¿Somos esclavos de nuestros genes?
Gonzalo Raso de Llanos
INTRODUCCIÓN
‘El gen egoísta’ es un libro de Richard Dawkins en el cual expone que el gen, y no el individuo, es la unidad evolutiva fundamental. Esto significa que los genes, o como él los llama, los replicadores, son los que realmente sufren la presión de la selección natural y los que son transmitidos a la descendencia, por lo que forman una línea inmortal en la cual van pasando de organismo en organismo mientras éstos perecen. Según esta teoría, los animales, plantas, bacterias y en conclusión, los organismos, solo son los ‘vehículos’ que usan estos genes para seguir avanzando en su línea inmortal, simples máquinas creadas por y para la supervivencia de los genes que contienen. Dawkins adjudica a estos genes el calificativo de egoístas por la característica principal que ha marcado la supervivencia de los seres vivos: el egoísmo. A lo largo de la historia de la evolución, desde el caldo primitivo hasta la actualidad, los genes han combatido en un mundo despiadado, en el cual la supervivencia ha sido el objetivo principal para mantener su línea inmortal. Por ello hemos visto cómo los genes incluso se han unido para formar máquinas que les permitieran luchar y sobrevivir por encima del resto. Éstos genes nunca ‘buscaron’ la supervivencia de los otros genes con los que se unieron, pues como buenos genes egoístas solo buscaban su propia supervivencia. Sin embargo, ‘vieron’ que las máquinas que formaban eran más aptas para la supervivencia, que incluía su propia supervivencia, por lo que entendieron que la cooperación era la mejor vía para alcanzar sus objetivos egoístas individuales. Nosotros somos el resultado de la cooperación de todos estos genes, que a lo largo de la historia de los seres vivos siguieron luchando para sobrevivir ante la presión incesante de la selección natural, tan dura y despiadada que hizo perecer a todas las máquinas que no se adaptaran a ella, y cuyo resultado hoy conocemos como evolución.
Soy consciente que esta idea de ser controlados y vivir a merced de nuestros genes puede generar mucha incredulidad en un principio, pues yo mismo fui muy escéptico al respecto antes de acabar el libro, sin embargo, debemos intentar que nuestra visión antropocéntrica de la vida no nuble nuestro pensamiento y recordar que solo somos arqueas evolucionadas, la punta del iceberg de la evolución, que los únicos que se mantienen desde los principios de la vida son los genes.
LA MÁQUINA DE GENES
Las máquinas de genes empezaron siendo simples receptáculos de genes, membranas que los protegían frente a la guerra química del caldo primitivo. Estas máquinas fueron desarrollándose por distintas ramas en los animales, las plantas o las bacterias, seres vivos capaces de aprovechar al máximo sus cualidades para sobrevivir. Sin embargo, debemos recordar que a pesar de que las máquinas se hayan perfeccionado enormemente siguen siendo máquinas construidas para la supervivencia de los genes egoístas, y como resultado de genes con fines egoístas surgen máquinas con los mismos fines egoístas: sobrevivir y prosperar incluso si tiene que ser a costa de la vida de los demás.
Las especies llevan luchando desde sus inicios contra el resto de especies y contra sí mismas, puesto que el fin de la máquina es la supervivencia de sus genes y no la de los demás. El amor y el bienestar de las especies carecen de sentido en cuanto a la evolución, e incluso los episodios que nosotros podríamos calificar de altruistas en el mundo animal no tienen por qué serlo realmente. Si por ejemplo viéramos una leona defendiendo a sus hijos, podríamos caer en el error de pensar que los protege porque los quiere, sin embargo esto es de nuevo resultado de nuestra visión antropocéntrica, ya que si realmente pasa esto es porque los recién nacidos tienen el 50% de los genes de la madre, por lo que a la madre le interesa mantenerlos con vida. Si la madre supiera que esos leones no son suyos, no tendría ningún reparo en dejarlos morir de la forma más cruel posible, y por eso nos cansamos de ver casos de abandonos, abortos, asesinatos y traiciones en el mundo animal.
