EL ÁNTRAX UN PROBLEMA DE AYER Y DE HOY
Isabel Pérez González e Isabel Serrano Fernández 1º Biología Sanitaria UAH
1.- En líneas generales:
La enfermedad del ántrax es una enfermedad infecciosa ocasionada por la bacteria Bacillus anthracis. Se denomina de múltiples formas: Carbunco bacteridiano o maligno, enfermedad de los laneros o traperos, úlcera de Siberia, etc.
La enfermedad posee como características principales una alta virulencia, elevada mortalidad y gran resistencia en el medio, debido a que es formador de esporas. Afecta a bastantes especies, siendo la de más relevancia, por gravedad e incidencia, los herbívoros, aunque afecta también a humanos.
En Europa, sólo se dan casos de ántrax en países en los que la enfermedad es endémica (Turquía, Albania, España, etc).
2.- Un poco de historia:
La enfermedad del ántrax fue descrita por primera vez por los griegos quienes la designaron como “antrhakis” que significa carbón, debido al parecido de dicha roca con las lesiones de color negro que se producen en la piel de quienes padecen la enfermedad en su forma cutánea. Por este mismo motivo los romanos la llamaron carbunco, que también significa carbón.
Los primeros estudios sobre la enfermedad del ántrax se remontan al siglo XVII. En 1681, el médico militar italiano G. B. Ramazzini (1633-1714) describió una enfermedad que afectaba a la piel y que presentaba lesiones negras. Posteriormente, en 1771, el doctor José Antonio Pavia (1735-1793) describió el síndrome característico de la fiebre del ántrax y el doctor Johann Heinrich Müller (1734-1815) describió las lesiones cutáneas que acompañaban a esta enfermedad.
No fue hasta el siglo XIX cuando, gracias a los experimentos de científicos como Pierre Rayer (1850), Casimir-Joseph Davaine (1862) y Tiegel y Klebs (1864), se demostró que la causa de la enfermedad del ántrax era una bacteria y que, por tanto, se trataba de una enfermedad infecciosa.
El Bacillus anthracis, bacteria causante de la enfermedad, fue objeto de investigación por parte de Robert Koch (1876) en el campo de la bacteriología, desmintiendo la teoría de la generación espontánea e introduciendo conceptos como gérmenes y agente microbianos. Koch fue también quien observó por primera vez el ciclo de vida de la bacteria y quien demostró que podía formar esporas altamente resistentes dentro de sí misma, especialmente en condiciones anaeróbicas. Cuando las condiciones eran favorables dichas esporas se volvían bacilos tratándose, por tanto, de un mecanismo de autoprotección.
Fue el botánico francés Louis Pasteur (1822-1895) quien en el año 1881, encontró la forma de destruir la bacteria del ántrax y fue el encargado de desarrollar la primera vacuna contra la enfermedad tras aislar una cepa atenuada.
3.- Forma y mecanismo de acción:
La enfermedad del ántrax es causada por la bacteria Bacillus anthracis que es una bacteria Gram positiva, aerobia, encapsulada, no móvil, formadora de endoesporas que le permiten sobrevivir en condiciones de falta de oxígeno (anaerobia facultativa), pero la formación de dichas esporas necesita de oxígeno. La forma infectiva son sus esporas, que es la forma resistente de este microorganismo y que tiene gran resistencia al medio e infectan a los herbívoros cuando estos se alimentan de pastos contaminados por esporas. Tales esporas son capaces de vivir más de 20 años y su letalidad es de un 80% cuando el individuo se infecta.
Cómo ya hemos dicho la enfermedad es causada por la bacteria Bacillus anthracis, más concretamente las características que ésta presenta en su cápsula y los componentes de la toxina que fabrica, son los factores que hacen que esta bacteria sea potencialmente peligrosa.
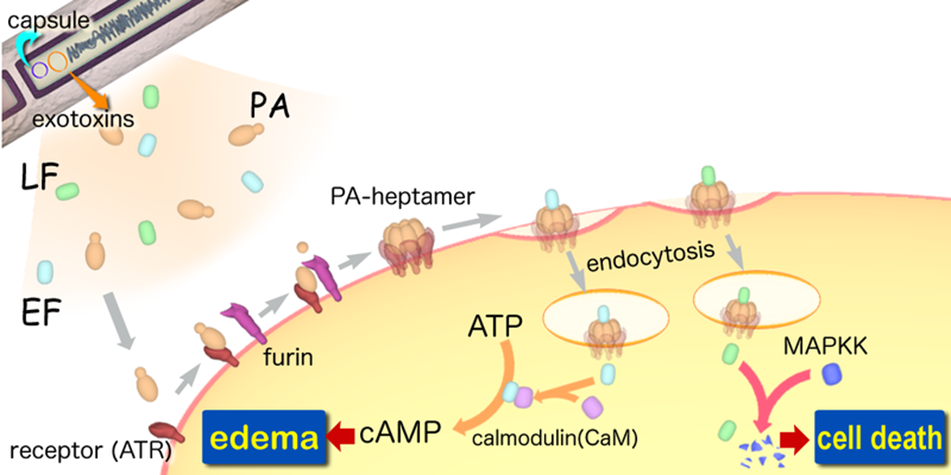
Imagen 1.: Ciclo patogenia ántrax
Cápsula:
La cápsula está compuesta por un polipéptido de ácido D-glutámico, no es tóxica en sí misma lo que permite evitar el sistema inmune del hospedador ya que no activa la respuesta inmune y, además, protege a la bacteria contra la fagocitosis.
