APP: la ladrona de recuerdos
Elaborado por Candela Martínez, María Orenes y Almudena López
INTRODUCCIÓN
La APP, proteína precursora del péptido beta-amiloide (Amyloid-beta Precursor Protein por sus siglas en inglés), es una glucoproteína transmembrana conservada evolutivamente desde Caenorhabditis elegans hasta los humanos.
En humanos, el gen está localizado en el cromosoma 21 (curiosamente, el cromosoma del síndrome de Down, hecho muy importante, como veremos más adelante) y formada por 770 aminoácidos repartidos en diferentes dominios proteicos. Se encuentra en diversos tejidos destacando el nervioso, pues se piensa que participa en la neurogénesis y maduración del cerebro, plasticidad, formación y reparación sináptica, además del transporte del hierro.
Es procesada por diferentes proteasas, las: α, β y γ. Los fallos en los cortes de sus dominios son los causantes de la formación de placas amiloides, que principalmente consisten en el cúmulo de beta-amiloide en el tejido cerebral, causando degeneración neuronal o más comúnmente llamado, enfermedad de Alzheimer (Cho et al., 2022).
ESTRUCTURA
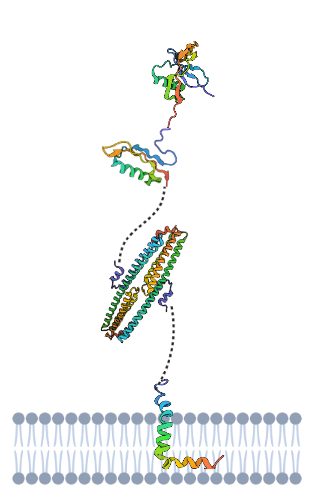
Tenemos que la APP es una proteína dividida en tres dominios, unidos por conectores flexibles formados por cadenas de aminoácidos sin estructura secundaria. Un dominio extracelular, otro transmembrana (el más importante en la formación de la enfermedad) y otro dominio hacia el medio intracelular. Estos dos últimos dominios forman un fragmento denominado C99 cuya fragmentación es decisiva en la generación de placas amiloides.
Cuando la proteína está intacta, actúa como proteína receptora enviando señales a otras proteínas del interior celular, además, es capaz de unirse a estructuras del exterior de la célula como la heparina y laminina, permitiendo la adhesión celular.
Por otro lado, la división en fragmentos de la misma también hace que sea funcional; división que se lleva a cabo por unas proteínas proteasas denominadas secretasas, que cortan a ambos lados del péptido transmembrana. Por un lado los dominios externos, de mayor tamaño, son liberados fuera de la célula ayudando a controlar el crecimiento de las neuronas, y por el otro, el dominio del interior celular se libera a este, ayudando a la síntesis de proteínas en el núcleo.
Y por último, el dominio transmembrana, el cual es el causante de la demencia, pues cuando se suelta de la membrana se introduce en la célula pudiendo cambiar su conformación a beta-lámina, y por acumulación de la misma se generan las placas amiloides. (Burley et al., 2017)
- Dominio más externo: PROTEÍNA DE UNIÓN A AZÚCAR:
- Es también llamado dominio N-terminal de la APP o estructura cristalina del dominio similar al factor de crecimiento N-terminal de la proteína precursora de Alzheimer.
- Esta es la estructura cristalina del dominio de unión a heparina. Entre sus funciones se encuentra la estimulación del crecimiento de neuritas.
- Se trata de una estructura muy cargada con capacidad de interaccionar con glucosaminoglicanos, mientras que su parte hidrófoba se estima que tiene función de dimerización o unión de ligandos.
- Dada la semejanzas estructurales entre la proteína y los factores de crecimiento que contienen cisteína se cree que este dominio N-terminal es capaz de llevar a cabo una función similar a uno de estos.
- Dominio intermedio: DOMINIO DE UNIÓN AL COBRE- REGULADOR DE LA HOMEOSTASIS NEURONAL DEL COBRE:
- El cobre es uno de los principales generadores de radicales libres en nuestro cerebro, también conocido como estrés oxidativo.
- Nuestras células han desarrollado sistemas para transportar estos metales, causantes de las especies oxidantes.
- En el caso de esta proteína, se encarga de regular la homeostasis del cobre neuronal, pues la unión de la APP con el cobre disminuye la formación de beta amiloide.
