LA PIRUVATO DESHIDROGENSA. LA ENZIMA INDISPENSABLE DEL METABOLISMO.
INTRODUCCIÓN
¿ES REALMENTE IMPORTANTE EL COMPLEJO DE LA PIRUVATO DESHIDROGENASA?
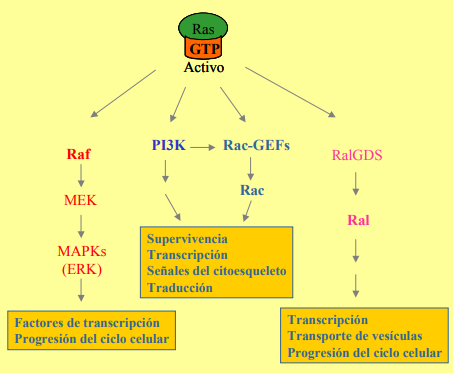
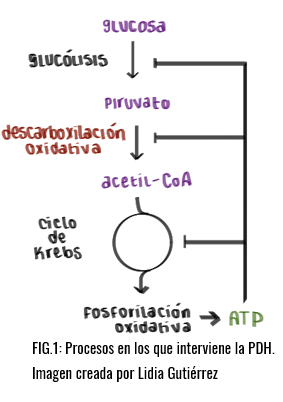
Sabiendo que la producción principal de energía se lleva a cabo mediante el Ciclo de Krebs y que comienza con el componente acetil-coa, podemos deducir que el complejo piruvato deshidrogenasa es altamente necesario e indispensable para poder transformar piruvato en acetil-coa, siendo este, el metabolito intermediario.
¿EN QUÉ PROCESOS Y DÓNDE APARECE?
Como ya hemos mencionado anteriormente es necesario para el funcionamiento del Ciclo de Krebs, actúa previamente en la preparación del acetil-coa, es decir, en la descarboxilación oxidativa del piruvato.
Este proceso convierte al piruvato, molécula de 3 carbonos en una molécula de 2, acetil-coa, unida a la coenzima a. Además de producir una molécula de NADH y otra de dióxido de carbono. Este proceso se lleva a cabo en eucariontes en la matriz mitocondrial, mientras que, en procariontes, se lleva a cabo en el citoplasma.
PAPEL BIOLÓGICO
¿QUÉ NOS APORTA UNA PROTEÍNA?
Las proteínas son moléculas muy grandes y complejas que van a realizar funciones críticas de nuestro organismo, encargándose de la mayor parte del trabajo de las células. Es decir, son estrictamente imprescindibles para el correcto funcionamiento de nuestro cuerpo.
En especial, en este caso, realizará una función principalmente enzimática, que se encargará prioritariamente de que se realicen las reacciones químicas en la célula y que se formen nuevas moléculas.
¿QUÉ APORTA EL PDH A NUESTRO ORGANISMO?
Es la forma en la que las células de nuestro cuerpo pueden recibir energía, siendo el piruvato el producto final de la glucólisis y la glucólisis el primer paso de la respiración celular. Por tanto, el PDH actúa como unión entre la glucólisis y el ciclo de Krebs. A partir de este punto, podemos deducir que el PDH es un componente crucial para la bioquímica, lo que implica que, si este no existiera o se presentará en una cantidad menor a la necesaria, se producirá una deficiencia de PDH (acidosis láctica) y por consiguiente de acetil-coa y como resultado final deficiencia de energía para las células de nuestro organismo.
