¿QUÉ ES LA PK ? Y ¿POR QUÉ PUEDE ACABAR CONTIGO?
Redactado por María Arranz Burón.
INTRODUCCIÓN
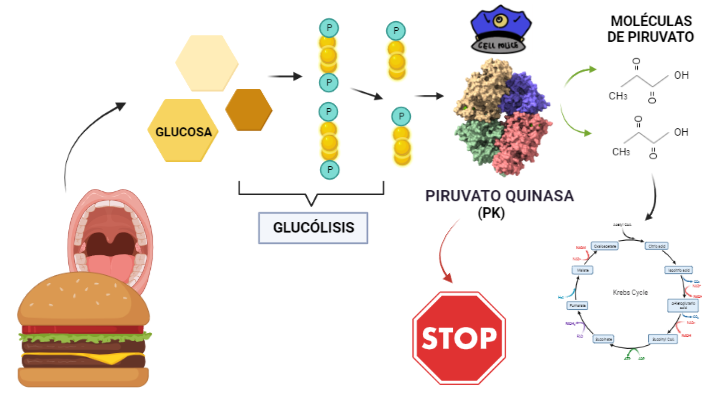
La Piruvato quinasa (PK) es una enzima que se encuentra al final de la ruta metabólica conocida como glucólisis y su función es permitir la salida del piruvato a la siguiente vía metabólica con el fin de obtener energía. Para entender la relevancia de esta molécula, quizás es conveniente explicar de forma concisa el recientemente popular término, conocido como “metabolismo”.
El metabolismo, al que hacen referencia desde dietistas y entrenadores hasta profesores de bioquímica, es quizás un proceso que para el ciudadano de a pie resulta abstracto en su relación con la ingesta de comida. Brevemente, se trata de un conjunto de reacciones que consisten en tomar una molécula grande (glucosa) con alto potencial energético y dividirla en paquetes más pequeños (piruvatos, en el caso de la glucólisis) con el fin de distribuir la energía por los distintos sistemas celulares y permitir que los organismos vivos se sigan considerando como tales. Esa glucosa, llega a las células por medio de la alimentación y se convierte en energía química útil mediante las distintas rutas metabólicas. De modo, que el correcto funcionamiento de la PK, es fundamental para que estos pequeños paquetes se introduzcan en la siguiente ruta metabólica (el ciclo de Krebs) que se encargará de distribuir la energía. De hecho, modificaciones de esta molécula podrían degenerar en condiciones tan serias como el Cáncer o el Alzheimer, dado que impediría que la energía alcanzase los destinos pertinentes.
PAPEL BIOLÓGICO
El rol que asume la PK a nivel biológico se traduce a la función que tiene por ejemplo el personal de seguridad a la entrada de un museo. Éste, en caso de que el turista no presente ninguna irregularidad (posesión de armas, comida, etc), procede a concederle la entrada al establecimiento; del mismo modo la PK permite el acceso al piruvato a la siguiente ruta, siempre que no suponga una amenaza contra el correcto funcionamiento de la célula. Al tratarse la PK de una quinasa, esto la convierte en una molécula con la capacidad de fosforilar (añadir grupos fosfato a otras moléculas). De modo que para concederle el acceso a la siguiente ruta, la PK retira los fosfatos de las triosas procedentes de la glucosa y los añade a moléculas de ADP, dando lugar a ATP (que es una molécula que transporta energía). Como resultado de este proceso, se sintetizan dos piruvatos, a los que finalmente les será concedido el acceso al ciclo de Krebs.