- Esto se ha observado en ratones que sobreexpresan esta proteína, y que poseen menor cantidad de cobre en el cerebro.
- Dominio más cercano a membrana: DOMINIO DE ADHESIÓN CELULAR:
- Se trata de un dímero antiparalelo del dominio E2 de la proteína.
- Hay estudios que han demostrado que la APP puede formar dímeros, siendo este dominio uno de los contribuyentes a la formación de esto.
- La estructura antiparalela se produce tras la dimerización de las dos subestructuras de E2 mediante la unión del N-terminal de un monómero con el C-terminal del segundo monómero.
- Se sugiere la idea de que la dimerización de esta viene inducida por la unión de heparina. (Lee S et al., 2011)
- Dominio transmembrana: PÉPTIDO BETA AMILOIDE. SIMILAR A UN DOMINIO DE FUSIÓN DE VIRUS:
- En términos generales, los principales causantes de la enfermedad del Alzheimer son los péptidos beta amiloides (Aβ o Abeta) que proceden de la escisión proteolítica de la APP.
- Se ha visto que in vitro, Abeta puede sufrir cambios conformacionales, pasando de estructura soluble a láminas beta fibrilares, las cuales se agregan, siendo neurotóxicas.
- Algunos estudios han visto que se trata de una estructura formada por dos tramos helicoidales, unidos por un puente flexible, pero de los cuales no se conoce con precisión la longitud y posición.
- La similitud que presenta esta estructura junto con la secuencia del resto C-terminal con el dominio de unión de hemaglutinina de influenza muestra la posibilidad de su capacidad neurotóxica. (Crescenzi, Orlando et al., 2022)
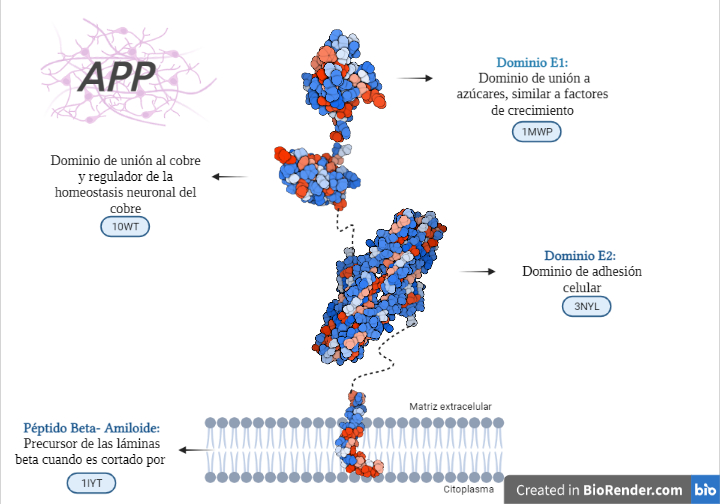
Estructura de los diferentes dominios de la APP con diferenciación entre partes polares (azules) y apolares (rojas). Creada por María Orenes a partir de ChimeraX y BioRender.com
FUNCIÓN BIOLÓGICA
Las funciones biológicas de la APP no han sido identificadas del todo desde el descubrimiento de esta proteína, pero sigue siendo objeto de numerosas investigaciones. Aunque su rol en la enfermedad del Alzheimer es la más conocida, cumple muchas otras funciones relevantes además de la vía patogénica tales como: la neurogénesis (nacimiento de nuevas neuronas), la maduración del cerebro, el crecimiento de las neuronas, la plasticidad, y la formación y reparación sináptica, de ahí que sea tan abundante en el cerebro en relación con otras zonas del cuerpo. También la interacción célula a célula, la adhesión célula-sustrato, apoptosis, el metabolismo de calcio o la transducción de señales a través de la membrana. Todas ellas continúan aún bajo estudio. (Zhang et al., 2011).
Una de las isoformas de la APP, como es la APP659, está involucrada en la maduración del cerebro durante la embriogénesis. Además, se requiere en la formación de la sinapsis neuromuscular; ya que se localiza con receptores de la acetilcolina. La abundancia de la APP durante la sinaptogénesis sugiere un papel funcional en la formación de la red neuronal y la participación en el crecimiento y restauración neuronal después de traumas cerebrales, ya que ayuda a la organización de las fibras neuronales. También interacciona con numerosas proteínas regulando el tráfico celular y el procesamiento de señales.
