La enzima telomerasa humana posee una subunidad catalítica (TERT), una subunidad de RNA (hTR, también llamado TERC o TER) que proporciona el molde para la adición de la secuencia telomérica (TTAGGG)n, y proteínas accesorias. Estas proteínas participan en la regulación de la biogénesis de la telomerasa, así como en su localización dentro de la célula y su funcionamiento in vivo. 5
Las proteínas accesorias que intervienen en la arquitectura de la telomerasa son5:
- Disquerina.
- NHP2.
- NOP10.
- Pontina/Reptina.
- GAR1.
- TCAB1.
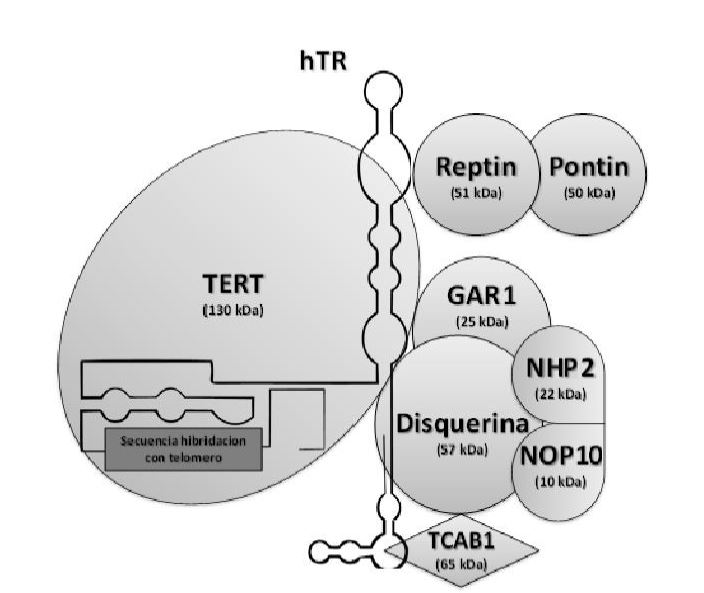
La disquerina, el NHP2 y la NOP10 son 3 de las proteínas accesorias implicadas en la estabilidad y la acumulación de TER (RNA de la telomerasa humana). Por otro lado, la disquerina y GAR1 asociados a TER, posibilitan que la telomerasa sea funcional.5
A su vez, nos encontramos con dos ATPasas (pontina y reptina), que se asocian a TERT (región catalítica) en la fase S del ciclo celular. Su ausencia, dificulta la acumulación de la telomerasa, por lo cual son dos proteínas muy importantes para el montaje de la enzima. Además, se ha visto que también ayudan a la estabilidad de TER durante el ensamblaje de la telomerasa.5
Se cree que una vez terminado el acoplamiento de todos los componentes de la telomerasa, la pontina y la reptina se disocian, dejando libre la enzima activa en su actividad catalítica.5
Se cree que TCAB1 sería la proteína accesoria encargada de la ubicar a la telomerasa dentro de la célula.5
Estructuras en alta resolución de los subdominios TER Y TERT
- TER
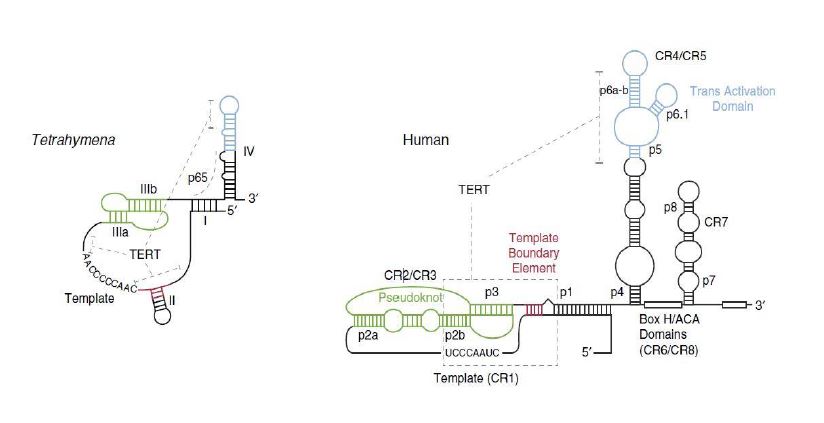
La longitud de TER puede variar dependiendo del organismo en el que nos fijemos: 150 nucleótidos en ciliados, 450 nucleótidos en vertebrados, y hasta 1300 nucleótidos en algunas levaduras. A pesar de esto, se ha descubierto que todas las subunidades TER contienen 2 estructuras secundarias conservadas: un dominio central de pseudonudo (“pseudoknot-template core domain”), catalíticamente esencial, y un tallo-bucle (“stem-loop element”), denominado CR4-CR5 en vertebrados. Dichas estructuras interaccionan directamente con TERT.9
- TERT
Gracias a la comparación de secuencias genéticas codificantes para TERT de distintos organismos, se ha descubierto la existencia de un dominio muy conservado en cuanto a su organización y tamaño (de unos 1100 aminoácidos).9
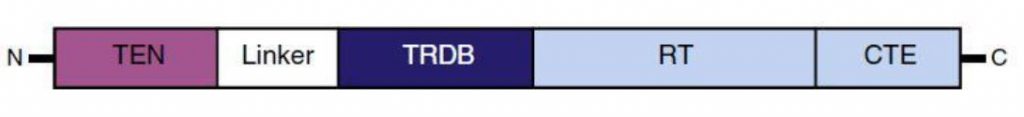
Dicha región de la subunidad catalítica TERT está formada desde el sentido N-terminal hasta el C-terminal por9:
-Un dominio esencial de extensión N-terminal (“essential N-terminal extension domain” o TEN), el cual tiene afinidad por el DNA telomérico monocatenario. A su vez, contacta directamente con TPP1/TIN2.