La PK en humanos, aparte de la función biológica general descrita anteriormente, también tiene una serie de isoformas cuya función e importancia biomédica es más específica. Las cuatro isoformas de la PK (PKM, PKK, PKR, PKL) fueron descubiertas en 1965 y reciben su nombre a partir del nombre tejido donde se encuentra cada una. La PKM (actualmente denotada como PKM1) se encuentra en los tejidos musculares (muscle, M) del corazón y en el cerebro, la PKK (actualmente denotada como PKM2) se encuentra en los tejidos de los riñones (kidneys, K), el intestino y las células cancerosas; la PKR en el tejido sanguíneo (red blood cells, R) y la PKL en el tejido del hígado (liver, L) y en los riñones también. La diferenciación (que ocurre tras la fase fetal, ya que todas provienen de la isoforma PKM2) favorece la aparición de patologías mucho más especializadas debido a ligeros cambios en la conformación proteica.
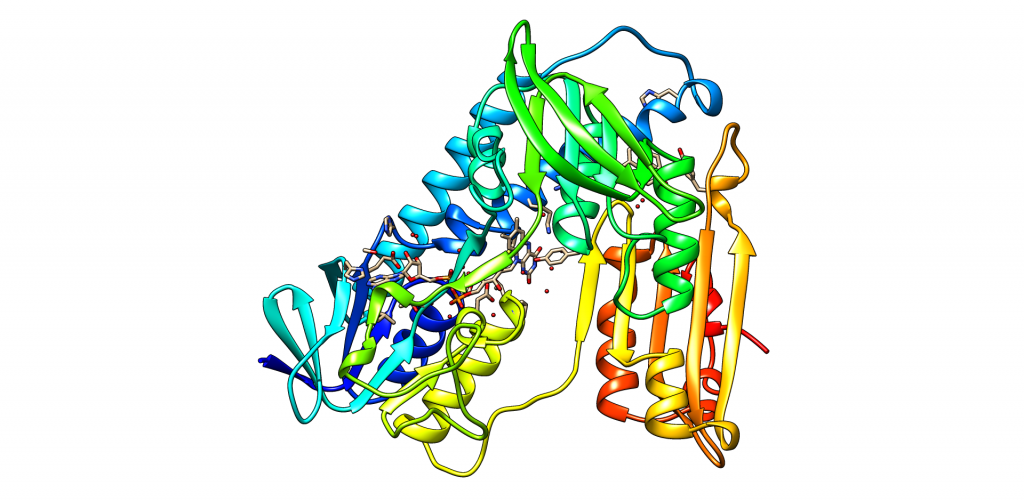
A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
ESTRUCTURA Y CÓMO FUNCIONA