La razón por la cual no he incluido todavía a los seres humanos en la concepción de ‘máquinas’ es por el nivel de desarrollo cultural e intelectual al que hemos llegado, que hace que ese mandato egoísta de los genes y ese ‘impulso animal’ se vea reprimido, entablándose un conflicto entre nuestra naturaleza biológica y la educación que recibimos, una lucha entre el egoísmo al que nos dirigimos por naturaleza y el altruismo que imponemos de forma artificial. De aquí surge la célebre frase de Dawkins: “El hombre es el único animal dominado por la cultura”.
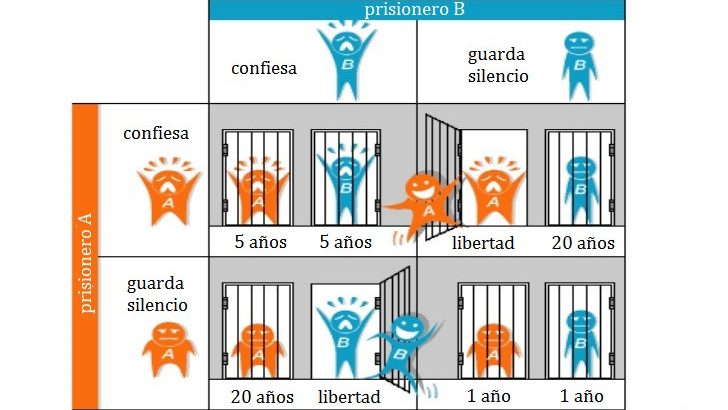
Sin embargo, en el mundo animal también podemos encontrarnos situaciones en las cuales los genes pueden alcanzar sus objetivos egoístas fomentando una forma limitada de altruismo. Un ejemplo para entender este falso altruismo que se da en animales es el dilema del prisionero.
El dilema del prisionero se basa en un interrogatorio hacia dos personas. En éste, se pregunta de forma separada si la otra persona ha realizado un crimen. Si ambos se niegan a acusar al otro, recibirán una pena de un año y si ambos acusan al contrario ambos recibirán una pena de cinco, ya que alguno de los dos tiene que estar mintiendo. En cambio, si uno niega que fuera el otro (de forma altruista) y el otro acusa al primero (de forma egoísta), el altruista recibirá la pena de 20 años y el egoísta saldrá libre por (supuestamente) delatar al culpable. Con unos simples cálculos matemáticos, podríamos concluir en que la estrategia más conveniente ante este dilema sería siempre acusar al contrario, ya que nunca te arriesgarías a afrontar la pena completa (20 años), a la que te expondrías si no delataras al contrario y este te traicionara delatándote. Sin embargo, en estas situaciones en las cuales lo mejor sería acusar siempre, surgen individuos que nunca acusan, y cuando todos los individuos se ponen de acuerdo en no delatar, todas las partes salen beneficiadas. Así, se camufla una actitud egoísta, basada en el interés de recibir la pena menor bajo la capa de un aparente altruismo por el cual no delatarías al contrario, ‘protegiéndolo’ de forma ‘altruista’
¿QUIÉN CONTROLA A QUIÉN?
Llegados aquí podemos ver de forma un poco más nítida que los genes son un factor decisivo para la conducta y toma de decisiones de los seres vivos, y siguiendo la premisa de que somos los vehículos que usan estos genes para propagarse, podemos deducir que nos ‘manipularán’ de forma que actuemos de la mejor manera para cumplir sus intereses.
Obviamente, llegados a un punto en la evolución, los genes no son lo suficientemente rápidos como para controlar lo que podemos hacer en cada momento, ni lo suficientemente veloces como para reaccionar a un estímulo a tiempo como lo hace nuestro sistema nervioso. Lo único que pueden hacer es darnos indicaciones por adelantado para fomentar nuestra supervivencia y reproducción (no porque les ‘importemos’, sino porque de ello depende su prosperidad).
Aquí, es probable que de nuevo nuestra visión antropocéntrica de la vida nos haga rechazar esta idea, ya que nos sentimos dueños de todo lo que pensamos y hacemos, y al final ¿Quién es nadie para decirnos que nuestra conducta no viene dada puramente por lo que nosotros decidimos? Por esto mismo recomiendo que volvamos a fijarnos en los animales, en los que la cultura y el desarrollo del intelecto no nubla su naturaleza biológica.