-Una región de enlace flexible (“linker”). Es el sitio de unión de TEN y TRBD.
-Un dominio de unión a RNA (“RNA binding domain” o TRBD). Interacciona con la región tallo-bucle o CR4-CR5 de TER.
-Un dominio central de la transcriptasa inversa (RT), el cual posee homología estructural y funcional con la transcriptasa inversa retroviral.
-Una extensión C-terminal (CTE).
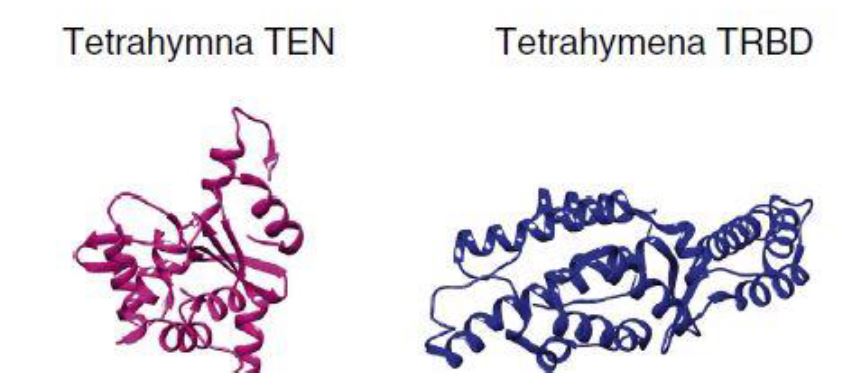
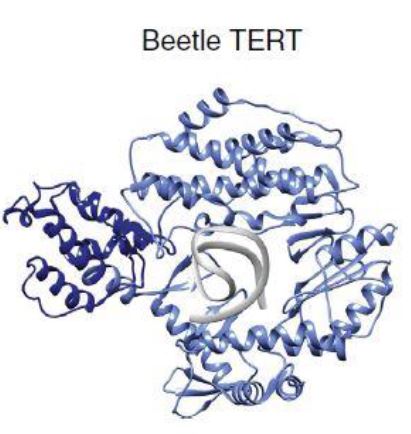
Para esclarecer la estructura tridimensional de TERT se han utilizado escarabajos (Tribolium castaneum). Teniendo en cuenta que no poseen el dominio TEN, la estructura resultante genera una forma de anillo gracias a que TRBD y CTE se acercan en el espacio y forman un túnel catalítico. El DNA se uniría a CTE y el molde de RNA a RT, posicionando el extremo 3’ del G-overhang en el túnel catalítico para la adición de nucleótidos.9
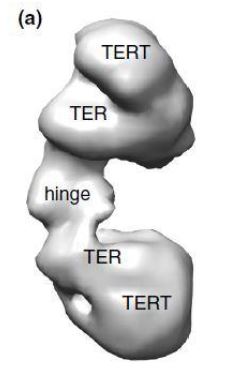
Estructuras tridimensionales de la telomerasa humana y de Tetrahymena
La estructura de las telomerasas se ha determinado mediante microscopía electrónica de partículas individuales (EM) en tinción negativa, con resolución de 25 Å.9
Se pueden observar diferencias a nivel de subunidades, a la vez que similitudes en la organización de TERT.9
La telomerasa humana consiste en un dímero, conteniendo cada monómero una subunidad TER y una subunidad TERT, además de proteínas accesorias. Dichos monómeros están unidos por una región de bisagra flexible. Los autores sugieren que la telomerasa humana debe ser dimérica para poder extender dos extremos teloméricos en paralelo, facilitando que las cromátidas hermanas presenten la misma longitud en sus telómeros.9
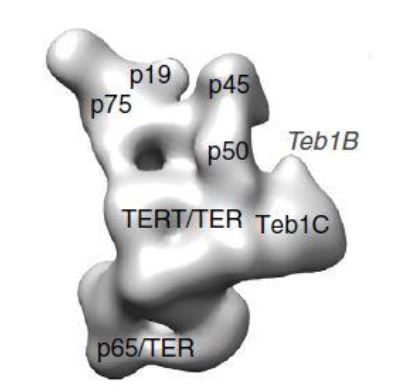
Por otro lado, la telomerasa de Tetrahymena es monomérica, y es funcional en esa forma. Contiene una subunidad TERT, una subunidad TER y proteínas accesorias. La subunidad TERT es próxima a la TER, y además, posee semejanzas estructurales en relación a la telomerasa humana.9
Funcionamiento de la telomerasa
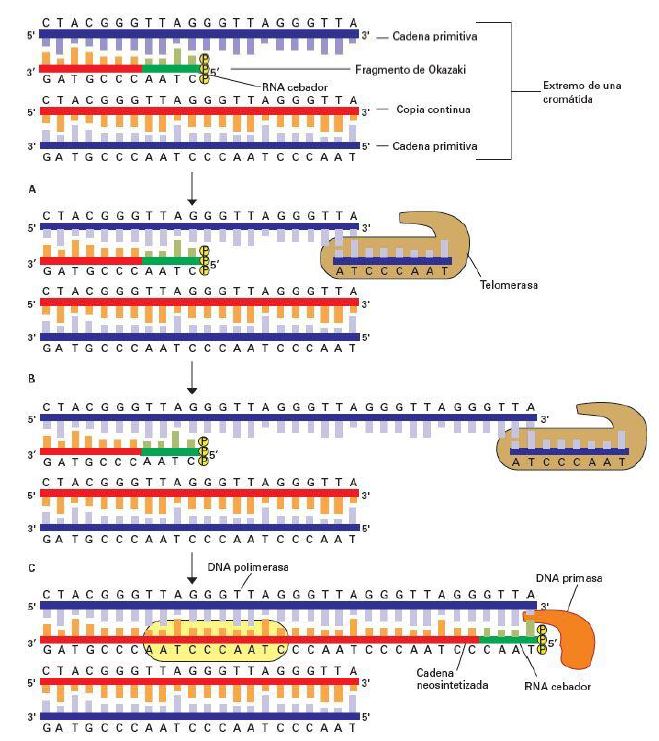
La replicación del DNA romosómico se realiza gracias a DNA polimerasas que pueden extender las cadenas a partir de un RNA cebador y en extremo 5’ → 3’. Dichos cebadores se reemplazarán por DNA y se unirán a las nuevas cadenas de DNA por las DNA ligasas.10
Cuando llegamos a los extremos del cromosoma se plantea un problema. La hebra cuyo molde es la cadena conductora 3’ → 5’, seguirá sintetizándose desde el último origen de replicación hacia el extremo final en dirección 5’ → 3’.10
Por otra parte, la hebra copiada a partir de la cadena retardada 5’ → 3’, contiene fragmentos de Okazaki. Se plantea la dificultad de que en el último fragmento de Okazaki, el RNA cebador no puede ser reemplazado por DNA, ya que al no haber una secuencia adyacente, no hay posibilidad de que actúen las DNA polimerasas. Debido a esto, la hebra sintetizada de la cadena retrasada, perderá entre 50 y 100 nucleótidos en su extremo 5’ con respecto a su molde.10