- Desglose estructural
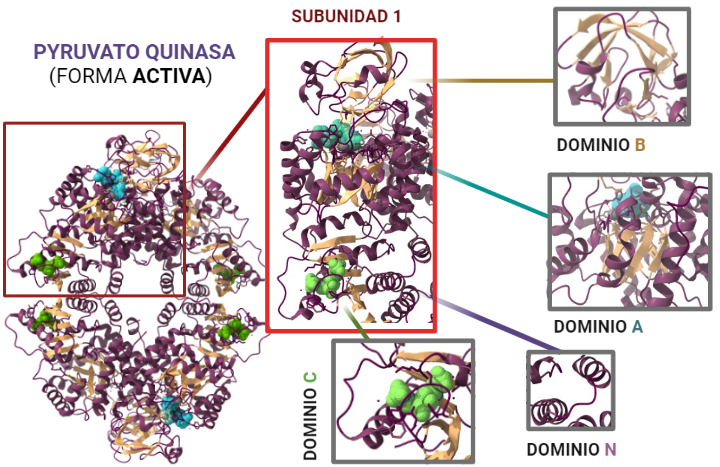
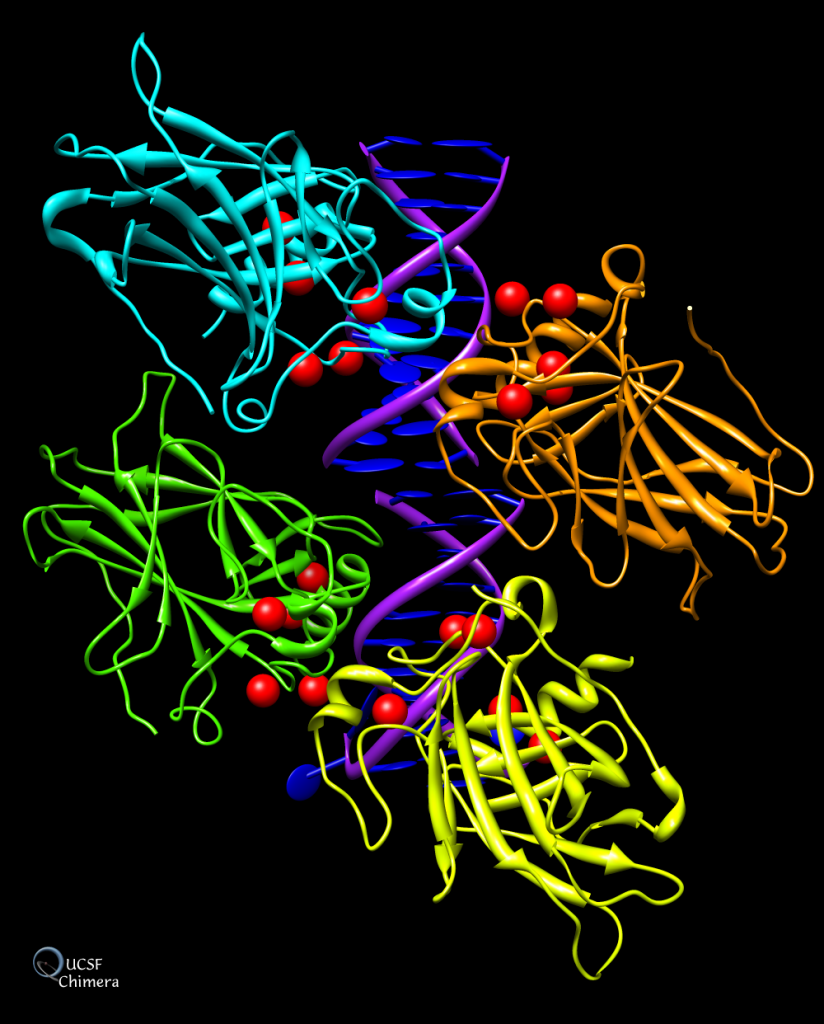
Para entender el papel que desempeña la PK es fundamental comprender su estructura, dado que de ésta dependerá el funcionamiento de la misma. ¿Qué constituye a la PK? La piruvato quinasa es una proteína conformada por 531 aminoácidos que dan lugar a un tetrámero, cuyas cuatro subunidades son iguales. Éstos están organizados en motivos de hélices alfa, láminas beta y bucles.
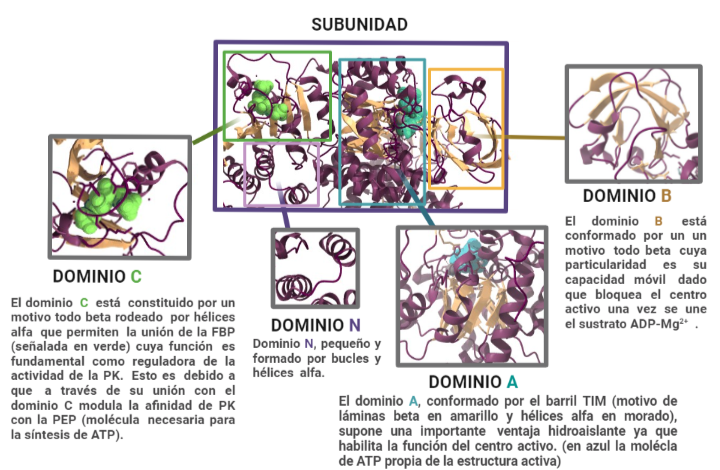
Estas subunidades se organizan a su vez en tres grandes dominios rotulados como A, B y C junto con un dominio N-terminal. El dominio A está conformado por un barril TIM α8/β8 cuyo centro activo se ubica entre el dominio A y el B, éste es además el dominio más grande de la subunidad. El dominio B sin embargo es móvil y bloquea el centro activo una vez que se le une el sustrato ADP-Mg2+. Finalmente, el dominio C contiene la fructosa-1,6-bifosfato (FBP) que es un potente activador alostérico.