Otra función neurotrófica atribuida a la APP es la propiedad ferroxidasa y de tráfico de hierro, por lo que puede que alguno de sus fragmentos se encuentren regulados por este metal, el cual actúa además en el metabolismo de la APP al regula la expresión de su ARNm. De igual manera actúa el cobre sobre la APP, sugiriendo que la APP participe en la homeostasis del cobre y sea junto con el hierro una posible diana terapéutica en el Alzheimer. (Nalivaeva NN & Turner AJ, 2013)
A continuación hablaremos con más profundidad sobre algunas de sus funciones:
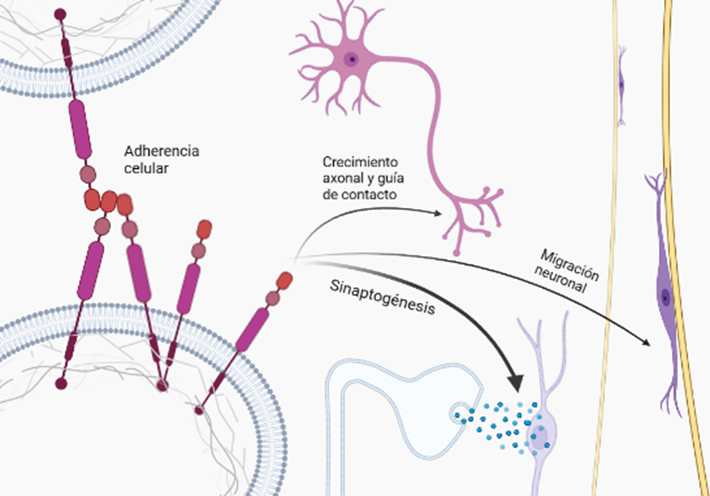
- Adhesión celular
Una de las funciones mencionada previamente es la de adhesión celular e interacción célula-célula, necesarias para diferentes etapas del neurodesarrollo, como la construcción de la red neuronal y madurez funcional de numerosas etapas complejas de migración de neuronas a la apropiada capa de la placa cortical y la acción conjunta de las CAM (moléculas de adhesión celular) y APP. Algunas CAM como las integrinas, se localizan en los conos de crecimiento y las dendritas neuronales, y la interacción con la APP proporciona la tracción necesaria para la generación de las espinas dendríticas y el cono de crecimiento (GC) en el axón, que detectará el entorno para la correcta formación de la sinapsis. (Sosa et al., 2017).
Espinas dendríticas. Imagen de Valencia Segura, R. K., Colín Barenque, L., & Fortoul van der Goes, T. I. (2018). Las espinas dendríticas, su función y algunas alteraciones. Revista de la Facultad de Medicina (México), 61(1), 46-55.

Cono de crecimiento. Imagen de https://www.researchgate.net/publication/318827764/figure/fig2/AS:522578831261696@1501603910083/Figura-2-4a-cono-de-crecimiento-en-el-extremo-de-un-axon-en-proceso-de-elongacion-b.png
Sosa et. al, The physiological role of the amyloid precursor protein as an adhesion molecule in the developing nervous system.
Esto implica un importante equilibrio y coordinación entre adhesión y movilidad, procesos mutuamente dependientes, ya que en caso de romperse esa relación con un exceso o insuficiencia de adhesión, se puede provocar una alteración en la organización normal de la red neuronal. En los casos de síndrome de Down (DS), donde el cromosoma 21 se encuentra duplicado y donde se localiza la APP sobreexpresada, da lugar a un desequilibrio que puede culminar en neurodegeneraciones similares al Alzheimer. Hay evidencias de que la APP presenta características importantes en sus dominios similares a las moléculas de adhesión celular (CAM), como la capacidad de generar una interacción con proteínas de andamio asociadas al citoesqueleto en el dominio citoplasmático, y de formar dímeros en el dominio extracelular. Esta capacidad de dimerización viene determinada por la capacidad del dominio extracelular de la APP (E2) de formar estructuras cristalinas junto con otros miembros de la familia APP, fundamental para la interacción transcelular. (Soba et al., 2005)
Por lo que la acción conjunta y mecánica de vinculación con elementos de la ECM (membrana extracelular) y la señalización, genera que la APP participe en la remodelación de las adherencias celulares así como en la organización dinámica del citoesqueleto subcortical, al coordinarse con lamelipodios y filopodios contenidos en este.