La cápsula también desempeña un papel importante durante el establecimiento de la infección y es la encargada de evitar la coagulación de la sangre del hospedador, también es la causante de las reacciones inflamatorias y necróticas.
Además, la presencia de esta cápsula condiciona la virulencia de Bacillus anthracis ya que es la encargada de la diferenciación de la bacteria en dos variantes: lisa (S) y áspera (R), siendo la variante R menos virulenta que la S.
Exotoxinas:
Las toxinas del ántrax son el principal factor de virulencia del Bacillus anthracis. Son producidas exclusivamente en los tejidos de los animales infectados y vienen codificadas en los plásmidos pXO1 y pXO2. Están conformadas por las distintas uniones de tres polipéptidos conocidos como: factor edema, factor letal y antígeno protector.
Individualmente ninguna de estas tres proteínas es tóxica pero cuando el antígeno protector se combina con el factor edema forman la toxina edema y cuando se combina con el factor letal forman la toxina letal, siendo estas toxinas las que producen la muerte del animal.
La combinación de las diferentes proteínas se corresponde al modelo de toxinas active-binding en el que hay un único componente central, en este caso PA, al que se pueden asociar dos componentes distintos (EF y LF). Según este modelo el PA es el componente que se une al receptor celular para introducir a los otros dos componentes (EF y LF).
– Factor edema: es una adenilato ciclasa dependiente de calmodulina su peso molecular es de 89 kDa y tiene una secuencia de 767 aminoácidos. Convierte, consumiendo ATP en el proceso, la adenosina trifosfato en adenosina monofosfato cíclico (AMPc), lo que causa la aparición de un edema. También inhibe la función de los neutrófilos y produce la lisis de los macrófagos. El EF depende de iones de calcio, magnesio y calmodulina para su actividad. En su región amino-terminal se encuentran los aminoácidos responsables de la unión a PA, mientras que en la región carboxilo-terminal se encuentra el sitio catalítico y el dominio de activación dependiente de calmodulina. El factor edema permanece asociado al endosoma tardío luego del ingreso a la célula, con su región catalítica expuesta hacia el citoplasma.
– Factor letal: tiene un peso de 90 kDa, está compuesta por 776 aminoácidos y es una metaloproteasa dependiente de zinc. Es altamente específica y dentro del macrófago induce un influjo de calcio y la inhibición de la síntesis de macromoléculas, además, promueve la apoptosis de los macrófagos. Su actividad radica en cortar la región amino-terminal de una familia de quinasas (MAPKK), inactivando de esta forma diferentes mecanismos de señalización celular. LF presenta cuatro dominios, el dominio 1 participa en la unión a antígeno protector, el dominio 2 está involucrado en la unión al sustrato MAPKK, el dominio 3 se encuentra dentro del dominio 2 pero tiene un plegado independiente y el dominio 4 contiene un centro catalítico.
Las proteínas EF y LF inhiben la secreción del Tumor Necrosis Factor-α (TNF-α) (22) y suprimen la respuesta inmunológica del hospedador permitiendo la propagación de Bacillus anthracis.
– Antígeno protector (PA): tiene un peso molecular de 83 Kd y está compuesto por 735 aminoácidos. Está formado por dos subunidades que se separan tras la unión del PA a la célula. Su función es la de adherirse a la superficie de la célula y facilitar la entrada de los otros dos factores. El antígeno protector posee cuatro dominios que son necesarios para que se produzca la intoxicación de la célula. En el dominio 1 amino-terminal se encuentra la región de corte para la activación, una secuencia de aminoácidos que estabiliza la estructura mediante la fijación de iones de calcio y la región de unión con LF Y EF. El dominio 2 se denomina dominio de heptamerización ya que es el implicado en la inserción del complejo en la membrana. Por otro lado el dominio 3 posee una zona hidrofóbica y participa en la oligomerización de PA a heptámero. Por último el dominio 4, carboxilo-terminal, es el que participa en la unión al receptor.
El proceso que sigue Bacillus anthracis para infectar a las células comienza con la unión del antígeno protector (PA), que es una proteína madura de 83 kDa, a dos receptores celulares: el marcador tumoral endotelial-8 y al gen de morfogénesis capilar 2. Tras esta unión el antígeno protector es clivado por una furina quedando su región carboxilo-terminal de 63 kDa unida a la superficie celular y liberándose la subunidad de 20 kDa. Para que se puedan unir los componentes EF y LF al antígeno protector es necesaria la eliminación del amino terminal.