A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
Esto significa, que a través de la unión de la FBP al dominio C, se facilitará la unión del fosfoenolpiruvato (PEP) que es fundamental para la regulación de la actividad de la PK, ya que ésta depende de la afinidad con el PEP. En ausencia de activadores alostéricos como la FBP, la PK tiene poca afinidad con el PEP. O sea, si la PK fuera un niño pequeño y su voluntad para realizar los deberes fuera análoga a la actividad de la quinasa, éste necesitaría una motivación para realizarlos. Si se impone la condición de recibir un caramelo a cambio de la tarea, éste cumplirá. De igual forma si la PK presenta la FBP unida al dominio C, ésta aumentará su afinidad a la PEP alterando su actividad. Además, la unión de FBP estabiliza la molécula en estado activo y promueve la tetramerización. Cabe destacar, que todas las isoformas de la PK se unen con la FBP exceptuando la PKM1 que debido a una discrepancia estructural es suficientemente estable por si sola (siendo además insensible a los moduladores alostéricos) y no presenta ni la región de unión a la FBP ni el interfaz dímero-dímero debido a éstas se expresan en los exones específicos de las isoformas.
A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
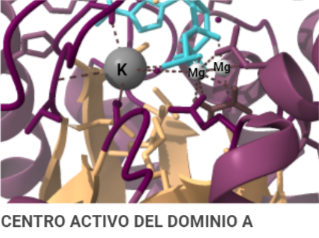
Otro detalle que cabe mencionar de la estructura de la PK es su capacidad de unión a cofactores (K+ y Mg 2+) cuya intervención en la activación de la molécula es esencial. En el caso del Mg 2+, se ha mencionado que está involucrado en bloquear el acceso al centro de activo (gracias al cambio conformacional que desplaza al dominio B) cuando éste forma un complejo de sustrato al unirse al ADP (formando el sustrato ADP-Mg2+). En el caso del K+ sin embargo, se ha observado que ante su presencia, el mecanismo cinético de la PK se mantiene desordenado (forma natural), esto supone que favorece la forma activa de la PK y permite que se unan el PEP o el complejo ADP-Mg2+ de forma independiente (mecanismo aleatorio). En ausencia de K+, por el contrario, el ADP no se pude unir al centro activo hasta que el PEP no haya terminado de formar un centro activo completamente funcional. De forma, que se deduce que el K+ es el encargado de inducir el cierre del centro activo y de que los residuos encargados de la unión al nucleótido adopten la conformación correspondiente.
A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
- Función ¿Qué hace?
Entonces, ¿Qué función tiene? Una vez conocida la estructura, es posible dilucidar qué función lleva a cabo, y cómo. La función catalítica de la PK consiste en fosforilar moléculas, debido a su condición de quinasa; en concreto, moléculas de ADP a partir de moléculas de PEP de la etapa anterior de la glucólisis. Todo ello para dar lugar a dos productos. Por un lado, piruvatos estables a partir de sus precursores PEP y por otro, obtener energía en forma de moléculas de ATP a través de la fosforilación del ADP. De modo, que el nombre surge del producto (piruvato) + tipo de enzima (quinasa). Para ello, al centro activo del dominio A se unen el PEP y el complejo ADP-Mg2+ dado que los cationes de Mg2+ median y facilitan la transferencia del grupo fosfato del PEP al ADP dando lugar al ATP y a los piruvatos. Todo ello es posible debido a la alta energía que libera PEP al ser hidrolizada. Al perder el fosfato, el PEP pasa a su forma de enolpiruvato que es menos estable, de modo que se llevará a cabo un proceso de tautomerización que consiste en que el enolpiruvato acepte un protón procedente de una molécula de agua convirtiéndose así en un piruvato estable y favoreciendo la fosforilación del ADP.