- Proteínas adaptadoras citoplasmáticas
Para determinar las funciones de la APP es importante saber con qué proteínas interactúa. Se ha encontrado que la familia de proteínas FE65 de los conos de crecimiento y sinapsis interactúan con ella a través de su C-terminal citoplasmático, por eso estas proteínas adaptadoras han sugerido roles para la APP en la dinámica del cono de crecimiento, la migración neuronal y la señalización intracelular. Además, FE65 asociada con el dominio intracelular (AICD) de la APP puede afectar a la transcripción de genes, ya que APP AICD-FE65 se libera de la membrana y se dirige al núcleo donde puede alterar la transcripción de genes. (Hoe et al., 2008). Aunque todavía quedan muchas dudas sobre los efectos en el tráfico y procesamiento de este tipo de proteínas adaptadoras en la APP, existen estudios que analizan este tipo de cuestiones (Borg et al. 1996; Parisiadou y Efthimiopoulos 2007).
- Unión de ligandos
Los procesos funcionales de la APP sugieren que funcione como un receptor transmembrana de determinados ligandos que afectan a su procesamiento. Exámenes recientes han demostrado que el principal factor genético de riesgo de la enfermedad del Alzheimer (EA) de inicio tardío es la apoliproteína E (ApoE) debido a las interacciones funcionales y estructurales con la APP, ya que estas asociaciones pueden afectar a la producción de la proteína beta amiloide (Aβ). En ausencia de la ApoE, la producción de Aβ, la secreción de APP y la estabilidad de los fragmentos de APP C-terminal se ven afectados. (Pietrzik et al., 2002)
La ApoE se encuentra mediada por una familia de receptores de lipoproteínas de baja densidad (LDLR) que se encuentran a su vez divididas en dos subgrupos. Actúan en la transducción de señales y la neurotransmisión, además, todos se encuentran dentro de las placas seniles y se han asociado genéticamente con la Enfermedad de Alzheimer.
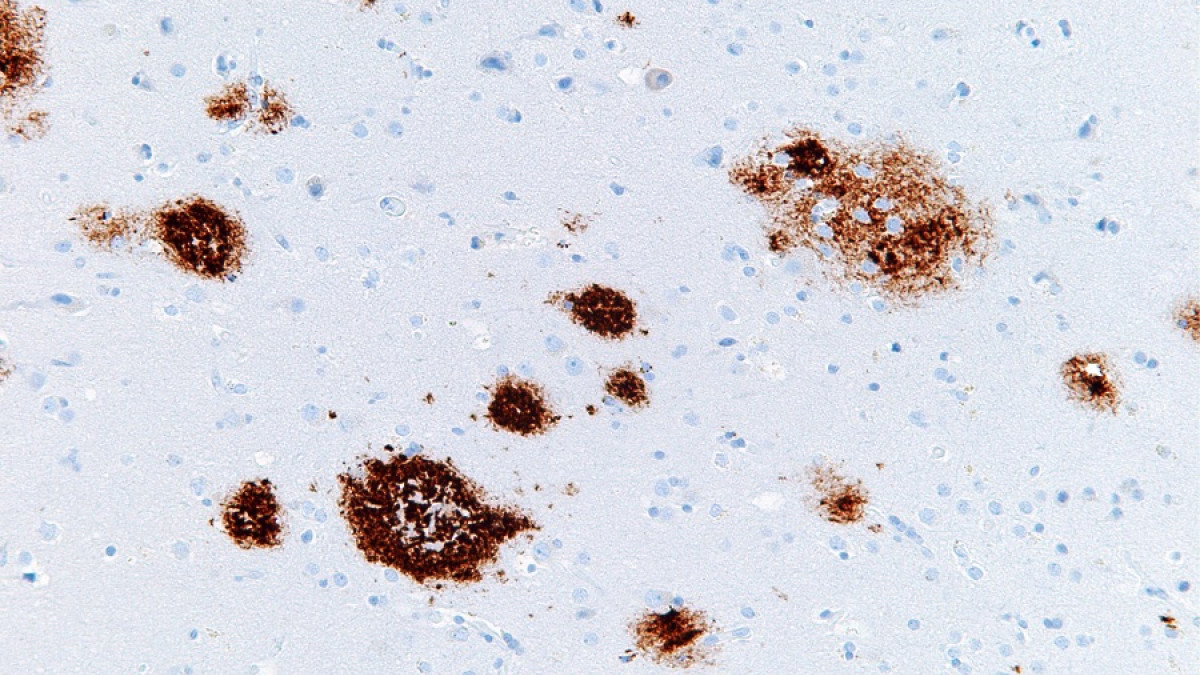
Placas seniles. Imagen de Curiosoando.com (Actualizado el 16 noviembre, 2018). «¿Qué son las placas neuríticas o placas seniles?». Disponible en https://curiosoando.com/que-son-las-placas-neuriticas-o-placas-seniles
La APP y los receptores de la apoE comparten una serie de rasgos estructurales y funcionales. Ambas son proteínas transmembrana de tipo I y presentan grandes dominios extracelulares y pequeños dominios citoplasmáticos, que en el caso de los receptores de la ApoE pueden ser numerosos mientras que en la APP observamos sólo uno para un número de proteínas adaptadoras (Stolt y Bock 2006). Además, comparten un patrón de proteólisis y de escisión superficial constitutiva e inducible que generan formas solubles que nos hace pensar cada vez más que la APP puede actuar como receptor. Una vez estudiemos y comprendamos cómo funciona la ApoE podremos quizás saber si la APP lo hace de manera similar.