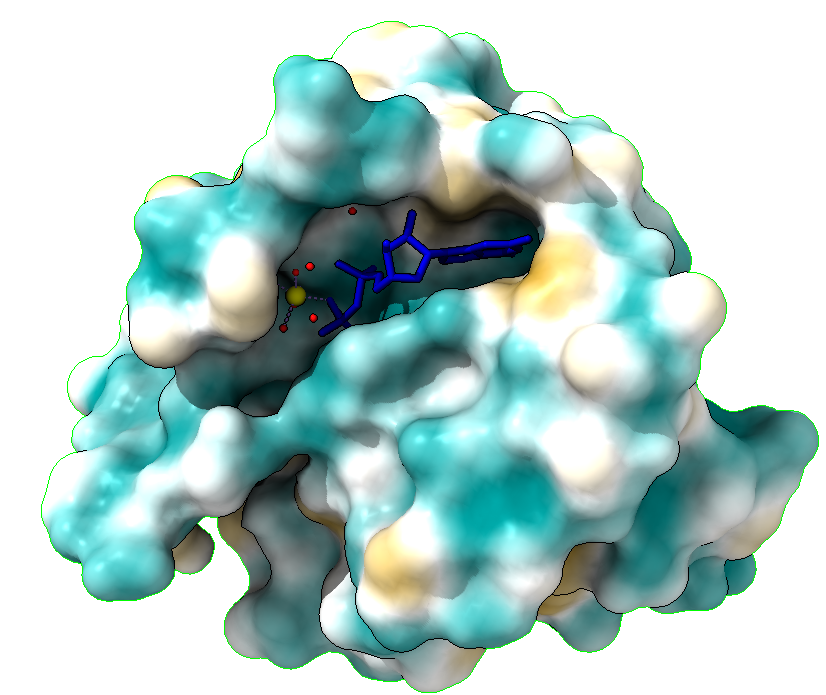
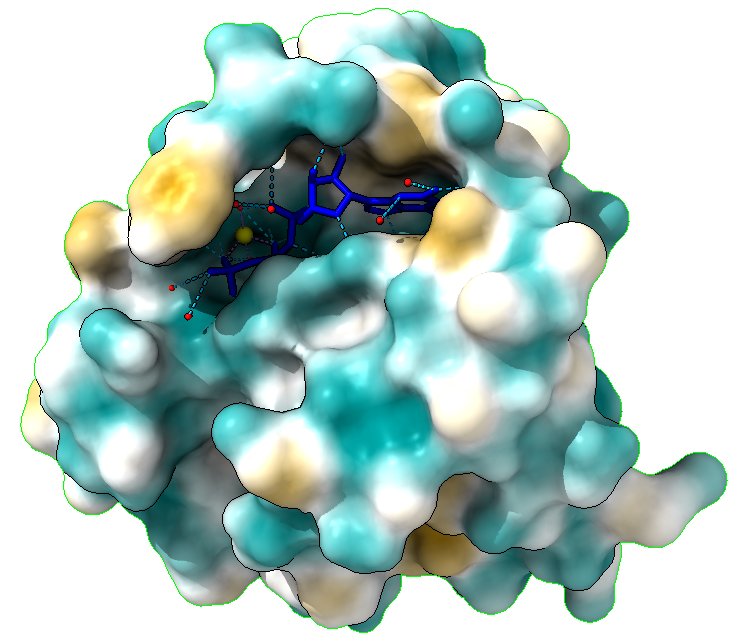
ESTRUCTURA Y MECANISMO
¿QUIÉN LO FORMA?
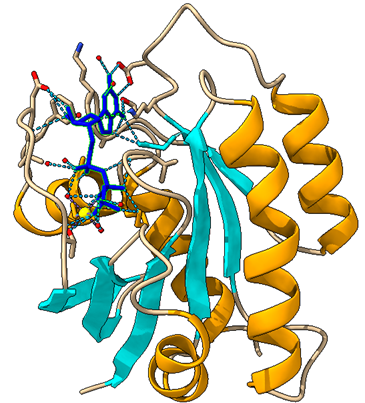
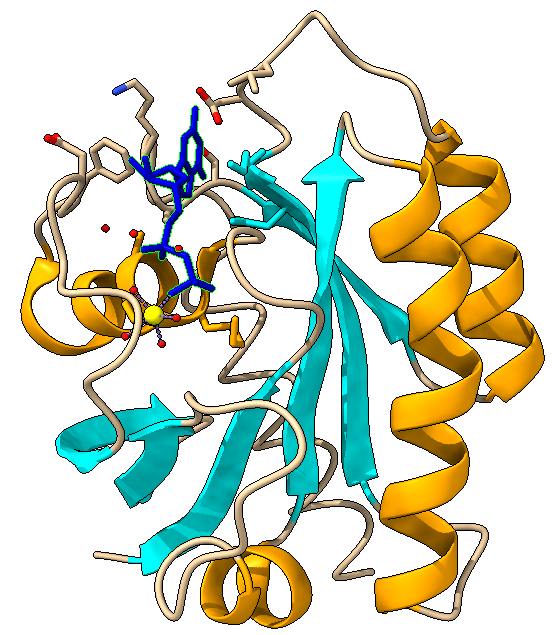
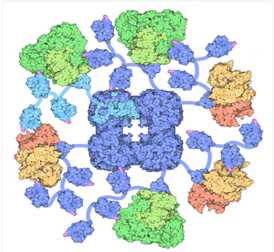
PDH es un complejo que, formado por la repetición de 3 proteínas con diferentes actividades enzimáticas (E1,E2,E3) y otras estructurales y reguladoras, además de diferentes coenzimas.