Para evitar la pérdida de información genética, ya se ha comentado que en los extremos 3’ de las hebras cromosómicas se encuentra el DNA telomérico rico en G, adyacente a la última secuencia de DNA que se puede replicar por la DNA polimerasa. Aunque el DNA telomérico contiene secuencias repetidas en tándem, podría desaparecer después de varios ciclos de replicación. Debido a esto, encontramos la enzima telomerasa, que va a prolongar la secuencia telomérica.10
En el primer paso de la síntesis de DNA telomérico, la telomerasa se recluta a través de la interacción de TPP1 de la shelterina con el dominio N-terminal de su subunidad catalítica TERT. Además, el extremo 3’ G-overhang del telómero se coloca en el sitio activo de TERT alineándose con el RNA (TER) a través de la formación de pares de bases.9 Este RNA es complementario a la cadena de DNA rica en G y aparea parcialmente con ella, proporcionando un molde para la síntesis de copias de la unidad repetida. Los desoxinucleótidos se añaden de novo al extremo 3’ de la cadena rica en G. Después de que se hayan añadido varios nucleótidos se produce una translocación de la telomerasa hacia el extremo del telómero y se reinicia el proceso.10
Se producen unos 10.000 pares de nucleótidos en el extremo 3’, siendo dicha hebra más larga que la complementaria. A su vez, esta elongación de la cadena retardada 5’ → 3’, sirve para producir un nuevo espacio para la creación de un fragmento de Okazaki: se sintetiza un RNA cebador, y la DNA polimerasa elonga la cadena en dirección 5’ → 3’.10
En humanos, la telomerasa está activa en las células embrionarias pluripotenciales y las células madre germinales, sanguíneas o de tejidos adultos en continua renovación. Por otra parte, está reprimida en el tejido somático, limitándose así su capacidad de división. Sin embargo, en los procesos tumorales esta enzima se reactiva, permitiendo su proliferación y desarrollo.6, 10
Los procariotas constan de un cromosoma circular, por lo que se podrá hacer una copia de todos los nucleótidos, ya que como la cadena no se interrumpe en ningún momento, el RNA cebador que se generó al comienzo de la copia, podrá ser sustituido por DNA una vez que la copia alcance el final de la cadena.10
Se ha visto que la actividad telomerasa puede encontrarse en las fases G1, S y G2 a lo largo de un ciclo celular. A su vez, se da una represión cuando las células entran en G0 debido a6:
- Una falta de factores de crecimiento.
- Inhibición por contacto de la división celular.
- Inducción de la senescencia por reversión en una línea celular inmortalizada.
- Diferenciación.