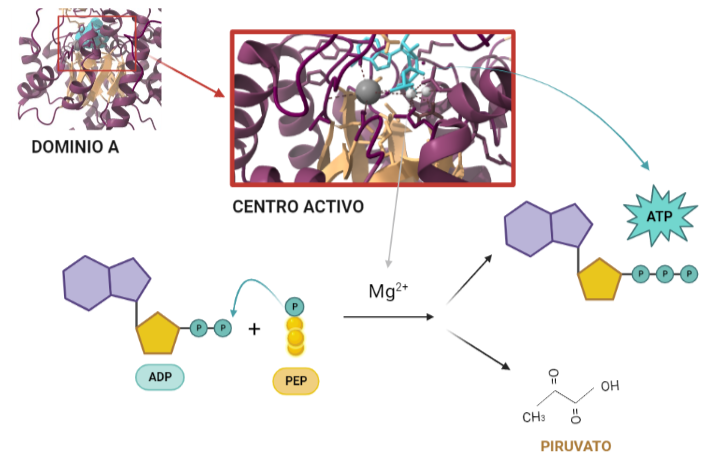
A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
En cuanto a la capacidad alostérica de la PK, aparte de la FBP, que favorece la unión del sustrato PEP, hay otras moléculas que alteran la actividad de la enzima. Por ejemplo, un inhibidor alostérico de esta enzima (PKM1, PKM2) sería la fenilalanina (Phe) cuya unión supone la disminución de afinidad con la PEP mediante la estabilización de la estructura inactiva de la PK. El lugar de unión de Phe también puede albergar a la alanina que actúa como inhibidor, pero solo ante la isoforma PKM2, y esto lo lleva a cabo favoreciendo la conformación dimérica, contraria a la tetramérica a la que se une FBP. A pesar de que en presencia de concentraciones normales de FBP la inhibición de la alanina queda mitigada. La serina sin embargo, también puede ocupar este centro de unión, pero con función activadora no inhibidora, en la PKM2. Dejando a un lado los aminoácidos, hormonas ,como la hormona tiroidea triyodo-L-tironina (T3), actúan también como inhibidor alostérico favoreciendo la conformación monomérica inactiva de la PK. Mientras que el oxalato puede actuar como activador de la PK mediante su interacción con el centro activo por ser análogo al enolpiruvato, en caso de que la concentración de PEP sea baja.
¿PORQUÉ PODRÍA ACABAR CONTIGO?
- EL PELIGRO RESIDE EN LA ISOFORMA
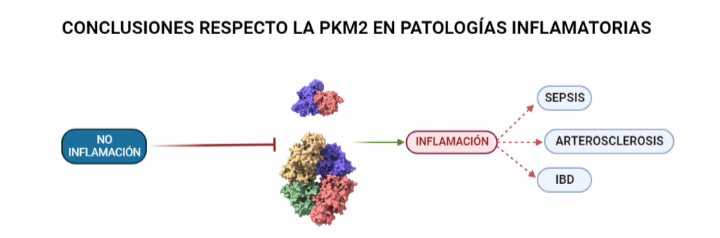
Las isoformas de una proteína son proteínas que provienen del mismo gen que la proteína original, dicho gen se duplica y comienza a acumular mutaciones para dar lugar a las distintas isoformas. En el caso de la PK, tras este proceso de duplicación y modificación por mutaciones se han obtenido 4 isoformas distintas: PKM1, PKM2, PKR y PKL. La importancia de las isoformas recae en que a pesar de realizar la misma función que la proteína inicial, cada una presenta ligeramente distintas: propiedades cinéticas, estructurales, de regulación o de localización en la célula. Estas ligeras diferencias atienden a las necesidades metabólicas del tejido al que pertenecen. O sea, las modificaciones que sufra PKL (L hace referencia a liver, hígado en inglés) afectarán en principio al hígado dado que la estructura de la PKL ha resultado ser la más eficaz a la hora de catalizar las reacciones que precisa este órgano. A pesar de esto, existe una isoforma que destaca en su implicación en numerosas patologías inflamatorias (como la Sepsis, IBD o Arterosclerosis) o enfermedades como el Cáncer o el Alzheimer. Ésta es la PKM2.