Tras esto se produce la oligomerización del factor antígeno que pasa a formar un heptámero al que se unen el factor edema y el factor letal, que, ya que tienen regiones homólogas entre sí generan que la unión sea por competencia.
Este complejo proteico penetra en la célula por endocitosis tras sufrir cambios conformacionales que le permiten insertarse en la membrana. Tras esto el factor letal es introducido en el citosol de la célula y el factor edema en la membrana del endosoma.
 |
|
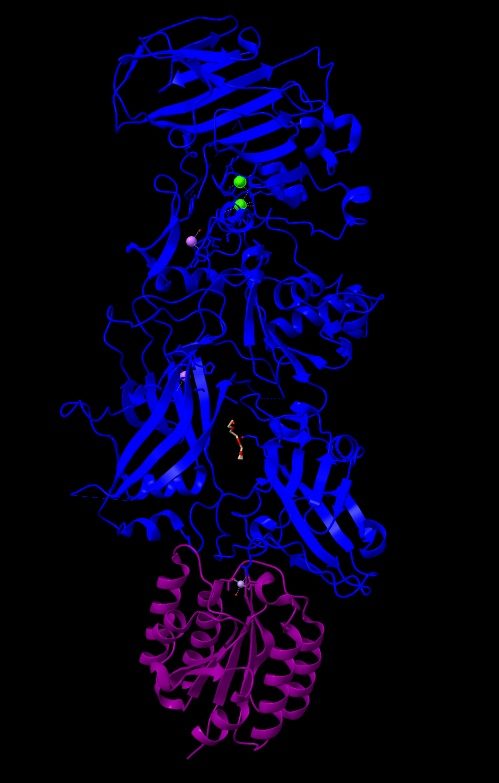
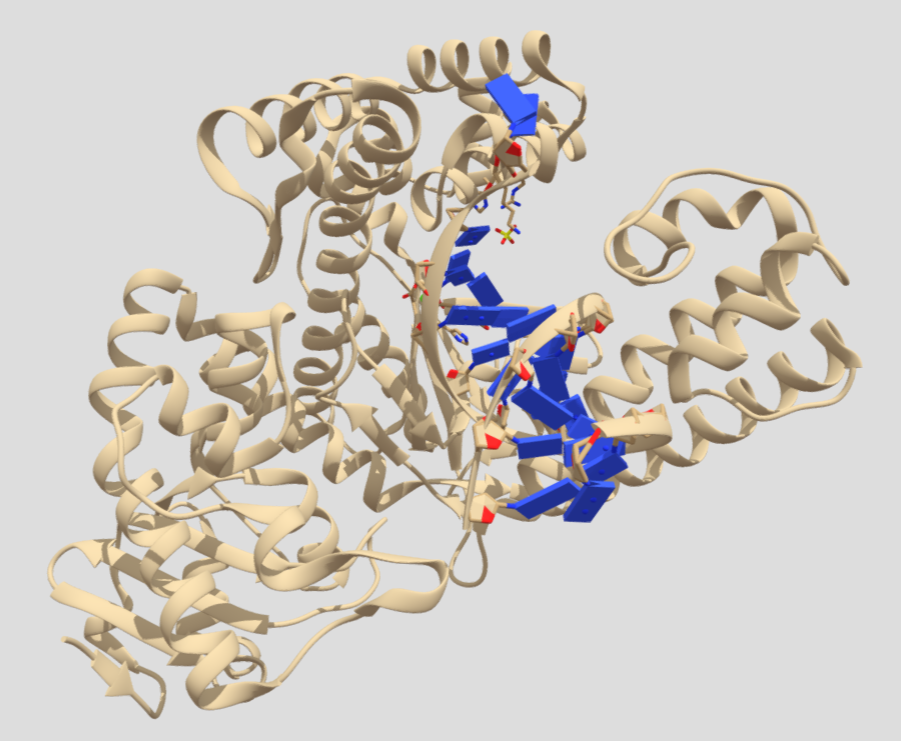
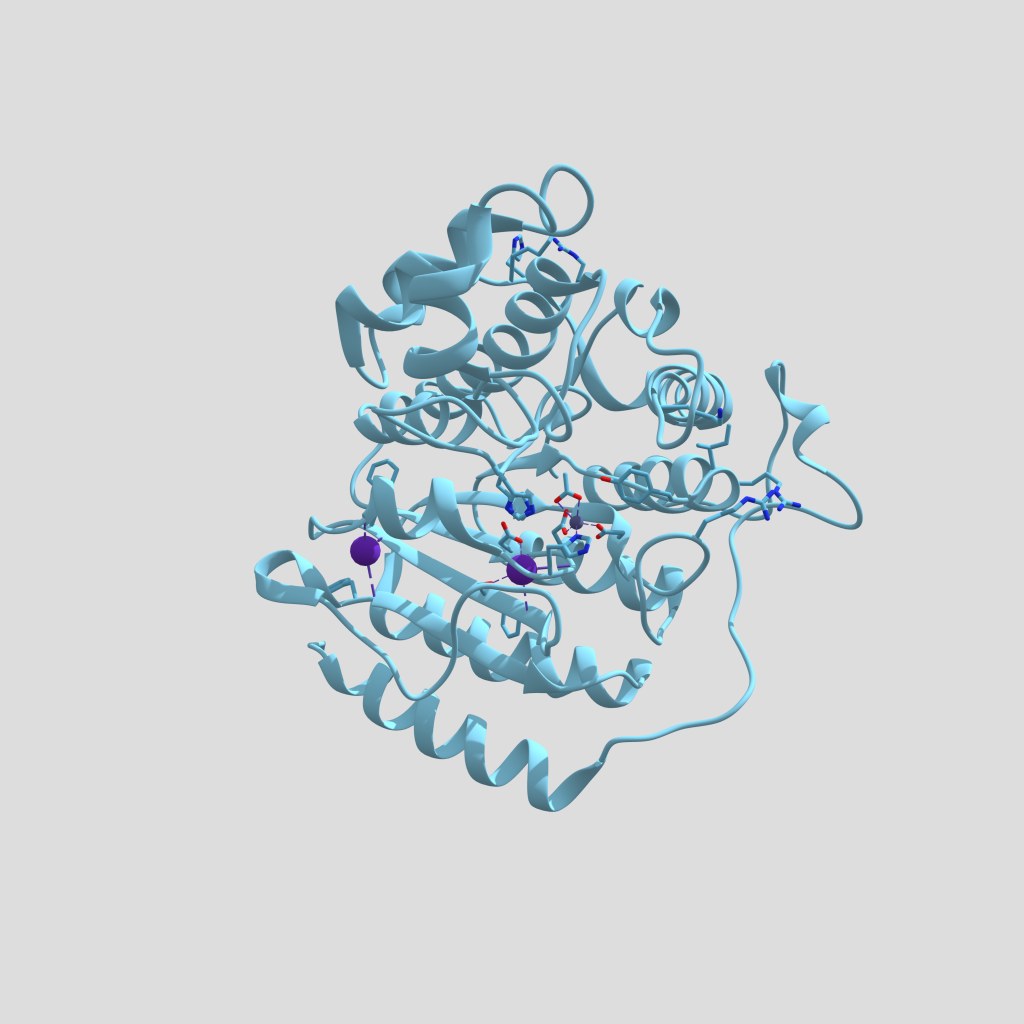
Imagen 2: Estructura cristalina del PA de B. anthracis complejado por receptor CMG2. Fuente: PDB 1T6B.