Regulación
La telomerasa se regula por factores genéticos, epigenéticos y ambientales.11
La regulación de la telomerasa se puede dar a través del factor TRF1, que actuaría como un supresor de la elongación telomérica. Su mecanismo de acción consiste en la unión al DNA telomérico de doble cadena, impidiendo la acción de la telomerasa. Dicho factor se encarga de establecer un feedback negativo que estabiliza la longitud telomérica en la interfase y la mitosis.12 Para que este proceso se lleve a cabo se ha descubierto que se necesita que TRF1 interactúe con TIN2.6
El cese de la actividad de la telomerasa en los tejidos embrionarios se puede dar a través de la represión del gen hTERT o a través de empalmes alternativos del transcrito hTERT, que daría lugar a proteínas sin acción transcriptasa inversa.13
Existen factores epigenéticos tales como metilación de islas CpG, metilación de histonas y acetilación, que son importantes para la transcripción de TERT.11
Cáncer
El cáncer es un conjunto heterogéneo de trastornos que se caracterizan por la presencia de células que no responden a los controles normales de la división, lo que hace que estas células cancerosas se dividan rápidamente y de forma continua, generando tumores que privan de nutrientes a los tejidos sanos.2
Varios autores afirman que en las células somáticas humanas, el potencial de proliferación restringido permite unas 50-70 divisiones celulares, alcanzando después la senescencia.5 El concepto de senescencia celular engloba a todas las respuestas que genera una célula mitóticamente competente frente a estímulos capaces de inducir su malignización, es decir, generar un cambio neoplásico en ella. Por tanto, se produce la transformación morfológica y funcional de la célula a un fenotipo senescente y se permite una detención de su crecimiento. Esto hace pensar que la senescencia es un mecanismo de seguridad que surgió para evitar la tumorigénesis en células que posean riesgos neoplásicos (como acortamiento/ alteración telomérica, daños DNA, proteína RAS mutada).14
Respecto al acortamiento telomérico mencionado anteriormente, se ha visto que en hongos, los telómeros cortos posibilitan el daño al DNA, la liberación factores de transcripción y la remodelación de la heterocromatina.14
Otros estudios han visto que si la reducción telomérica supera un límite mínimo de longitud, se crean impedimentos para una correcta separación cromosómica en la mitosis, debido a la aparición de asociaciones teloméricas (tas). Esto provoca inestabilidad cromosómica, que podrá dar lugar a errores genéticos, importantes en procesos neoplásicos como amplificación génica y pérdida de heterocigosidad. Las células que vayan acumulando tas pueden acabar perdiendo regiones repetitivas ricas en guanina, favoreciendo así dichos mecanismos neoplásicos.6
Las tas consisten en la unión de los extremos de dos o más cromosomas, pero sin pérdida aparente de material genético. Pueden producirse en una cromátida o en ambas (simple o doble cromátida).6
Algunos autores proponen que la formación de tas estaría asociada a replicaciones defectuosas de los telómeros, traduciéndose en pérdidas de las secuencias teloméricas tras cada ciclo, aumentando así la probabilidad de producir fusiones cromosomales. Los mecanismos por los cuales esto ocurre apuntan al fallo de la enzima telomerasa, tal y como se ha visto en células de ratones.6
Las tas se han descrito tanto en tumores sólidos, como en neoplasias hematológicas. A su vez también se han encontrado en células infectadas por virus, células con fenotipo senescente y patologías genéticas como síndrome de Turner. Sin embargo, no se ha visto este fenómeno en células normales, ya que están protegidas de la unión entre cromosomas mediante sus telómeros.6
La mayor parte de los tumores malignos presentan la enzima telomerasa activa, lo que hace que las células adquieran la capacidad de proliferar de manera ilimitada. Sin embargo, esto no ocurre en los tumores benignos, que se caracterizan por la ausencia de la telomerasa.15
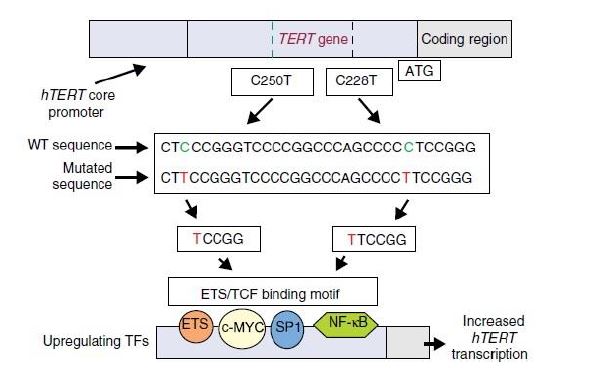
El promotor del gen de hTERT es rico en guanina y citosina y presenta numerosos sitios de unión para múltiples proteínas, por lo que su expresión está muy controlada. Por ejemplo, este gen presenta sitios para la unión de los dedos de zinc del factor de transcripción SP1 y para la unión de otras proteínas como USF1/2, MYC, MAX, MXD1 o el factor de transcripción TFII-I. Algunos factores que producen una estimulación de la expresión de hTERT son c-MYC, SP1 y los factores de transcripción ETS.16
Las mutaciones del gen hTERT se pueden producir en dos sitios de la secuencia en los que tienen lugar transiciones C-T, y ambas dan lugar a un mismo ácido nucleico de 11 pares de bases, que presenta una secuencia para la unión de los factores ETS. Aunque el papel de estos factores todavía no está muy claro, se sabe que la unión del ETS-2 al promotor de hTERT está asociada con la activación de la telomerasa mediada por el factor de crecimiento epidérmico (EGF) en el cáncer de pulmón.16
