El plegamiento de proteínas y las mutaciones del código genético
C. Menor-Salván ,oct 2022. V2 Nov 2023
En el año 1969 ya se conocía mucho sobre la estructura de las proteínas y hacía 10 años que había nacido la Biología Molecular moderna. Ya se entendía la relación entre el código genético, la secuencia de aminoácidos de un péptido y la proteína final. Poco tiempo antes, el bioquímico Christian Anfinsen había descubierto que una proteína desnaturalizada podía recuperar su estructura y actividad en cuestión de milisegundos, por lo que quedaba demostrado que la estructura estaba, de alguna manera, codificada en su secuencia.
Para Cyrus Levinthal esto era paradógico: una secuencia de aminoácidos tiene un número enorme de posibilidades estructurales, por lo que las diferentes conformaciones posibles no pueden tener la misma probabilidad y el plegamiento no es resultado de la búsqueda de la conformación nativa, sino que debía ser dirigido por un camino específico y predeterminado. La resolución de la paradoja de Levinthal, es decir, entender cómo se pliegan las proteínas, es uno de los problemas más complejos de la Bioquímica y la Biofísica, y sólo ahora, gracias a la inteligencia artificial y a los métodos computacionales modernos, estamos en condiciones de resolverla completamente.

El plegamiento proteico es un problema muy complejo, incluso para las células que generan las proteínas: los fallos o, simplemente, las diferentes soluciones al plegamiento estable de un péptido están detrás de enfermedades como el Alzheimer ,las enfermedades por priones o las amiloidosis. Pero, aunque sea tan complejo, podemos señalar algun aspecto interesante, como es el papel esencial de los aminoácidos hidrofóbicos y su relación con la evolución del código genético.

El colapso hidrofóbico
El proceso de plegamiento proteico es gradual, formándose regiones, llamadas foldones, que van adquiriendo conformaciones similares a la forma nativa final. Estos foldones son cooperativos y, cuando empiezan a formarse, favorecen el plegamiento del resto de la proteína. Esto se denomina la hipótesis del foldón.
En la formación de las regiones plegadas es esencial la estabilización energética que proporciona el efecto hidrofóbico, es decir, la expulsión del agua y el secuestro de los grupos R de los aminoácidos hidrofóbicos en el interior de la estructura plegada, dejando los grupos polares y cargados expuestos al solvente. Este efecto es uno de los responsables de la extraordinaria capacidad de las enzimas como catalizadores: al tener un núcleo hidrofóbico, la exclusión del agua y la desolvatación de los sustratos de las enzimas promueven la actividad enzimática.
Esta fase de plegamiento de la proteína, en la que adquiere su estructura nativa en regiones y se ha producido el colapso hidrofóbico, es muy dinámica: la estructura se mueve, los foldones se forman y reconvierten de un modo fluido. Este estado se denomina glóbulo fundido. Finalmente, la proteína alcanza un estado similar a un sólido tridimensional y se estabiliza, formando el estado nativo de la proteína. En este estado, aparecen otras interacciones, como los puentes disulfuro, el stacking de residuos de aminoácidos aromáticos y la unión de cofactores, que terminan de ‘fijar’ la estructura nativa.
En estas condiciones, tenemos la proteína plegada y funcional. Así puede cristalizar, lo cual es muy importante para conocer la estructura. Pero el estado nativo no es totalmente sólido: la proteína puede desnaturalizarse, o volver al estado de glóbulo fundido y cambiar sus conformaciones en respuesta a algún estímulo.