En la naturaleza podemos ver cómo la reproducción es la meta de todos los seres vivos, y esto se da porque que es la vía por la cual prosperan los genes que ‘llevamos’. Cabría preguntarnos cuál es la motivación de los animales, los vehículos que transportan estos genes, para darlo todo por la reproducción si no es para que sus genes prosigan en la línea inmortal. ¿Por qué conociendo la efimeridad de sus vidas deciden seguir luchando? Prefiero no contestar esta pregunta, que divagaría demasiado en otro plano más filosófico, sobre todo por el posible paralelismo que se pueda generar con el ser humano, pero sin duda los genes tienen que tener un papel fundamental en influir a sus vehículos de tal forma que aseguren su continuidad.
GENES BUENOS, GENES MALOS
Un dato que sorprende muchas veces es que los genes que pasan la prueba de la selección natural no tienen por qué ser siempre buenos para nosotros. Sería razonable pensar que la presión de la selección natural selecciona los mejores individuos con los mejores genes, sin embargo, esto no tiene por qué darse así. Para empezar la presión de la selección natural no es regular ni se direcciona siempre hacia el mismo lado, por lo que a veces no se seleccionan ni los individuos con características ‘buenas’ ni los mejores en esa característica, sino que se seleccionan solo los que en ese caso se hayan adaptado mejor al medio. Los genes actúan en conjunto, creando numerosas interacciones enormemente complejas. Por esto mismo, a la hora de enfrentarse a la presión de la selección natural, no importa si un gen es malo para nosotros, importa lo aptos que sean en conjunto. Una analogía para entender esto sería la de una carrera entre botes de 10 remeros. El bote más rápido, que se correspondería con el animal que sobrevive, no tiene por qué tener todo remeros buenos, solo necesita ser más rápido que el resto. Esto podría darse en un bote con 8 remeros decentes y dos remeros malos (8 genes decentes y 2 malos), que sería más rápido que un bote con 2 remeros excelentes y 8 remeros malos (2 genes excelentes y 8 malos). Este es el ejemplo claro de que no siempre tienen por qué seleccionarse los mejores genes, ya que en este caso los genes excelentes son descartados como si fueran malos.
Cabe destacar que siempre que hablemos de genes buenos o malos para nosotros no debemos pensar en genes benevolentes o malvados. Obviamente los genes en sí no tienen conciencia ni nos ‘quieren’ hacer ni bien ni daño. El hecho de que se seleccionen realmente solo depende de las matemáticas, del porcentaje de éxito reproductor del vehículo que lo porte, de la probabilidad de muerte no deseada del vehículo que lo porte, etc.
MEDAWAR Y LA SENESCENCIA
La senescencia ha sido desde siempre uno de los temas más abordados desde el sector científico, se ha estudiado desde numerosos puntos de vista, y desde luego, en ‘El gen egoísta’, Dawkins también ofrece su visión al respecto. Como hemos dicho anteriormente los genes no tienen por qué ser siempre buenos para nosotros (Genes buenos, genes malos) pero como su intención es la de transmitirse a la descendencia (Quién controla a quién), suelen protegernos temporalmente (La máquina de genes). Sin embargo, una vez hemos cumplido nuestra función como vehículos, reproduciéndonos y pasando nuestros genes a la siguiente generación, podemos ver cómo con la edad aumenta nuestra probabilidad de sufrir enfermedades graves, que se manifiesten genes letales o que fallen nuestros sistemas de reparación de ADN, como si de repente los genes que nos estaban ‘protegiendo’ se desvanecieran.