Por otro lado, también se cree hTERT podría tener otro papel en el cáncer, además de intervenir en el alargamiento de los telómeros. Se ha observado que el aumento de la expresión de TERT daba lugar a linfomas de linfocitos T en células del timo de ratones sin cambios significativos en la longitud de los telómeros. Asimismo, se ha visto que TERT se asocia indirectamente con genes como el de la IL-6, la IL-8 o el TNFα (factor de necrosis tumoral), los cuales intervienen en los procesos de inflamación y de progresión del cáncer.17
Por tanto, una de las mutaciones que se producen en muchos cánceres, como en glioblastomas, liposarcomas o melanomas primarios, es la mutación del promotor proximal del gen de TERT de la telomerasa. Sin embargo, no se conoce por qué la frecuencia de esta mutación es menor en otros cánceres bastante comunes, como el cáncer de pulmón, el cáncer de colon, el cáncer de ovario o el cáncer de próstata. Por tanto, se piensa que no es necesario que las células presenten telomerasa activa para que se desarrolle el cáncer, sino que solo se requiere algún mecanismo que mantenga la longitud de los telómeros.15
De hecho, en algunos cánceres en los que no se detecta la telomerasa existe un mecanismo alternativo de alargamiento de los telómeros, el cual recibe el nombre de ALT (alternative lengthening of telomeres). Además, los tumores telomerasa-positivos pueden pasar a tener el mecanismo ALT y, por tanto, convertirse en resistentes a los fármacos dirigidos frente a la telomerasa. Este mecanismo ALT se basa en copiar la secuencia telomérica mediante recombinación homóloga. Las células que lo presentan suelen tener una longitud de los telómeros muy variada y gran cantidad de ADN telomérico extracromosómico.18
El alargamiento alternativo de los telómeros se ha encontrado en osteosarcomas, tumores pancreáticos neuroendocrinos, tumores astrocíticos, leidomiosarcomas y sarcomas pleomórficos indiferenciados. Se cree que este mecanismo se activa cuando se producen mutaciones en las proteínas ATRX o DAXX y/o en la histona H3.3 o por fallos en su expresión, lo cual se ha observado en tumores del sistema nervioso central y en tumores pancreáticos neuroendocrinos.18 La histona H3.3 se incorpora a regiones teloméricas y sitios de transcripción y se asocia con cromatina activa, para lo cual requiere la presencia de ATRX y DAXX, lo que impide la recombinación telomérica. Si existe una mutación en una de estas proteínas, se alterará la incorporación de la H3.3 a la cromatina, lo que modificará la heterocromatina telomérica. Esto conducirá a una desestabilización de los telómeros y a un aumento de la recombinación homóloga en ellos, facilitándose así el mecanismo ALT. 18, 19, 20 Además, la mutación de los genes que dan lugar a ATRX o a DAXX puede aumentar la expresión del RNA TERRA, que se trata de un RNA no codificante que inhibe la actividad de la telomerasa. Este RNA TERRA también estimula la formación de un “loop R” en los telómeros, lo que induce ALT.17
Envejecimiento.
Uno de los procesos que contribuye al envejecimiento es el acortamiento de los telómeros.2 Se propone que el DNA telomérico, que protege los extremos cromosómicos de la recombinación, actuaría como un “reloj mitótico”, el cual induciría la salida del ciclo celular mediante la senescencia una vez que los telómeros se acorten.6

Para la comprensión del proceso del envejecimiento, se han llevado a cabo estudios en los que se han empleado ratones que no tenían un gen funcional de la telomerasa, por lo que en ellos no se expresaba esta enzima. Se observó que los telómeros de estos ratones se iban acortando progresivamente y, tras varias generaciones, los animales presentaban signos de envejecimiento prematuro (caída del pelo, aparición de canas o retraso en cicatrización de heridas).2
Además, en el año 2012, científicos de Reino Unido realizaron una investigación sobre la longitud de los telómeros en pinzones y observaron que los pájaros que tenían telómeros más cortos eran menos longevos que los pájaros que tenían telómeros más largos.2
Otros estudios han revelado que la disfunción telomérica (relacionada con las proteínas de respuesta al daño del ADN) aumentan con la edad en preparaciones in vivo en la piel de primates y el hígado, tracto digestivo y pulmones de ratones.21
Por tanto, según estos resultados, podemos afirmar que hay cierta relación entre el envejecimiento y la longitud de los telómeros.
La longitud inicial de los telómeros puede variar entre distintos individuos. En mamíferos, la tasa de acortamiento de los telómeros es igual en todos los tejidos del mismo organismo (entre 50 y 200 nucleótidos por duplicación).6, 22