- PKM2 Y UN COMPENDIO DE LO QUE PUEDE SALIR MAL
- PKM2 en el Cáncer
Ante el desarrollo de células neoplásicas, la PKM2 tiene un comportamiento que podría clasificarse como moonlighting. Esto se debe a que en condiciones normales la PK transforma PEP en piruvato y este sigue la ruta metabólica normal hacia el ciclo de Krebs, mientras que, ante una situación de estrés, como puede ser el desarrollo de células cancerígenas, ésta altera su forma tetramérica natural y pasa a su forma dimérica. Al dimerizarse mediante la fosforilación de su tirosina 105 la proteína deja de realizar su función natural y divierte el proceso de la glucólisis hacia la síntesis de metabolitos necesarios para la síntesis de serina. Esto se debe a que dicho aminácido regula a mTORC1 ( mammalian target of rapamacyn complex 1), que es fundamental para favorecer la proliferación celular, característica de las células cancerígenas. Otra de las modificaciones que sufre, es la acetilación de su lisina 305, junto con la oxidación de su cisteína 358 que provoca una alteración en la ruta de la glucólisis haca la PPP (pentose phosphate pathway, vía de la pentosa fosfato) que favorece la síntesis de nucleótidos para sufragar los efectos de la interrupción de la ruta glucolítica.
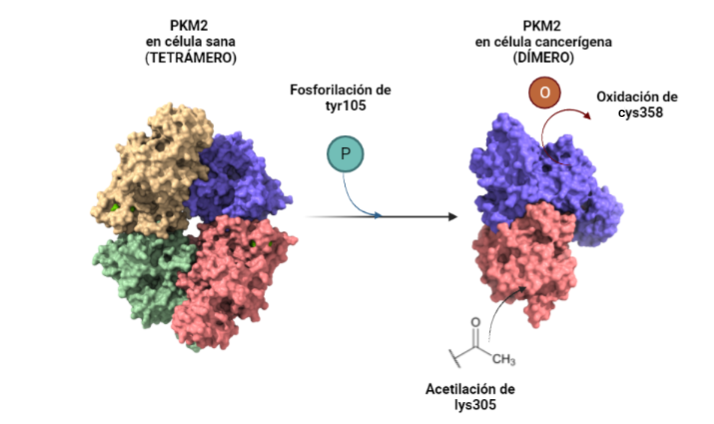
A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
Estas modificaciones de la PK se conocen como el efecto Warburg o Glucólisis Aeróbica. El efecto Warburg supone que las células cancerígenas conviertan el piruvato en lactato, disminuyendo su producción de ATP. La disminución de síntesis de ATP se soluciona mediante los procesos mencionados anteriormente, mientras que el aumento de lactato supone la aparición de un ambiente tumorigénico dado que es excretado, reduciendo así el pH extracelular (esto favorece las condiciones para la proliferación celular) y provocando inflamación. El aumento de lactato también se utiliza para acceder a un recurso de energía alternativo, como es la glutamina. Esto es posible, dado que se disminuye la entrada de piruvato en el ciclo de Krebs, por lo que comienzan a introducirse metabolitos de glutamina en su lugar y aumenta por lo tanto la síntesis de lípidos y aminoácidos. Finalmente, la PKM2 es translocada al núcleo donde se une a HIF1-α (hypoxia-inducible factor-1 alfa) promoviendo la transactivación de HIF1 que favorece la aparición de un ambiente tumorigénico y la desviación de la glucólisis hacia la vía de la pentosa fosfato.