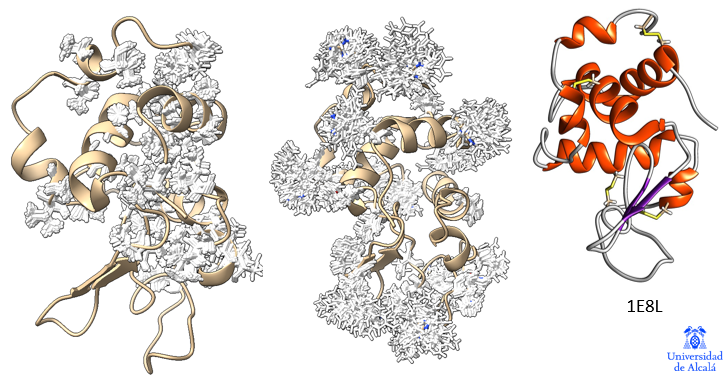

La idea del colapso hidrofóbico puede parecer contraintuitiva: como es posible que una fuerza débil, como la fuerza de Van der Waals entre grupos apolares, sea lo que dirige el plegamiento de la proteína. ¿cual es el papel de los enlaces de hidrógeno?. Al principio se pensaba que los enlaces de hidrógeno, mucho mas fuertes, eran esenciales en la estabilización de la estructura proteica. Actualmente sabemos que los enlaces de hidrógeno juegan un papel director durante el proceso de plegamiento, ya que dirigen la formación de diversas estructuras secundarias, que se agrupan en motivos y van dando lugar al proceso cooperativo de plegamiento. Sin embargo, no sólo no son esenciales en la estabilización de la estructura nativa, sino que, en ocasiones, son desestabilizadores que favorecen los cambios de conformación. La clave no es la fuerza de las interacciones hidrofóbicas per se, sino el efecto termodinámico, como la variación de entropía asociada al efecto hidrofóbico, que favorece el proceso.
Dada la importancia del colapso hidrofóbico, está claro que los aminoácidos hidrofóbicos van a ser claves en la evolución de las estructuras. Ello ha condicionado la evolución del código genético.
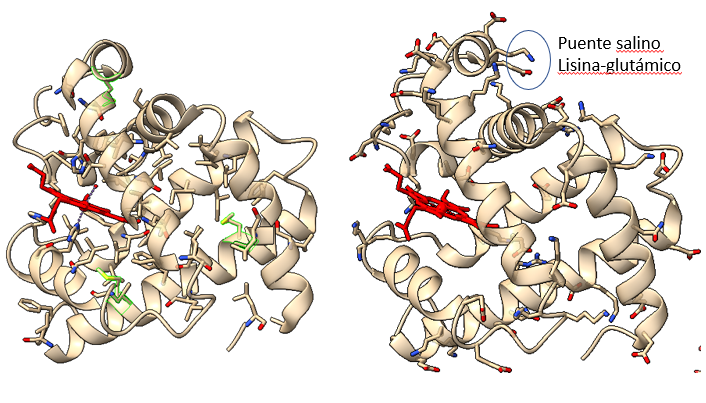
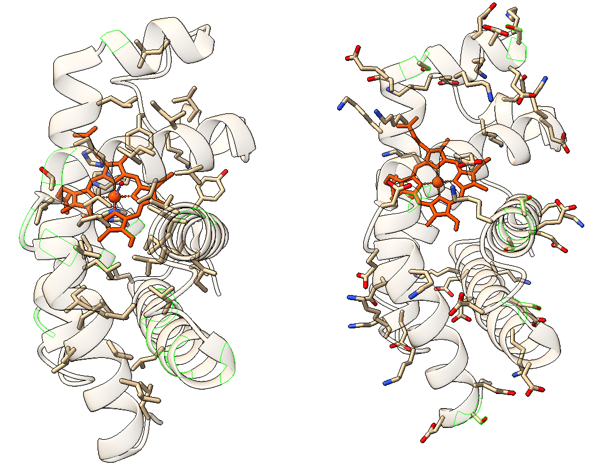
Código genético, mutaciones y la preservación de la estructura proteica
La secuencia de bases del ADN sufre constantes cambios: mutaciones que cambian bases en la secuencia y que se traducen en cambios en la secuencia de una proteína. Estos cambios pueden ser deletéreos, pero también silenciosos o incluso beneficiosos, dando lugar a nuevas funciones. El código genético ha evolucionado de tal forma que las mutaciones mas comunes, llamadas mutaciones missense, en las que un par de bases sustituye a otro, limiten sus efectos sobre las proteínas resultantes. Afortunadamente, el código genético, que se compone de unas ‘palabras’ de tres letras, llamadas codones, esta degenerado. Esto quiere decir que los 64 posibles codones (‘palabras’ de tres letras formadas con las bases A, G, C y U, es decir 43) van a codificar para tan solo 20 aminoácidos distintos. Ello implica que algunos aminoácidos van a tener hasta 6 codones sinónimos. Esto es posible gracias al balanceo o wobble del RNA de transferencia: la tercera posición del anticodon puede reconocer diferentes bases en el codón, gracias a la formación de pares de bases no Watson-Crick, o la presencia de bases no canónicas en esa posición.