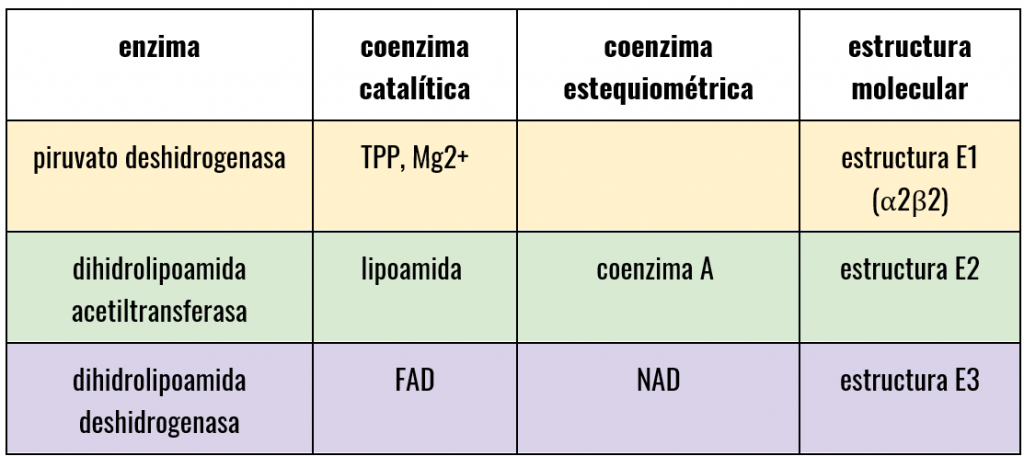
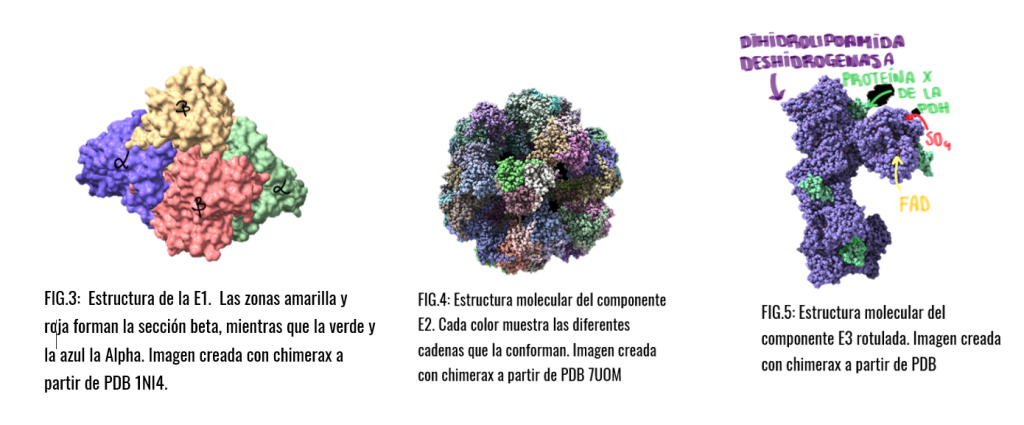
¿QUÉ FUNCIÓN PRESENTA CADA COMPONENTE?
- La estructura E1 va a depender de pirofosfato de tiamina y se va a encargar de catalizar 2 etapas, la primera, la descarboxilación del piruvato que dará como resultado un componente intermedio (hidroxietil-tiamina-difosfato), y la segunda, la acetilación reductora del grupo lipoílo (unido a E2).
- Seguidamente la estructura E2 se va a encargar de catalizar la transferencia del grupo acetilo al CoA.