En esta imagen podemos observar en azul el antígeno protector (PA), que posee 5379 átomos y 676 residuos. De color púrpura el receptor CMG2 que tiene 1316 átomos y 170 residuos. El peso total de la estructura es 103.77 kDa, y revela una superficie muy grande de interacción entre PA y CMG2 que, este a su vez tiene 2 dominios con PA y modela el heptámero (PA63), actuando como sistema sensible al pH y, asegurando una integración perfecta en la membrana.
Toda la estructura posee 4 ligandos no estándares:
1.- Ion Calcio (Ca++); nos encontramos 2 átomos, con un peso molecular de 40.08 u, unidos fuertemente por enlaces covalentes al PA. En la siguiente imagen los veremos de color amarillo.
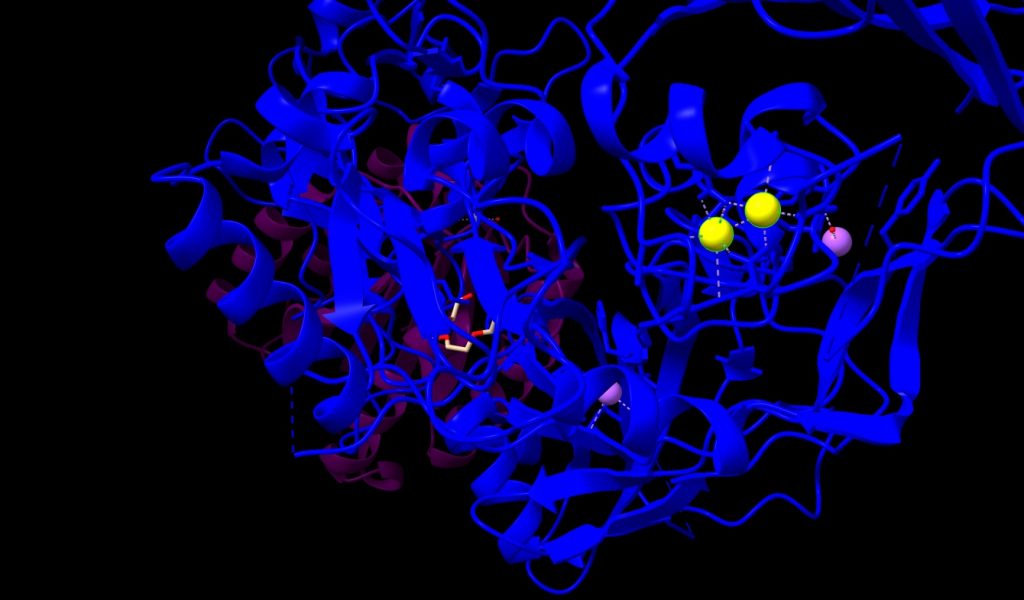
Imagen 3: átomos de Ca++, en estructura de PA de Bacillus anthracis. Fuente: PDB 1T6B.