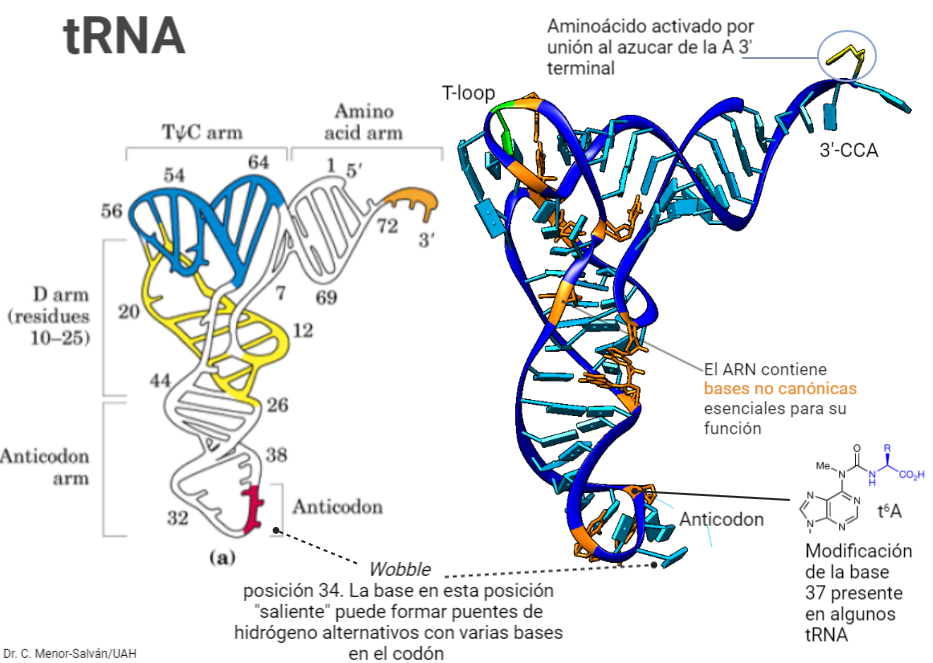
Gracias al balanceo, hay tan solo 32 diferentes tRNA para todos los codones, y estos transportan sólo 20 aminoácidos (es decir, hay más de un tRNA por aminoácido en algunos casos). Por ejemplo, un tRNAarg de las levaduras tiene el anticodón GCI (donde I es inosina, una base que usualmente no está presente en el DNA ni el RNA). La posición I es de balanceo y puede unirse a los codones CGA, CGU y CGC. Ahora, imagina que hay una mutación y un codón CGU se transforma en un codón CGC. Esta sería una mutación silenciosa, pues, al traducirse el gen, seguiría codificando para una arginina y no se producirían cambios en la proteína. La degeneración del código genético hace posible que cambie el aminoácido sólo en un 25% de las mutaciones de este tipo. Ello aporta estabilidad al genoma: durante la evolución, solo perduraron los genomas capaces de mantener sus estructuras funcionales en un ambiente con muchos cambios. Así, la degeneración del código genético y la limitación del número de aminoácidos pudo aportar una ventaja selectiva a los organismos, reduciendo los efectos de las mutaciones. Un potencial organismo con otro código que implicara más aminoácidos debió ser inviable con las mismas tasas de mutación.
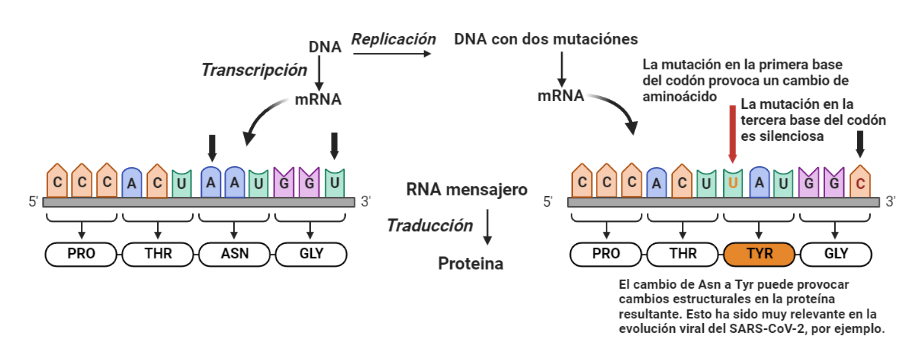
Aun así, el efecto del wobble no es suficiente; es necesario proteger la función de las proteínas, y, para ello, es clave mantener su estructura. Así, los aminoácidos hidrofóbicos están especialmente protegidos, debido a su paper fundamental en el plegamiento proteico. Cuando se produce una mutación en la primera posición del codón, se va a producir un cambio de aminoácido. Pero, en general, el cambio da lugar a la sustitución de un aminoácido por otro de propiedades similares. Por ejemplo, la valina, un aminoácido hidrófobo, esta codificada por un codón GUU. Si hay una mutación en la tercera posición y se transforma en GUC, sigue codificando para valina, y estamos ante una mutación silenciosa. Pero si hay una mutación en la primera posición y se convierte en AUU, el aminoácido cambia a isoleucina, que también es hidrófobo y tiene propiedades similares. Si la mutación lo convierte en CUU, el aminoácido cambia a leucina, que, de nuevo, es hidrófobo y tiene propiedades similares. Estos cambios no alteran significativamente el plegamiento proteico, ya que el colapso hidrófobo va a seguir sucediendo, por lo que, a pesar de las mutaciones, el plegamiento (y por tanto, la función) se va a mantener.