- Por último, la estructura E3 realizará la función de transferencia de electrones en dos fases, la primera a FAD y la segunda a NAD, para regenerar el lipoílo oxidado y pueda ser reciclado en próximos ciclos.
¿CÓMO SE LLEVA A CABO SU MECANISMO?
Reacción producida:
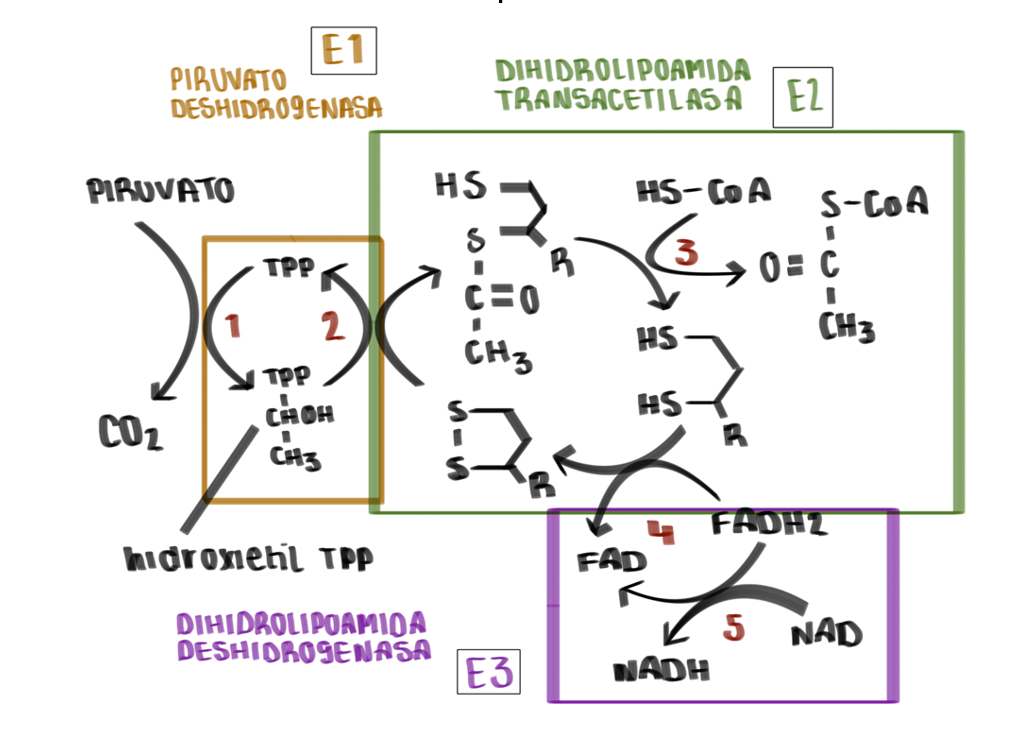
Pasos que se llevan a cabo:

¿DE QUÉ SE ENCARGAN LAS COENZIMAS EN ESTE PROCESO?
Sabemos que en este proceso participan 5 coenzimas mencionadas anteriormente, las cuales se clasifican según la función que desempeñan.
- Transporte de electrones, se encarga la coenzima FAD y NAD.
- Transporte de carbonos, en el cual participan la TPP y la coenzima A.
- Transporte mixto (electrones y carbonos), en el que solo encontramos el ácido lipoico.
¿CÓMO ACTÚAN?
Primeramente, el FAD es capaz de intercambiar electrones con versatilidad, es un transportador fijo, se asocia establemente a la enzima con la que trabaja.
Su estado oxidado es FAD y reducido es FADH2.
En segundo lugar, el NAD es un transportador móvil, se va moviendo para recoger los electrones dispuestos en los centros activos de las enzimas.
Su estado oxidado es NAD+ y reducido es NADH.
Seguidamente, el TPP, pirofosfato de tiamina, se encarga de unir los carbonos al suyo formando enlaces C-C, es un transportador fijo ya que está unido fuertemente a la enzima.
A continuación, la coenzima A es un transportador móvil de carbono que puede actuar tanto de dador como dador de los mismos.
Por último, el ácido lipoico es transportador fijo ya que está unido estrechamente a la enzima
BIOMEDICINA
¿QUÉ RELACIÓN PUEDE PRESENTAR CON LA BIOMEDICINA?