2.- Ion Manganeso (Mn++); esta estructura posee 1 átomo, su peso molecular es de 54.94 u, unido al receptor de membrana CMG2, y muy próximo a PA. Lo veremos de color naranja, en la imagen que mostramos a continuación.
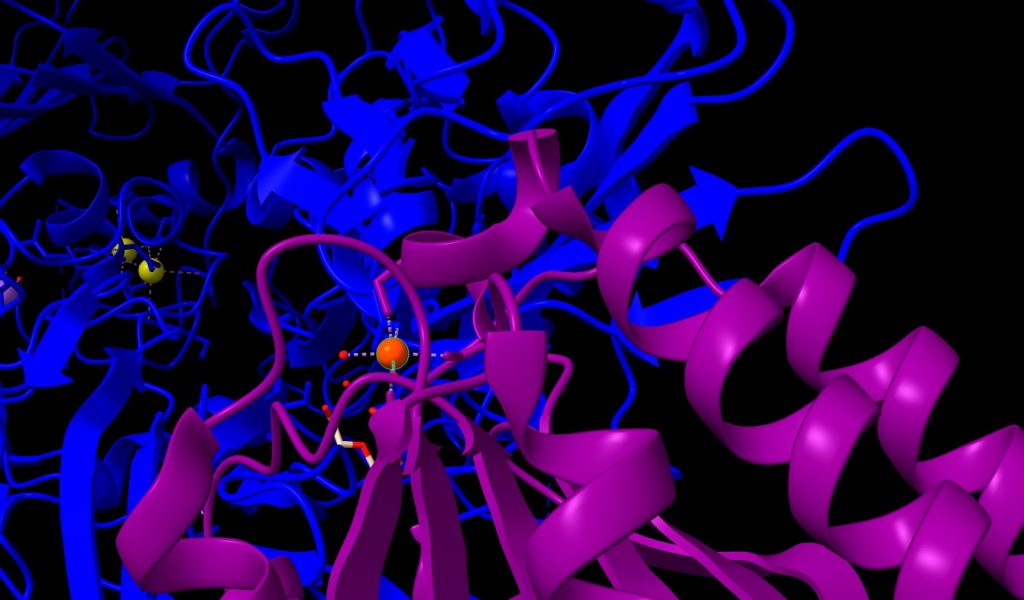
Imagen 4: átomo de Mn++ en receptor de membrana CMG2. Fuente: PDB 1T6B.
3.- Ion Sodio (Na+); podemos ver 2 átomos de Na+, unidos a PA con un peso molecular de 22.99 u. Los identificaremos de color verde, en la imagen siguiente.
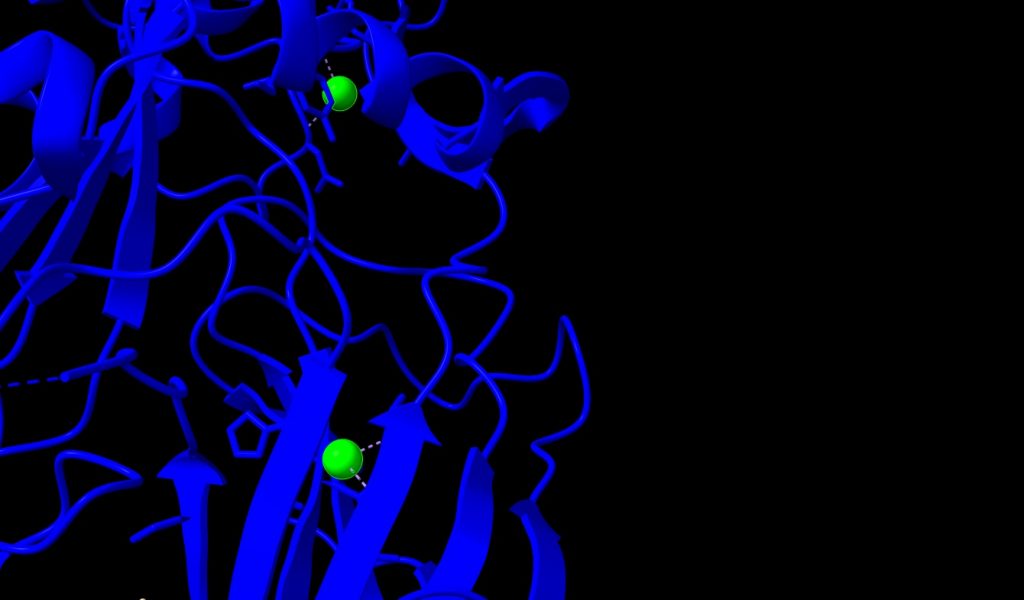
Imagen 5: átomos de Na+ unidos a PA de Bacillus anthracis. Fuente: PDB 1T6B.