- Apoptosis
Aunque no se sabe con claridad los mecanismos de muerte celular en la enfermedad del Alzheimer (EA), se asocia con la pérdida general de neuronas y mutaciones en el gen de la APP que conducen a un aumento de producción de la Aβ. La APP-BP1, identificada como una proteína de unión a la APP, es la subunidad reguladora de la enzima E1 activadora de una proteína similar a la ubiquitina NEDD8 que participa en una vía para la ubiquitinación. (Chen et al., 2003), y juega un papel importante en la progresión del ciclo celular y la supervivencia.
APP-BP1 es un gen cuyas proteínas codificadas al interactuar con la APP impulsa la transición de la fase S a la M en células neuronales en división y causa apoptosis en neuronas por la vía de conjugación de NEDD8 (neddilación). La sobreexpresión de la APP-BP1 en neuronas primarias que conducen a la apoptosis puede ser bloqueada con la inhibición de la neddilación y la coexpresión de un péptido que compite con la APP-BP1 por unirse a la APP. Se observa una sobreexpresión de la APP-BP1 en el hipocampo cerebral de las personas con EA, ya que participa como hemos mencionado antes, en la activación de NEDD8. (Chen et al., 2000)
MECANISMOS DE ACCIÓN
- Formas tempranas de la EA:
Se han descubierto tres genes causantes de formas tempranas de la Enfermedad del Alzheimer: el gen que codifica para la proteína precursora del péptido β-amiloide (APP), el gen de la presenilina 1 (PSEN1) y el de la presenilina 2 (PSEN2).
- Gen codificante para APP:
Vía no amiloidogénica:
Por esta vía tiene lugar el corte de alfa-secretasa (entre los aminoácidos 687 y 688). Este da lugar a dos fragmentos: uno de ellos es sAPPα, el cual es soluble y se expulsa al exterior celular; mientras que el otro permanece en la membrana, y se conoce como C83.
Y es este último fragmento el que posteriormente será procesado por la γ-secretasa, cortando por los aminoácidos 712, 714 o 715, que corresponden con los residuos 40, 42 o 43 del péptido Aβ.
Esta vía es conocida como no amiloidogénica, es decir, no patológica, ya que como producto final no se obtiene un agregado peptídico.
Vía amiloidogénica:
Por otra parte, existe otra vía donde intervienen la beta-secretasa y la γ-secretasa.
El corte de β-secretasa se produce entre los aminoácidos 671 y 672, liberando al exterior celular el fragmento, sAPPβ, el cual también es soluble.
Al igual que en la vía anterior, en la membrana permanece otro fragmento, llamado C99, es procesado también por la γ-secretasa, cortando por los mismos aminoácidos 712, 714 o 715, dando lugar a tres posibles péptidos. Estos péptidos se conocen como: Aβ40, Aβ42 o Aβ43, y pueden formar agregados que forman las fibras insolubles de los depósitos de amiloide.