En parte, esto se puede explicar con la teoría de la senescencia de Medawar. Esta teoría se basa en que los genes letales que se expresan de forma tardía en nuestra vida no sufren la misma presión de la selección natural que sufren los que se expresan en una edad temprana. Esto ocurre porque si el gen letal se expresa tempranamente matará al individuo antes de que pueda reproducirse y propagar sus genes, por lo que se corta la línea inmortal del gen letal, disminuyendo su presencia en el acervo genético hasta extinguirse. En cambio, si el gen se expresa de forma tardía, habrá dado tiempo a que se propague ese gen letal a la descendencia antes de que acabe con el individuo, por lo que esos genes letales no son desechados por la selección natural. El problema de estos genes es que como no son desechados, se van acumulando generación tras generación, lo que podría ser una causa de que con el tiempo haya cada vez más propensión a desarrollar enfermedades letales en las etapas de la vida posteriores a la edad reproductiva. Esto, es un ejemplo más del ‘egoísmo’ de los genes que portamos, que solo buscan su transmisión a la siguiente generación, independientemente del resto, de los demás vehículos e incluso de su propio vehículo.
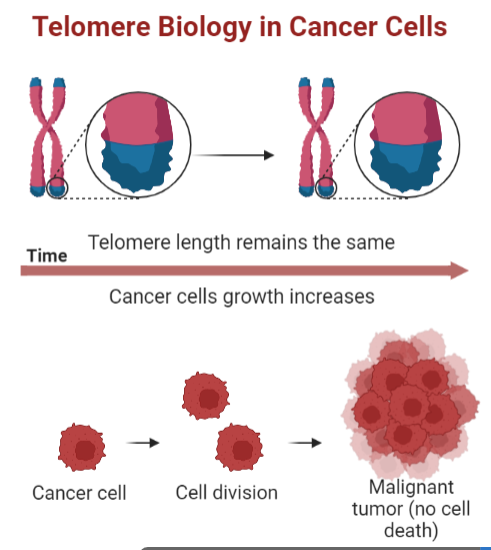
Estos genes de ‘expresión tardía’ han sido muy estudiados por más investigadores además de Medawar y Dawkins. En 2021, un estudio demostraba que estos genes sufrían mutaciones de forma mucho más frecuente que los que se expresaban de forma temprana, de tal forma que era hasta 150% más probable que pudieran conducir a un cáncer (Fig.4).
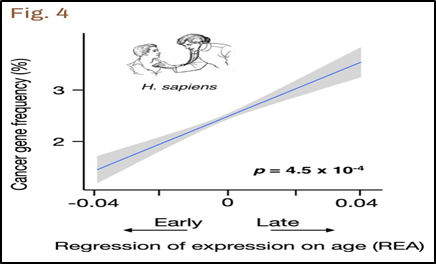
Otros estudios han demostrado que esta falta presión evolutiva para los genes en la etapa post-reproductiva también ha favorecido la aparición de la llamada pleiotropía antagónica. La pleiotropía antagónica se basa en que un gen pueda ser favorable para la salud en la vida temprana pero nocivo en una edad más avanzada. Esto genes pueden ser seleccionados como ‘buenos’ por la selección natural ya que al dar una buena salud durante la edad reproductiva, permite que se propaguen estos genes a la descendencia. La pleiotropía antagónica ha sido ampliamente estudiada ya que en numerosas ocasiones se ha propuesto que un ejemplo de ésta podría ser la enfermedad de Alzheimer, en la cual distintos genes podrían ser beneficiosos para la salud cerebral en edades pre-reproductivas y reproductivas pero nocivos para las neuronas en las edades más avanzadas. Esto es, de nuevo, otro ejemplo de cómo los genes nos ‘utilizan’, protegiéndonos solamente cuando les somos útiles.
Además, se ha comprobado que muchos de estos genes pleiotrópicos pueden interaccionar con el sistema de ‘proofreading’ del ADN o con la proteína MYC, lo cual se ha visto que dispara las probabilidades de sufrir cáncer y otras muchas patologías.