4.- Tetraetilenglicol (C8H18O5); es una cadena de 13 átomos, con 8 C, y 2 grupos OH terminales, está unido al PA y todo el conjunto de átomos tiene un peso molecular de 194.23. En la imagen que se muestra a continuación, veremos los O de color rojo y los C de color azul turquesa.
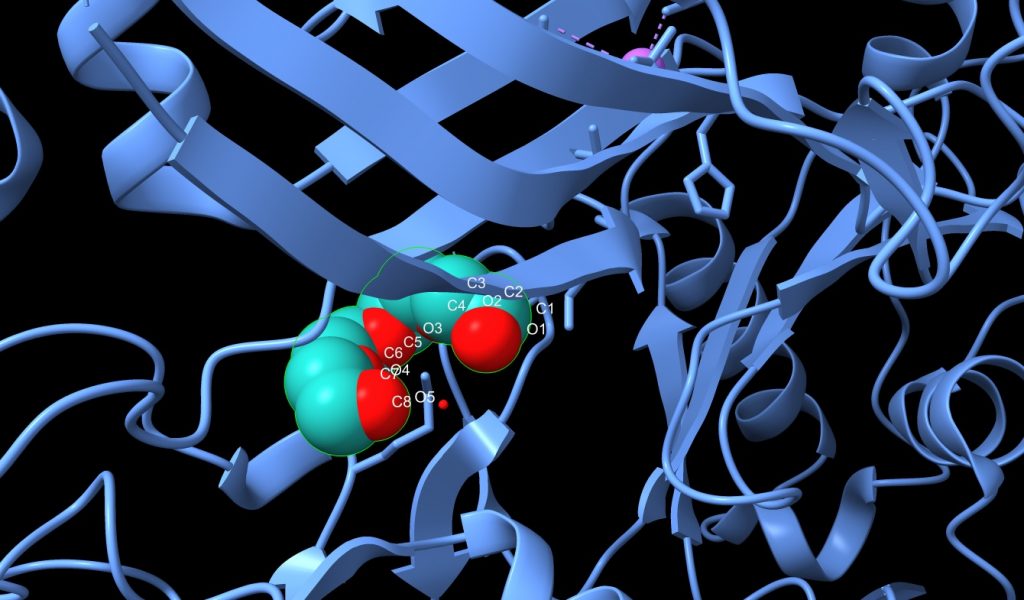
Imagen 6: cadena de Tetraetilenglicol unido a PA de Bacillus anthracis. Fuente: PDB 1T6B.
4.- Etiopatogenia e identificación:
La aparición de la enfermedad por Bacillus anthracis se da tras un período de incubación de unos 20 días (varía dependiendo del tipo de infección). La transmisión se da, generalmente, por vía oral cuando los animales herbívoros comen pastos contaminados. También es posible el contagio por vía respiratoria, cuando se inhalan las esporas, aunque se considera poco frecuente. Algunos estudios hablan, que también es posible la vía indirecta, a través de vectores como los tábanos, que pueden portar la bacteria tras haber estado en contacto con algún individuo infectado.
En el ser humano, el Ántrax puede manifestarse de 3 formas distintas:
- Forma cutánea; aparece entre el 1º y 7º día tras haber estado expuesto. Es la forma más común, y la vía de entrada del microorganismo es, a través de heridas en la piel. Comienza como una lesión sin prurito, con eritema y edema. Se desarrolla una vesícula, que después se rompe y da lugar a una placa necrótica. Suelen aparecer en cabeza, manos y piernas. Su pronóstico es favorable, pero en el caso de no tratarse, las lesiones pueden llegar a evolucionar a una septicemia generalizada, y producir la muerte en un 10% de los casos.