La formación de la enzima piruvato deshidrogenasa es un proceso completamente regulado. Por ello un cambio en las concentraciones por déficit o exceso ocasionan patologías metabólicas en el organismo
¿QUÉ PATOLOGÍAS ESTÁN RELACIONADAS CON EL PDH?
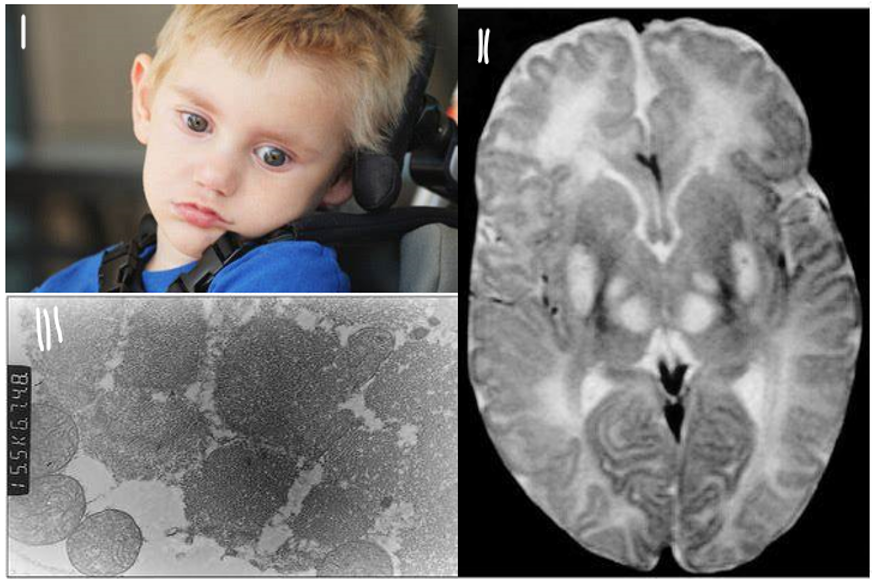
DÉFICIT DE PDH o ACIDOSIS LÁCTICA: como hemos dicho anteriormente, el PDH está relacionado con la obtención energética por vía aeróbica. La deficiencia del complejo provoca un bloqueo de esta vía, impidiendo así la transformación del piruvato en el metabolito intermediario. Por ende, no se inicia el ciclo de Krebs, disminuye la producción de energía y se produce la acumulación de lactato.
Las patologías clínicas varían de gravedad, pero consisten en acidosis láctica y malformaciones del sistema nervioso central y otras alteraciones posnatales, como retraso intrauterino.
Además, se puede producir síndrome de Leigh (enfermedad neurodegenerativa progresiva hereditaria) y/o atrofia cerebral.
La forma tardía se caracteriza por ataxia ( “signo neurológico que consiste en la falta de coordinación voluntaria de los movimientos musculares que puede incluir anomalías en la marcha, cambios en el habla y anomalías en los movimientos oculares”) o fatigabilidad y/o debilidad muscular.
Se ha demostrado correlación entre la edad y la concentración de PDH: la actividad de la PDH mitocondrial disminuye con la edad en el corazón, así como en ciertas regiones del cerebro (el cuerpo estriado y el tronco encefálico). Según un estudio de cardiología Marín-García, J. (2002, 1 diciembre).
La mitocondria y el corazón | Revista Española de Cardiología. https://www.revespcardiol.org/es-la-mitocondria-el-corazon-articulo-13040595
Por ello, un diagnóstico y tratamiento precoces puede mejorar la calidad de vida de algunos pacientes.
AUMENTO SIGNIFICATIVO DE PDH, un aumento de la pdh promueve el envejecimiento, además puede causar senescencia celular inducida por oncogenes.