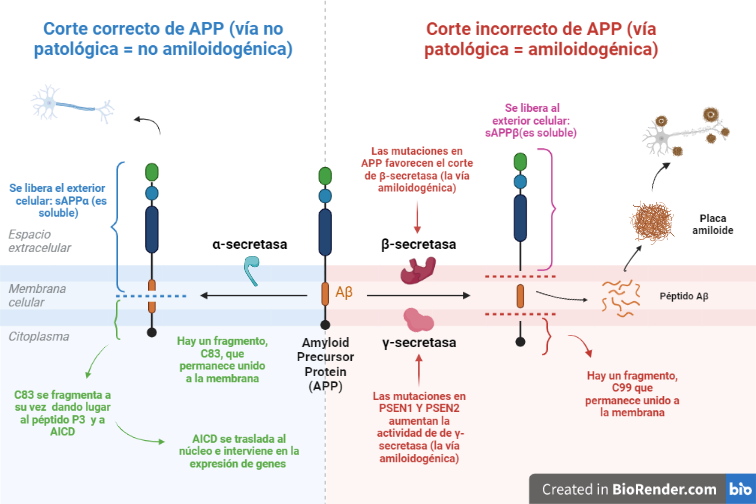
- Genes de las presenilinas: PSEN1 y PSEN2
En 1995 se clonó el gen responsable del 50% de los casos de la EA. Este se llamó inicialmente S182 pero poco después pasó a llamarse presenilina 1 (PSEN1). Hoy en día, existen 177 mutaciones en PSEN1, las cuales causan EA a edades muy tempranas.
Posteriormente, se localizó en el cromosoma 1 una secuencia genética muy similar a PSEN1. Este gen, conocido como presenilina 2 (PSEN2), es responsable de una proporción muy pequeña de los casos de EA autosómicos dominantes. Además, actualmente se sabe que las presenilinas son un cofactor del complejo multiproteico de la γ-secretasa.
- Formas tardías de EA:
Alrededor del 95% de los casos de EA aparecen a edades avanzadas.
Por el momento, el único gen que se ha relacionado con la EA tardía es el que codifica para la apolipoproteína E (APOE). El gen APOE, se localiza en el cromosoma 19 y codifica para tres isoformas : ε2, ε3 y ε4, las cuales se diferencian entre sí por la presencia de los aminoácidos cisteína o arginina en las posiciones 112 y 158 de la APOE.
De forma que si las frecuencias de cada una de las isoformas son comparadas entre pacientes, atendiendo a criterios de edad, se percibe un incremento del alelo ε4 en pacientes con EA tardía respecto a controles sanos de edades avanzadas. Esto quiere decir, que un paciente que contenga una copia del alelo ε4 tiene un riesgo de contraer la enfermedad mayor, y este será aún mayor si posee dos copias del alelo. (Setó-Salvia et al., 2010)
RELACIÓN CON LAS DROGAS
El consumo en exceso de drogas por el abuso de opiáceos es un problema extendido por todo el mundo. Sin embargo, a pesar de no conocerse gran información sobre la neuropatología que causa este consumo, estudios han concluido que la neuroinflamación que se genera podría desembocar en una neurodegeneración prematura de los cerebros de los consumidores.
Tras investigaciones de los cerebros de jóvenes consumidores , se aplicó un análisis inmunohistoquímico detallado del hipocampo, el tronco encefálico y los ganglios basales para tau hiperfosforilada, β-amiloide, proteína precursora de β-amiloide (βAPP) y ubiquitina, el cual denotó un exceso de ovillos neurofibrilares (NFTs); los cuales son causantes de la degeneración neuronal y disfunción sináptica.
A diferencia de los no consumidores, en quienes los NFTs solamente se localizaron en la corteza entorrinal; en aquellos que sí consumían drogas también se localizaron en el subículo, la neocorteza temporal, el núcleo basal de Meynert y el locus coeruleus.
Además, los consumidores de drogas dieron positivo en βAPP en mayor proporción que en los no consumidores, aunque prácticamente no se localizaron placas de beta-amiloide. (Ramage et al., 2005)
IMPLICACIÓN DE LA PROTEÍNA TAU

Papel de la proteína TAU en la Enfermedad del Alzheimer. Creada por Almudena López a partir de BioRender.com (Vallés-saiz, L., 2022)
DAÑO CEREBRAL
La encefalopatía traumática crónica (ETC) es una enfermedad neurodegenerativa que se asocia con lesiones cerebrales traumáticas leves repetitivas. De forma patológica se caracteriza por la acumulación anormal de la proteína TAU.