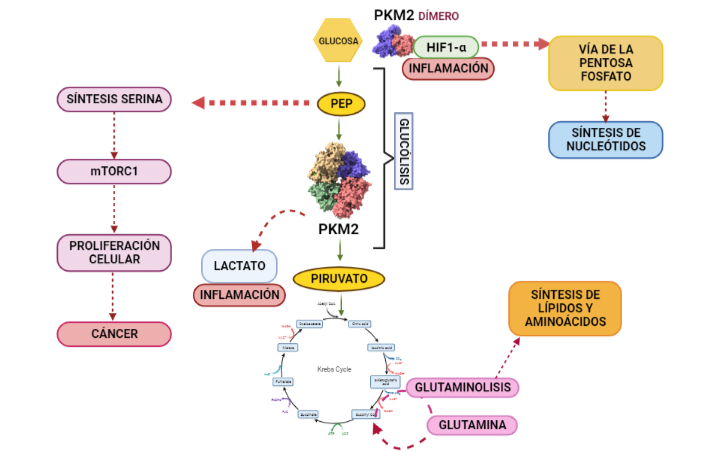
A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
- PKM2 en el Alzheimer
Debido a que el origen y funcionamiento del Alzheimer todavía no se ha esclarecido, se están explorando nuevas vías de investigación. Entre ellas, destacan factores como el estrés oxidativo, fallos en el transporte y metabolismo de la glucosa, y la inflamación. Dado que la piruvato quinasa juega un papel central en el metabolismo de la glucosa, su función se ve afectada por la enfermedad. Como consecuencia de la alteración de la ruta metabólica de la glucosa (debida a la acumulación de β-amiloides), se da la inactivación HIF1-α lo que supone que la PKM2 en dímero no puede unirse a ella, quedando inhibida. Otra forma en la que la PKM2 queda inhibida (en presencia de esta enfermedad) es mediante la inactivación o la regulación insuficiente de PPIasa (peptidil-prolil cis-trans-isomerasa) o la elevada concentración de ROS (especies reactivas de oxígeno), ya que ambas impiden la translocación de la PKM2 al núcleo, y consecuentemente esta no se une a HIF1-α, dando lugar en este caso a un desequilibrio en el metabolismo mediante una cascada de PEP. Respecto al papel de la PKM2 en la respuesta inflamatoria, la PKM2 regula la respuesta de los macrófagos, que son esenciales a la hora de retirar los amiloides para evitar la formación de placas. Esto se debe a que ante la inflamación, los macrófagos adoptan un fenotipo que favorece la síntesis de ATP y se ha observado que algunos incluso activan la Glucólisis Aeróbica (efecto Warburg), que se da cuando hay una sobreactivación de la PKM2. La Glucólisis Aeróbica mediada por PKM2 también promueve la activación de inflamasomas mediante la fosforilación de EIF2AK2 (factor 2 de iniciación de traducción de la α quinasa 2). Por otro lado, mediante la interacción con HIF1-α se promueve la transcripción de genes que activan la glucólisis en macrófagos. Finalmente, la PKM2 también es responsable de la glucólisis de la α-sinucleína que es una proteína presináptica asociada a la fisiopatología del Alzheimer.