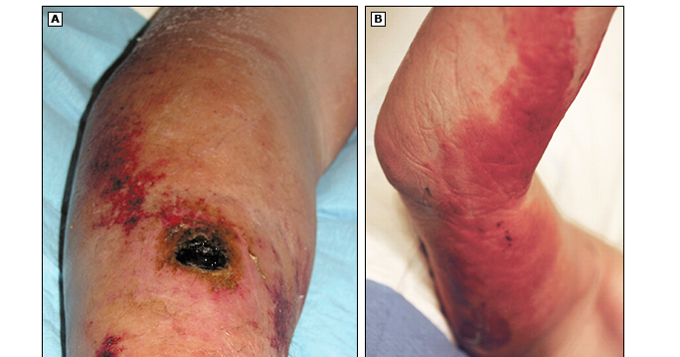
Imagen 7: Carbunco cutáneo en brazo de humano
- Forma intestinal; la vía de infección se da por el consumo de alimentos contaminados. mal cocinados o tratados. El período de incubación de esta forma es desde horas, hasta 1 o 2 semanas. La tasa de mortalidad de esta forma es de un 25-60%. Los síntomas son vómitos, diarrea severa, fiebre, dolor abdominal, etc.
- Forma pulmonar; la vía de entrada del microorganismo se da por la inhalación de las esporas del ántrax, pudiendo desarrollar ántrax pulmonar. El período de incubación es de 1 semana aproximadamente. Las profesiones proclives a padecer este tipo de afección son, aquellas que trabajan en mataderos, procesadoras de lana, curtidores, etc, cuyos productos pueden venir de animales infectados.
Según el CDC, solo alrededor del 10-15% de las personas con ántrax pulmonar, que no reciban tratamiento, sobreviven. Sin embargo, un 55% aproximadamente, con un tratamiento intensivo, puede salvarse. Los síntomas son parecidos a los de una gripe, fiebre, tos, disnea, dolor muscular, etc.

Imagen 8: Radiografía torácica de paciente con ántrax pulmonar
Aparte del ser humano, también se infectan otras especies, que tiene relevancia destacar por salud pública.
Generalmente, se da en herbívoros y, en concreto: bovinos, ovinos, caprinos, equinos, porcinos y carnívoros.
Cabe destacar el carácter zoonósico de la enfermedad, aunque los casos son extremadamente raros, ya que, la seguridad alimentaria es exhaustiva en países desarrollados.
5.- Diagnóstico
El diagnóstico del Carbunco se realiza con muestras de sangre de individuos que se piensen estén infectados o también con hisopos de las mucosas de individuos que hayan muerto por dicha enfermedad.
Las técnicas a realizar para la identificación del Bacillus anthracis son:
- Cultivo bacteridiano; el medio de cultivo más empleado es el agar sangre de caballo u oveja al 5-7%. Las colonias se mostrarán como blanco-grisáceas, no hemolíticas y superficie mate, siendo pegajosas al contactar con asa de siembra. Mediante Tinción de Gram, se observarán bacilos Gram +, abastonados, encapsulados no móviles y con esporas en su interior. También se puede realizar mediante PCR, para identificar y diferenciar cepas patógenas y vacunales.
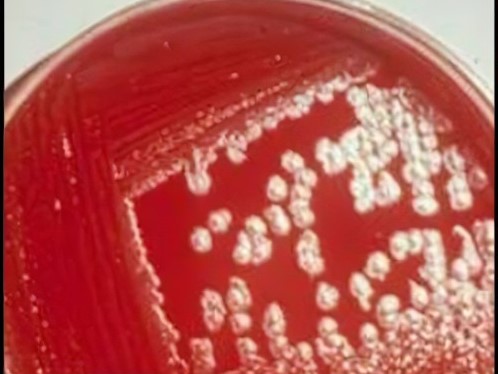
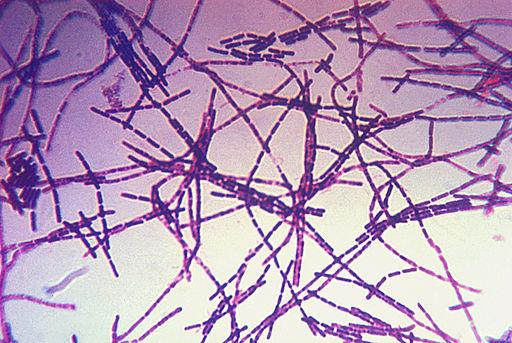
Imagen 9.1: Agar sangre con colonias de Imagen 9.2: Tinción de Gram de Bacillus
Bacillus anthracis, anthracis.
- Visualización de la cápsula; la cápsula del microorganismo se puede visualizar en un frotis sanguíneo, realizando una tinción de azul de metileno policrómico, lo que confiere a la cápsula un color rosáceo, mientras que los bacilos se tienen de un color azul oscuro. Los bacilos se agrupan en pares o cadenas cortas en bisel, conocido como “vagones de ferrocarril”.