Los telómeros se van acortando progresivamente en cada ciclo celular y esto hace que llegue un momento en el que no se pueda unir el complejo de la shelterina, por lo que se desestabiliza el “loop T”. Esto provoca la exposición del extremo del cromosoma, el cual es reconocido por la maquinaria de reparación del DNA como una rotura de la doble cadena de DNA. De hecho, algunos estudios han demostrado que la eliminación del TRF2 del complejo de la shelterina provocaba el reclutamiento y la activación de proteínas como 53BP1, ATM o la histona H2AX, que están implicadas en la respuesta al daño del DNA. Además, se ha visto que esta respuesta al daño, se produce específicamente en la región telomérica cuando POT1 se separa del telómero. Por otro lado, dicha maquinaria reparativa del DNA también puede dar lugar a la activación de la proteína p53, que estimula la reparación del DNA, la detención del ciclo celular, la senescencia y la apoptosis.21
Se han hallado evidencias de que la senescencia puede estar relacionada con el envejecimiento a través de la acumulación de células con fenotipo senescente en los tejidos, ya que éstas pueden resistir más los estímulos apoptóticos. A su vez, dichas células expresan en mayor frecuencia algunas moléculas de secreción (metaloproteinasas de la matriz junto a otras enzimas capaces de degradar, factores de crecimiento, citocinas inflamatorias), las cuales pueden alterar el entorno local del tejido al que se vierten. La acumulación de dichas células y su hipersecreción provocan la destrucción de la integridad y la funcionalidad tisular.14
Debido a la perturbación tisular, se ha sugerido que las células senescentes puedan facilitar la expresión de fenotipos neoplásicos de otras células mutadas.14
La extensión telomérica ha sido diana de numerosos estudios. Se ha visto que esta longitud telomérica es muy variable entre individuos de la misma edad. Además, dicha longitud pasará a ser más heterogénea entre los diferentes tejidos en la vejez.11
Otro dato de interés que se ha encontrado es que, aunque no haya diferencias de longitud telomérica entre los recién nacidos varones y mujeres afroamericanos y caucásicos, a medida que estas poblaciones pasan a edad adulta, los individuos afroamericanos conservan unos telómeros más extensos que los caucásicos. Además, generalmente los varones adultos tienen los telómeros más cortos que las mujeres adultas. Se piensa que la tasa de acortamiento telomérico es menor en la población femenina debido a la acción estimulante del estrógeno sobre la telomerasa, tal y como se ha comprobado in vitro.11
La longitud de los telómeros estará determinada por factores genéticos y ambientales. Al poseer muchos residuos de G, los telómeros son más susceptibles al estrés oxidativo. Además, se ha comprobado que en células como las del endotelio, el estrés oxidativo disminuye en gran medida la actividad telomerasa. Por lo tanto, si se añaden antioxidantes, se enlentece el acortamiento telomérico al promover a la enzima telomerasa (comprobado en cultivos celulares).11
De esto se deduce lo siguiente: si evitamos el estrés psicológico, se produce menos estrés oxidativo, y por lo tanto prolongamos más la actividad telomerasa, así como los telómeros. Estudios han demostrado que eventos adversos, maltrato infantil, enfermedades crónicas, también parecen ser la causa de unos telómeros más cortos de cara al futuro.11
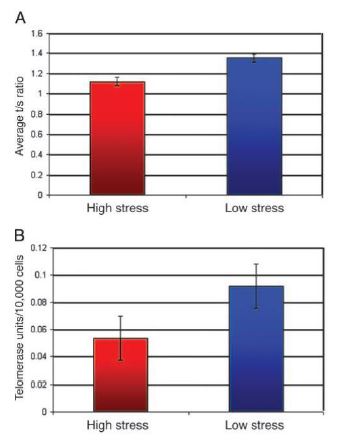
Existen datos sobre los hábitos de vida, que también están relacionados con la longitud telomérica11:
- Los fumadores tienen más estrés oxidativo, por tanto, menor longitud telomérica.
- El deporte influye de manera positiva sobre dicha longitud.
- Personas que, debido a su dieta, presentaban niveles más bajos de ácido docosahexaenoico y ácido eicosapentaenoico, sufrían de un acortamiento telomérico más veloz.

Patologías relacionadas
Disqueratosis congénita/ Síndrome de Zinsser-Engman-Cole
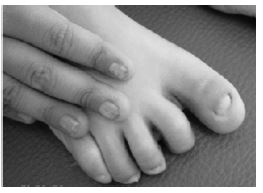
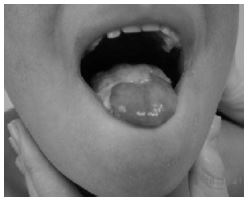
La disqueratosis congénita fue la primera patología en la que se identificaron mutaciones en la telomerasa humana. Dicha enfermedad cursa con: pigmentación anormal de la piel, distrofia de las uñas, y leucoplasia oral (placa blanca localizada en la mucosa oral que puede ser un factor de riesgo para el cáncer oral23). Además hay otro síntomas como tales como retraso en el desarrollo, atrofia testicular, pérdida prematura del cabello e incapacidad funcional de algunos órganos (siendo la deficiencia de la médula ósea, la principal razón de mortalidad prematura).5
En esta patología se ha detectado la mutación del gen DKC1, situado en el cromosoma X, dando lugar a la disqueratosis congénita ligada al cromosoma X. La consecuencia de esto es una sustitución de aminoácidos en la posición 353 de Alanina por Valina, afectando a la disquerina. Dicha proteína es 1 de las 3 de las proteínas accesorias implicadas en la estabilidad y la acumulación de TER (RNA de la telomerasa humana). Debido a esto los niveles de TER están disminuidos, y por tanto, sus telómeros están más reducidos que sus respectivos controles normales.24,25