REFERENCIAS
Attention Required! | Cloudflare. (s. f.). https://metabolicas.sjdhospitalbarcelona.org/ecm/deficiencia-piruvato-deshidrogenasa-pdh/info/funcion-tiene-pdh
Bank, R. P. D. (s. f.). RCSB PDB – 1EAA: ATOMIC STRUCTURE OF THE CUBIC CORE OF THE PYRUVATE DEHYDROGENASE MULTIENZYME COMPLEX. https://www.rcsb.org/structure/1eaa
Castaño, M. J. (2005, 1 febrero). Síndrome de Leigh con déficit de los complejos I, III y IV de la cadena respiratoria mitocondrial | Anales de Pediatría. https://www.analesdepediatria.org/es-sindrome-leigh-con-deficit-complejos-articulo-13071315
Complejo de la piruvato deshidrogenasa. (s. f.). https://biomodel.uah.es/metab/Krebs/PyrDH.htm
Database, A. P. S. (s. f.). AlphaFold Protein Structure Database. https://alphafold.com/entry/W1YPD9
Demczko, M. (2023, 1 febrero). Trastornos del metabolismo del piruvato. Manual MSD versión para profesionales. https://www.msdmanuals.com/es-es/professional/pediatr%C3%ADa/trastornos-hereditarios-del-metabolismo/trastornos-del-metabolismo-del-piruvato
La oxidación del piruvato (artículo). (s. f.). Khan Academy. https://es.khanacademy.org/science/biology/cellular-respiration-and-fermentation/pyruvate-oxidation-and-the-citric-acid-cycle/a/pyruvate-oxidation
Libretexts. (2022, 2 noviembre). 17.4: Reacción de difosfato de tiamina, lipoamida y piruvato deshidrogenasa. LibreTexts Español. https://espanol.libretexts.org/Quimica/Qu%C3%ADmica_Org%C3%A1nica/Qu%C3%ADmica_Org%C3%A1nica_con_%C3%89nfasis_Biol%C3%B3gico_(Soderberg)/17:_La_qu%C3%ADmica_org%C3%A1nica_de_las_vitaminas/17.04:_Reacci%C3%B3n_de_difosfato_de_tiamina,_lipoamida_y_piruvato_deshidrogenasa
Marín-García, J. (2002, 1 diciembre). La mitocondria y el corazón | Revista Española de Cardiología. https://www.revespcardiol.org/es-la-mitocondria-el-corazon-articulo-13040595
PDB101: Molecule of the Month: Pyruvate Dehydrogenase Complex. (s. f.-a). RCSB: PDB-101. https://pdb101.rcsb.org/motm/153
PDB101: Molecule of the Month: Pyruvate Dehydrogenase Complex. (s. f.-b). RCSB: PDB-101. https://pdb101.rcsb.org/motm/153
PDB101: Molecule of the Month: Pyruvate Dehydrogenase Complex. (s. f.-c). RCSB: PDB-101. https://pdb101.rcsb.org/motm/153
Regulación de la respiración celular (artículo). (s. f.). Khan Academy. https://es.khanacademy.org/science/biology/cellular-respiration-and-fermentation/variations-on-cellular-respiration/a/regulation-of-cellular-respiration
Universidad CEU Cardenal Herrera. (2019, 5 junio). Proyecto Innovación: Complejo piruvato deshidrogenasa Reacciones mas importantes del ciclo de krebs [Vídeo]. YouTube. https://www.youtube.com/watch?v=peAQoeIb8GQ
User, S. (s. f.). Pruebas genéticas – Piruvato deshidrogenasa, Deficiencia de . . .; Deficiencia del complejo de piruvato-deshidrogenasa; Acidosis láctica por deficiencia de piruvato deshidrogenasa (Pyruvate dehydrogenase deficiency; pyruvate dehydrogenase complex deficiency. IVAMI. https://www.ivami.com/es/pruebas-geneticas-mutaciones-de-genes-humanos-enfermedades-neoplasias-y-farmacogenetica/1159-pruebas-geneticas-acidosis-lactica-por-deficiencia-de-piruvato-deshidrogenasa-deficiencia-de-piruvato-deshidrogenasa-gen-pdp1
Wikipedia contributors. (2023a, enero 17). Leigh syndrome. Wikipedia. https://en.wikipedia.org/wiki/Leigh_syndrome
Wikipedia contributors. (2023b, enero 29). Ataxia. Wikipedia. https://en.wikipedia.org/wiki/Ataxia
Wikipedia contributors. (2023c, enero 29). Pyruvate dehydrogenase. Wikipedia. https://en.wikipedia.org/wiki/Pyruvate_dehydrogenase
Realizado por Lidia Gutiérrez Miguel y Sofía Gómez García.
Grado de Biología Sanitaria, Universidad de Alcalá de Henares.