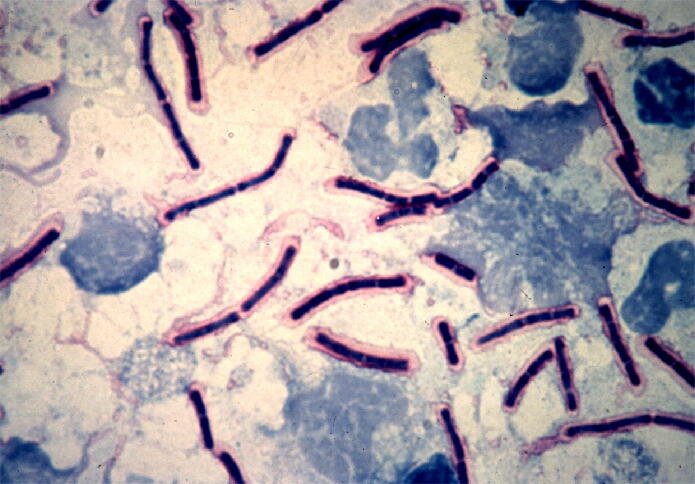
Imagen 10: Tinción de azul de metileno policrómico en frotis sanguíneo.
- Pruebas inmunológicas; realizando técnicas de inmunofluorescencia se puede visualizar las cápsulas, aunque no es una técnica rutinaria para la detección del Bacillus anthracis.
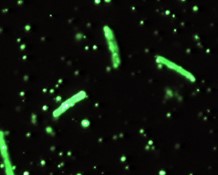
Imagen 11: Anticuerpos fluorescentes de la cápsula de Bacillus anthracis.
- Otras pruebas; en 1991 se desarrolló un método para la detección del microorganismo, empleando un antisuero pero, no presentaba mucha especificidad, porque es común a otras especies de Bacillus. También se puede emplear la lisis del fago gamma y la sensibilidad a la penicilina.
6.- Tratamiento contra ántrax
El tratamiento de elección para tratar el Carbunco, son los antibióticos. Los recomendados son Ciprofloxacino o la Doxiciclina por vía intravenosa, hasta conocer los resultados del antibiograma.
Para tratar el ántrax, se recomienda usar 2 o más antibióticos por la letalidad de este microorganismo. A su vez, las penicilinas, cefalosporinas o cotrimoxazol, están contraindicadas por que las cepas suelen ser resistentes a estos antibióticos.
7.- Ántrax como arma biológica
Debido a las propiedades y características del ántrax, este ha sido utilizado como arma biológica en numerosas ocasiones desde hace unos 80 años. La técnica utilizada es la liberación de esporas de ántrax en forma de aerosol ya que de las tres formas en las que se puede manifestar la enfermedad, cutánea, digestiva y respiratoria, esta última es la más letal. En 1979 tuvo lugar la liberación accidental de esporas de ántrax en Sverdlovsk donde se infectaron 79 personas de las que murieron 68.
De los numerosos ataques con ántrax uno de los más recientes tuvo lugar en 2001 cuando varios políticos y periodistas de EEUU fueron infectados con esporas de ántrax presentes en su correspondencia.
La Organización Mundial de la Salud estimó que, la liberación de 50 kilos de ántrax en una población urbana de cinco millones afectarían a 250 000 habitantes, de los cuales 100 000 morirían, demostrando así la letalidad de la bacteria.
El Bacillus anthracis en España, se encuentra incluido en el Anexo II del R.D. 664/1997 y, está clasificado como agente biológico del grupo 3, que es aquel que puede causar una enfermedad grave en el ser humano y, presenta un serio problema para los trabajadores, con riesgo de propagarse a la colectividad, existiendo profilaxis o tratamiento eficaz. En España es una enfermedad de declaración obligatoria (EDO).
8. Bibliografía
- Campos Perelló, L., & Pastor Armendariz, M. E. (2015). Caracterización analítica de las toxinas de Bacillus anthracis cepa Sterne 34F2.
- Cabezas Sánchez, C., Vargas Herrera, J., Suárez Moreno, V., Herrera Bernuy, S., Mostorino Elguera, R., Morales de Santa Gadea, S., & Guillen Oneeglio, A. (2006). El ántrax: un problema de salud pública vigente.
- PAVAN, MARÍA E., & PETTINARI, MARÍA J., & CAIRÓ, FABIÁN, & PAVAN, ESTEBAN E., & CATALDI, ANGEL A. (2011). Bacillus anthracis: una mirada molecular a un patógeno célebre. Revista Argentina de Microbiología, 43(4),294-310.[fecha de Consulta 16 de Enero de 2022]. ISSN: 0325-7541. Disponible en: https://www.redalyc.org/articulo.oa?id=213021188010
- Department of Health and Human Services. (2014). Resumen del ántrax U.S. Department of Health and Human Services Centers for Disease Control and Prevention Guía básica para comprender el ántrax. Mayo 2019, de Centers for Disease Control and Prevention
- https://www.oie.int/fileadmin/Home/esp/Health_standards/tahm/3.01.01_Carbunco_bacteridiano.pdf































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}