Se crea inestabilidad cromosómica que afecta más a tejidos de rápida proliferación, tales como: médula ósea, piel y mucosa gastrointestinal.24
Anemia aplásica
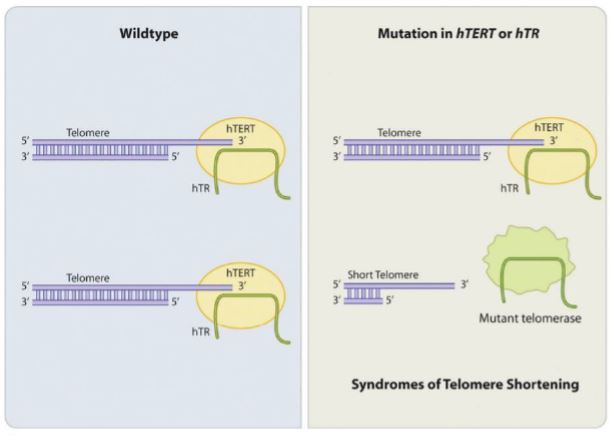
Es un síndrome hereditario que cursa en la disqueratosis congénita autosómica dominante.26
Aparte de los síntomas descritos anteriormente de una disqueratosis congénita, la anemia aplásica produce un trastorno hematológico que viene dado por: la reducción de los eritrocitos, fallo de médula ósea, y patologías hepáticas y pulmonares.5
Este tipo de anemia es ocasionado por una mutación en el gen hTR que codifica para TER.25,26 También se han visto mutaciones en TERT en la disqueratosis congénita autosómica dominante.27
Fibrosis pulmonar idiopática
Es una patología crónica y progresiva que cursa con una fibrosis pulmonar irreversible.5 Desde el diagnóstico, los pacientes viven de promedio 3 años.27
Las bases patológicas radican en la mutaciones de genes codificantes para TER y TERT.27
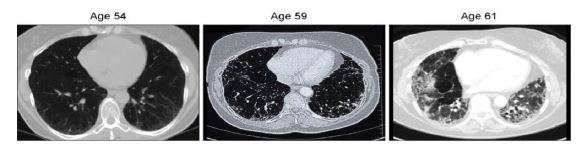
Dicha patología también se asocia con la disqueratosis congénita.27
Síndrome de Werner
El síndrome de Werner es producido por una mutación del gen WRN, que codifica para la helicasa Recq, que es necesaria para la replicación de los telómeros. Cuando esta enzima es defectuosa, los telómeros se acortan de forma prematura, lo que hace que aparezcan signos de envejecimiento en la adolescencia y primeras etapas de la adultez, como piel arrugada, encanecimiento, cataratas o atrofia muscular.2
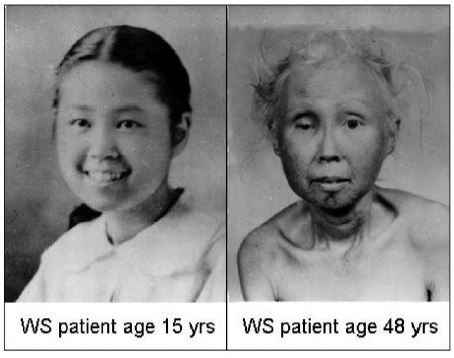
En conclusión, las enfermedades mencionadas anteriormente poseen telómeros más cortos que los controles (sin patologías) con los que son comparados.
Dianas farmacéuticas
Las posibles dianas farmacéuticas en situaciones de cáncer (supresión de telomerasa) y de envejecimiento (activación de la telomerasa), vendrían resumidas en la siguiente imagen28:

La telomerasa es una buena diana terapéutica ya que está muy expresada en las células cancerígenas en comparación con el resto de las células (por ejemplo, las células somáticas tienen expresión casi nula o nula de la telomerasa) . Además, los procedimientos terapéuticos van encaminados a la subunidad catalítica TERT.28
Algunas potenciales terapias son28:
- Inhibidores de oligonucleótidos: son oligonucleótidos antisentido o ácidos nucleicos modificados químicamente. Actúan inhibiendo la telomerasa, concretamente sobre la subunidad TER o TERT, o sobre proteínas asociadas. El acortamiento telomérico produce apoptosis o senescencia. Un ejemplo bastante prometedor es el Imetestalt, cuya secuencia oligonucleotídica es complementaria a TER (hTR o TERC) de la telomerasa, inhibiendo dicha subunidad. Dicho compuesto ha sido testado con éxito en glioblastoma (tumor cerebral).28
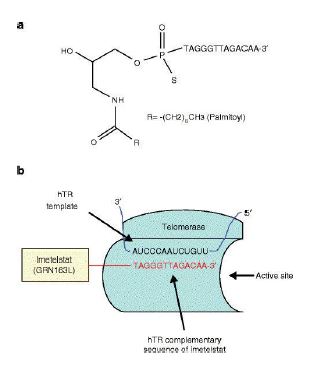
- Inhibidores de molécula pequeña: a partir de inhibidor natural epigalactocatequina-3-galato (EGCG). Por ejemplo, la rapamicina, inhibidor de mTOR, una proteína serín/treonín quinasa encargada de la localización de la subunidad TERT.29
- Terapia génica dirigida a la telomerasa: dicha terapia se dirige al promotor de genes de la telomerasa de células cancerígenas. Se usan Adenovirus, que al usar el promotor de hTERT, son capaces de replicarse y matar a la célula cancerígena infectada.28
- Fitoquímicos: moléculas naturales de plantas que tienen efecto inhibitorio de la telomerasa en algunos cánceres (alicina, curcumina, sibilina, etc…). El mecanismo de acción no se conoce del todo, pero se sugiere que afecta a TERT, ya sea en su expresión, actividad o disociación de la Hsp90 co-chaperona.28
Otro fármaco de importante mención es la telomestatina, un potente inhibidor de la telomerasa extraído de Streptomyces anulatus.30
Dicha molécula posee similitud estructural con el G-cuadruplexo. Su habilidad inhibitoria permite crear con mayor facilidad los G-cuadruplexos o estabilizarlos intramolecularmente, en caso de que ya estuvieran formados.30
De este modo, la telomerasa no puede alargar el telómero porque no es capaz de desorganizar los G-cuadruplexos, y se produce senescencia de tipo Hayflick, que es el envejecimiento celular provocado por desgaste de telómeros.
Conclusión
Los telómeros y la telomerasa son unas estructuras moleculares muy importantes a la hora de comprender procesos naturales como el envejecimiento, y procesos patológicos como el cáncer.
La integridad de los cromosomas en sus extremos está mantenida por la telomerasa, y el acortamiento de dichas zonas produce senescencia, envejecimiento o cáncer.
En muchas ocasiones, el cáncer presentará aumentada la actividad telomerasa, por lo que dicha enzima es una diana terapéutica bastante prometedora a la hora de intentar erradicar patologías de origen tumoral.
Hay que tener en cuenta que la telomerasa es un elemento destacado en todos los procedimientos relacionados con los telómeros, pero no hay que olvidar la interacción que ejercen otros factores conocidos y los que faltan por descubrir.