El problema surge cuando se produce un cambio de un aminoácido a otro de un tipo diferente. Esto puede provocar un cambio más o menos drástico en la estructura o propiedades de la proteína. Es el caso, por ejemplo, de la anemia falciforme, en la que hay una mutación de una base en el gen de la cadena beta de la hemoglobina. El aminoácido mutado cambia el codón para un ácido glutámico CTC por el codón para la valina CAC.
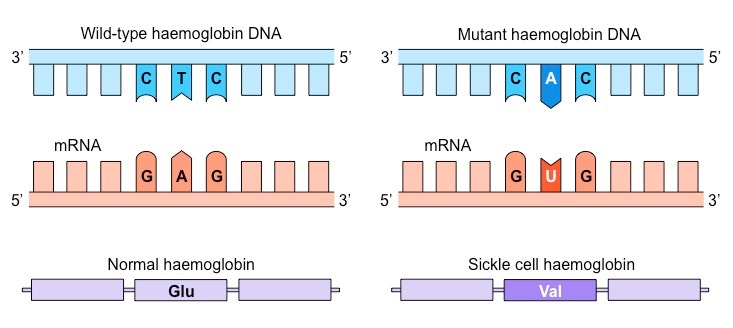
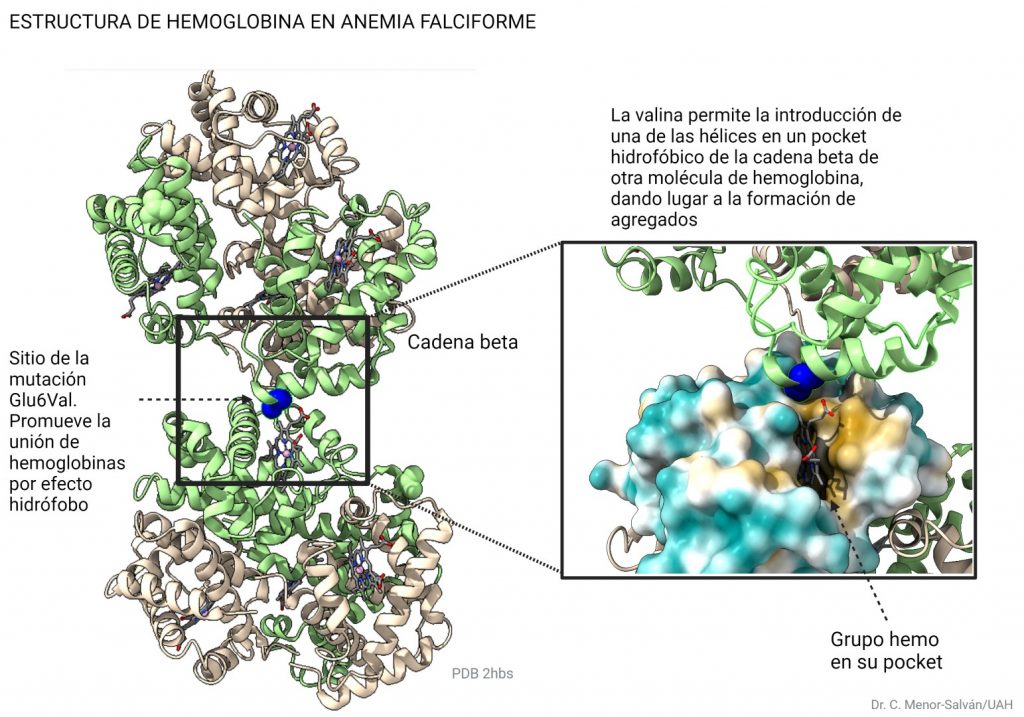
El cambio elimina un aminoácido cargado, el glutámico, por un aminoácido hidrófobo, la valina. Al desaparecer la carga, desaparece la repulsión electrostática entre unidades de hemoglobina, y se sustituye por un aminoácido hidrófobo superficial, que provoca una interacción hidrófoba entre cadenas. Como resultado, la hemoglobina precipita en forma de fibras. De nuevo, los aminoácidos hidrófobos son claves en la estructura. Aquí, la introducción del aminoácido hidrófobo valina tiene consecuencias relevantes, provocando la enfermedad de la anemia falciforme.