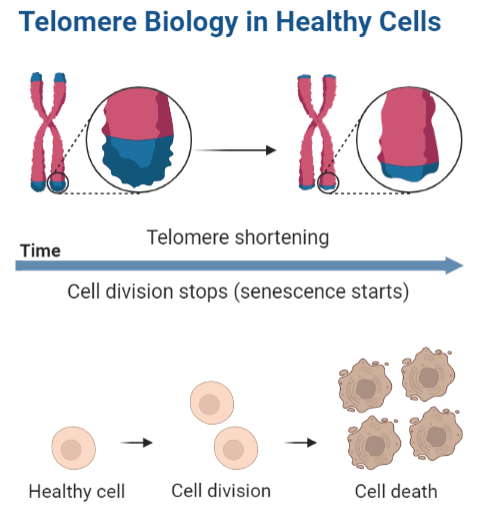
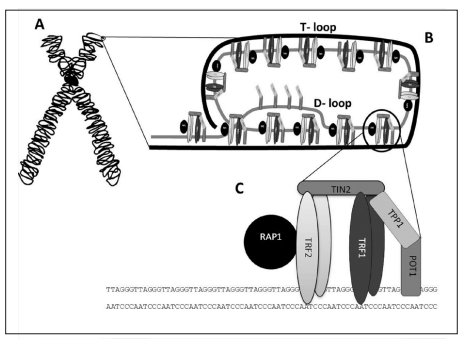
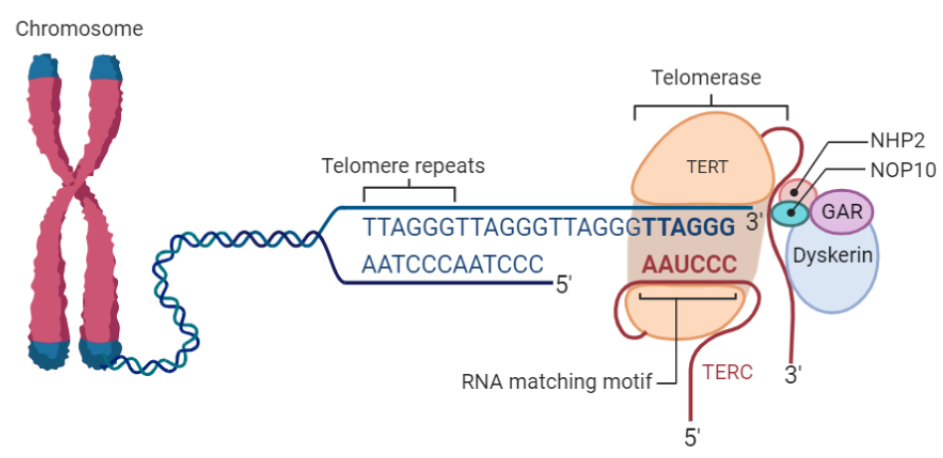
Naturalmente, no se puede achacar todo esto a la teoría de Medawar, ya que existen otros muchos factores que hacen que estas enfermedades y patologías se manifiesten con mayor frecuencia en una edad avanzada. No podemos olvidarnos de que, por ejemplo, la senescencia Hayflick afecta gravemente a varios sistemas como el inmunitario de forma progresiva según los individuos van avanzando en edad. Otro ejemplo es el acortamiento de los telómeros, que puede causar el colapso de las células por cómo afecta a la telomerasa o al sistema de las shelterinas.
REFERENCIAS
- El gen egoísta: las bases biológicas de nuestra conducta, (1994). Salvat.
- Miao, Y., Qian, N., Shi, L., Hu, F. y Min, W., (2021). 9-Cyanopyronin probe palette for super-multiplexed vibrational imaging. Nature Communications [en línea]. 12(1). [Consultado el 8 de enero de 2023]. Disponible en: doi: 10.1038/s41467-021-24855-6
- Gleeson, S. K., (1984). Medawar’s theory of senescence. Journal of Theoretical Biology [en línea]. 108(3), 475–479. [Consultado el 8 de enero de 2023]. Disponible en: doi: 10.1016/s0022-5193(84)80048-x
- Fukuda, M., Taguchi, T. y Ohashi, M., (1999). Age-dependent changes in DNA polymerase fidelity and proofreading activity during cellular aging. Mechanisms of Ageing and Development [en línea]. 109(2), 141–151. [Consultado el 8 de enero de 2023]. Disponible en: doi: 10.1016/s0047-6374(99)00034-2
- Austad, S. N. y Hoffman, J. M., (2018). Is antagonistic pleiotropy ubiquitous in aging biology? Evolution, Medicine, and Public Health [en línea]. 2018(1), 287–294. [Consultado el 8 de enero de 2023]. Disponible en: doi: 10.1093/emph/eoy033