De cara al futuro, la investigación en este campo permitirá una mejor comprensión global de los telómeros, con importantes repercusiones clínicas.
Bibliografía
- Chuaire, L. (2006). Telómeros y telomerasa: breve recuento de una historia iniciada por Hermann Müller y Barbara McClintock. Colombia Médica, 37(4), 332-336.
- Pierce, B. A. (2016). Genética: Un enfoque conceptual. Madrid, España: Ed. Médica Panamericana.
- Hernández Fernández, R. A. (1999). Telómeros y telomerasas. Revista Cubana de Investigaciones Biomédicas, 18(2), 121-129.
- Blackburn, E. H. (1991). Structure and function of telomeres. Nature, 350(6319), 569.
- Gómez, D. E., Armando, R. G., & Farina, H. G. (2014). Telomerasa y telómero: su estructura y dinámica en salud y enfermedad. Medicina (Buenos Aires) 74, 69-76
- Cottliar, A. S., & Slavutsky, I. R. (2001). Telómeros y actividad de telomerasa: su participación en el envejecimiento y el desarrollo neoplásico. Medicina (Buenos Aires), 61, 335-42.
- Maciejowski, J., & de Lange, T. (2017). Telomeres in cancer: tumour suppression and genome instability. Nature reviews Molecular cell biology, 18(3), 175.
- Shay, J. W. (2018). Telomeres and aging. Current opinion in cell biology, 52, 1-7.
- Sandin, S., & Rhodes, D. (2014). Telomerase structure. Current opinion in structural biology, 25, 104-110.
- Paniagua, R., Nistal, M., Sesma, P., Álvarez-Uría, M., Fraile, B., Anadón, R. & Sáez, F. J. (2002). Citología e histología vegetal y animal. Madrid, España: McGraw-Hill Interamericana.
- Zhu, H., Belcher, M., & Van Der Harst, P. (2011). Healthy aging and disease: role for telomere biology? Clinical science, 120(10), 427-440.
- Van Steensel, B., & De Lange, T. (1997). Control of telomere length by the human telomeric protein TRF1. Nature, 385(6618), 740.
- Ulaner, G. A., Hu, J. F., Vu, T. H., Giudice, L. C., & Hoffman, A. R. (1998). Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer research, 58(18), 4168-4172.
- Pardo Andreu, Gilberto, & Delgado Hernández, René. (2003). Senescencia celular y envejecimiento. Revista Cubana de Investigaciones Biomédicas, 22(3), 204-212.
- Shay, J. W. (2016). Role of telomeres and telomerase in aging and cancer. Cancer discovery, 6(6), 584-593.
- Jafri, M. A., Ansari, S. A., Alqahtani, M. H., & Shay, J. W. (2016). Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome medicine, 8(1), 69.
- Akincilar, S. C., Unal, B., & Tergaonkar, V. (2016). Reactivation of telomerase in cancer. Cellular and Molecular Life Sciences, 73(8), 1659-1670.
- Shay, J. W., Reddel, R. R., & Wright, W. E. (2012). Cancer and telomeres—an ALTernative to telomerase. Science, 336(6087), 1388-1390.
- Heaphy, C. M., De Wilde, R. F., Jiao, Y., Klein, A. P., Edil, B. H., Shi, C., … & Offerhaus, G. J. (2011). Altered telomeres in tumors with ATRX and DAXX mutations. Science, 333(6041), 425-425.
- Schwartzentruber, J., Korshunov, A., Liu, X. Y., Jones, D. T., Pfaff, E., Jacob, K., … & Hovestadt, V. (2012). Driver mutations in histone H3. 3 and chromatin remodelling genes in paediatric glioblastoma. Nature, 482(7384), 226.
- Victorelli, S., & Passos, J. F. (2017). Telomeres and cell senescence-size matters not. EBioMedicine, 21, 14-20.
- Bernadotte, A., Mikhelson, V. M., & Spivak, I. M. (2016). Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging (Albany NY), 8(1), 3.
- Moles, M. G., & González-Ruiz, L. (2018). Leucoplasia oral, una revisión de los aspectos esenciales de su diagnóstico y tratamiento. Actualidad médica, 103(803), 44-46.
- Smoje, G., Dal Borgo, A., Cuevas, M., Núñez, L., Bolte, C., & Martinez, W. (2004). Disqueratosis congénita ligada al cromosoma X. Revista chilena de pediatría, 75(6), 547-550.
- Núñez Quintana, A., Nordet Carrera, I., Menéndez Veitía, A., & González Otero, A. (2004). Neutropenias congénitas. Revista Cubana de Hematología, Inmunología y Hemoterapia, 20(1)
- Vulliamy, T., Marrone, A., Dokal, I., & Mason, P. J. (2002). Association between aplastic anaemia and mutations in telomerase RNA. The Lancet, 359(9324), 2168-2170.
- Armanios, M. (2012). Telomerase and idiopathic pulmonary fibrosis. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 730(1-2), 52-58.
- Jäger, K., & Walter, M. (2016). Therapeutic targeting of telomerase. Genes, 7(7), 39.
- Miwa, S., & Saretzki, G. (2017). Telomerase and mTOR in the brain: the mitochondria connection. Neural regeneration research, 12(3), 358.
- Kim, M. Y., Vankayalapati, H., Shin-Ya, K., Wierzba, K., & Hurley, L. H. (2002). Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular G-quadruplex. Journal of the American Chemical Society, 124(10), 2098-2099.