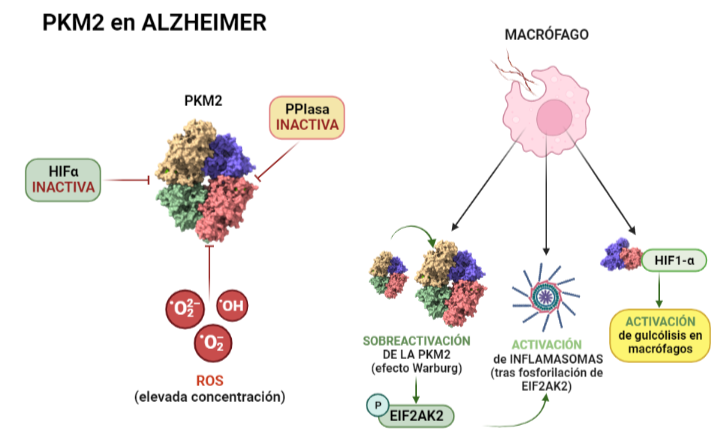
A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
- PKM2 en diversas enfermedades inflamatorias
La respuesta inflamatoria, como se ha expuesto en los dos casos anteriores, requiere de un alto consumo de energía. Células del sistema inmune, como los macrófagos, activan mecanismos similares al denominado efecto Warburg en el cáncer para aumentar la síntesis de energía y poder responder de manera eficaz. Esto está íntimamente relacionado con la glucólisis por lo que la implicación de la PK es crucial. En el caso de la Sepsis la implicación de la PKM2 en la glucólisis anaeróbica que adoptan las células bajo la patología supone un perjuicio para la supervivencia del individuo. Esto se ha observado tras inhibir su actividad comprobando que de este modo también quedaban inhibidas moléculas como el inflamasoma NLRP3 y HMGB1 (proteína de alta movilidad del grupo 1) cuya represión favorece la supervivencia del individuo. En el caso de la Arterosclerosis se ha observado que la inhibición de PKM2 en linfocitos B y T favorece la atenuación de la inflamación. Esto está unido a que parte de los productos que se sintetizan durante la glucólisis aeróbica (adoptada por las células del sistema inmune bajo condiciones de estrés) producen inflamación como se expone en la segunda imagen de la PKM2 en el cáncer. Incluso en la IBD (enfermedad inflamatoria de Bowel) la PKM2 se está valorando como posible bioindicador de la enfermedad según estudios recientes. Una vez más, la supresión de la PKM2 favoreció el control sobre la inflamación ya que en su versión nuclear dimérica contribuye a la adaptación de las necesidades metabólicas de los linfocitos. Esto significa que la presencia (en muestras) de esta proteína en estado dimérico podría ayudar a identificar la enfermedad. También cabe mencionar, que se ha observado que la PKM2 regula a Bcl-xL impidiendo la apoptosis de las células del epitelio intestinal.
A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
- REFERENCIAS
Boutet E, Lieberherr D, Tognolli M, Schneider M, Bairoch A.
UniProtKB/Swiss-Prot
Methods Mol. Biol. 406:89-112 (2007)
Encyclopedia of Cancer (Second Edition)2002Pages 535-547 Paschal A. Oude Weernink Gerard E. J. Stoal Gert Rijksen
February 2004, David Goodsell
doi:10.2210/rcsb_pdb/mom_2004_2
Israelsen, W. J., & Vander Heiden, M. G. (2015). Pyruvate kinase: Function, regulation and role in cancer. Seminars in cell & developmental biology, 43, 43–51. https://doi.org/10.1016/j.semcdb.2015.08.004
June 2022, Faiza Ahmed, Jonathan Ash, Thirth Patel, Auriel Sanders, David Goodsell, Shuchismita Dutta
doi:10.2210/rcsb_pdb/mom_2022_6
Oria-Hernández, J., Cabrera, N., Pérez-Montfort, R., & Ramírez-Silva, L. (2005). Pyruvate kinase revisited: the activating effect of K+. The Journal of biological chemistry, 280(45), 37924–37929. https://doi.org/10.1074/jbc.M508490200
Patel, S., Das, A., Meshram, P., Sharma, A., Chowdhury, A., Jariyal, H., … & Shard, A. (2021). Pyruvate kinase M2 in chronic inflammations: a potpourri of crucial protein–protein interactions. Cell Biology and Toxicology, 37(5), 653-678.
The RCSB PDB «Molecule of the Month»: Inspiring a Molecular View of Biology D.S. Goodsell, S. Dutta, C. Zardecki, M. Voigt, H.M. Berman, S.K. Burley (2015) PLoS Biol 13(5): e1002140. doi: 10.1371/journal.pbio.1002140
Yang, L., Venneti, S., & Nagrath, D. (2017). Glutaminolysis: a hallmark of cancer metabolism. Annual review of biomedical engineering, 19, 163-194.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}