Aunque un cambio de aminoácido no tenga gran efecto estructural, influyen en la variabilidad genética y pueden tener importancia. Los SNP o polimorfismos de nucleótido único, son cambios en la secuencia de genes que se traducen en modificaciones de algún aminoácido en las proteínas. Estos SNP crean muchas variaciones entre individuos de la misma especie, y, normalmente, no tienen ningún efecto.
Sin embargo, en algunos casos, los cambios afectan a aminoácidos que alteran ligeramente la función de la proteína (haciendo una enzima mas activa o menos activa, o modificando la afinidad de una proteína por un sustrato). Aunque un cambio de afinidad sea pequeño, tienen una gran importancia farmacológica, explicando la diferente respuesta a algunos fármacos entre individuos. Entre diversas especies, los cambios son mayores para la misma proteína (o para la proteína ortóloga: proteína que ejerce la misma función en otro organismo y proviene del mismo ancestro común, y que normalmente son estructuralmente equivalentes). Esto permite trazar la evolución de las especies y de las propias proteínas.
Referencias
Alas-Guardado, S. de J., Rojo, A. and Merino, G. (2011) ‘La paradoja de Levinthal: cuando una contradicción se vuelve lógica’, Educación Química, 22(1), pp. 51–54. doi: 10.1016/S0187-893X(18)30114-9.
Baldwin, R. L. & Rose, G. D. (2013) ‘Molten globules, entropy-driven conformational change and protein folding’, Current Opinion in Structural Biology, 23(1), pp. 4–10. doi: 10.1016/j.sbi.2012.11.004.
Braakman, R. & Smith, E. (2013) ‘The compositional and evolutionary logic of metabolism’, Physical Biology, 10(1). doi: 10.1088/1478-3975/10/1/011001.
Englander, S. W. & Mayne, L. (2014) ‘The nature of protein folding pathways’, Proceedings of the National Academy of Sciences of the United States of America, 111(45), pp. 15873–15880. doi: 10.1073/pnas.1411798111.
Francoeur, E. (2002). Cyrus Levinthal, the Kluge and the origins of interactive molecular graphics. Endeavour, 26(4), 127–131. doi:10.1016/s0160-9327(02)01468-0
Honig, B. (1999) ‘Protein folding: From the levinthal paradox to structure prediction’, Journal of Molecular Biology, 293(2), pp. 283–293. doi: 10.1006/jmbi.1999.3006.
Karplus, M. (1997) ‘The Levinthal paradox: yesterday and today’, Folding and Design, 2, pp. S69–S75. doi: 10.1016/S1359-0278(97)00067-9.
Kurata, S., Weixlbaumer, A., Ohtsuki, T., Shimazaki, T., Wada, T., Kirino, Y., Takai, K., Watanabe, K., Ramakrishnan, V. & Suzuki, T. (2008) ‘Modified uridines with C5-methylene substituents at the first position of the tRNA anticodon stabilize U·G wobble pairing during decoding’, Journal of Biological Chemistry, 283(27), pp. 18801–18811. doi: 10.1074/jbc.M800233200.
Pak, D., Root-Bernstein, R. & Burton, Z. F. (2017) ‘tRNA structure and evolution and standardization to the three nucleotide genetic code’, Transcription, 8(4), pp. 205–219. doi: 10.1080/21541264.2017.1318811.
Root-Bernstein, R., Kim, Y., Sanjay, A. & Burton, Z. F. (2016) ‘tRNA evolution from the proto-tRNA minihelix world’, Transcription, 7(5), pp. 153–163. doi: 10.1080/21541264.2016.1235527.