Para determinar el alcance de la extensión del depósito del péptido β amiloide (Aβ), se realizaron estudios en atletas y militares veteranos con diagnóstico de ETC. Gracias a estos se descubrió que más del 52% de los pacientes diagnosticados con CTE portaban depósitos del péptido Aβ. Además, esta acumulación de este péptido tenía lugar a una velocidad mayor que en aquellos que no diagnosticados con CTE. E incluso, un subgrupo cumplió con los criterios de diagnóstico tanto para CTE como para EA.
En conclusión, estos resultados proponen que existe una alteración y aceleración en el depósito de Aβ en un conjunto de individuos con CTE si lo comparamos con el envejecimiento normal, y que Aβ mantiene una fuerte relación con la progresión patológica y clínica de CTE, independientemente de la edad. (Stein et al., 2015)
TRATAMIENTOS
- Antioxidantes:
Se ha demostrado que el estrés oxidativo, es decir, la acumulación de productos derivados de la cadena de transporte de electrones, tales como: el radical hidroxilo, superóxido o el peróxido de hidrógeno (localizados en la mitocondria) son responsables de estrés oxidativo. Este es a su vez precedente de las lesiones que se generan en la Enfermedad de Alzheimer. Además, los radicales libres podrían ser los causantes de cambios en pares de bases del ADN, o incluso de daños en las cadenas de ADN. Es por esto que uno de los tratamientos se basa en combatir este estrés oxidativo para lograr paliar el deterioro cognitivo de la enfermedad.
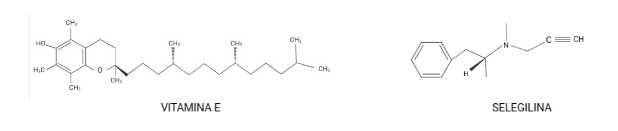
Durante un trabajo realizado por el Alzheimer’s Disease Cooperative Study, en el que se usaron los antioxidantes: α-tocoferol (mayormente conocido como vitamina E) y selegilina, se señaló un posible efecto positivo de la vitamina E en la EA.
Una de las principales conclusiones fue la prolongación del tiempo en el que ocurrían procesos, como por ejemplo la pérdida de la capacidad para realizar algunas actividades de la vida cotidiana (basándose en la escala de puntuación clínica de la demencia) era más largo en aquellos que tomaban vitamina E que en comparación con quienes tomaban selegilina, ambos fármacos o placebo. Sin embargo, el uso de vitamina E o selegilina no causó alteraciones en el deterioro cognitivo. Estos fármacos se seleccionaron porque la vitamina E es un “scavenger” (destructor) de radicales libres; y por su parte la selegilina inhibe la desaminación oxidativa. (Shah et al., 2008)
- Agentes nootrópicos:
Los agentes nootrópicos son derivados cíclicos del ácido γ-aminobutírico (GABA), este es un aminoácido no proteico que se localiza en altas concentraciones en el SNC de los mamíferos, cuya su función principal es actuar como un neurotransmisor inhibidor. Algunos de estos ejemplos pueden ser : piracetam, pramiracetam, aniracetam u oxiracetam. En concreto el primero de ellos, ha manifestado beneficios a corto y largo plazo, principalmente en los relacionado a las funciones de la atención y la memoria. Asimismo, estudios en animales suscitan que estimulan la síntesis y liberación de AcH y que desarrollan una función sobre los receptores de glutamato AMPA. (Santos-Espinosa et al., 2018)
El glutamato es el principal neurotransmisor excitatorio en las neuronas corticales e hipocampales.
La glutamina sintetasa, se oxida en los cerebros de las personas con EA, lo que conduce a un exceso de glutamato. La activación excesiva de los receptores NMDA por el glutamato, aumenta la vulnerabilidad de las neuronas del SNC; lo que concluye en degeneración neuronal.
Un ejemplo de estos fármacos es la memantina, esta se emplea para el bloqueo de los canales NMDA controlados por glutamato, inhibiendo así su activación patológica, pero manteniendo la activación fisiológica.
- Inhibidores de la acetilcolinesterasa:
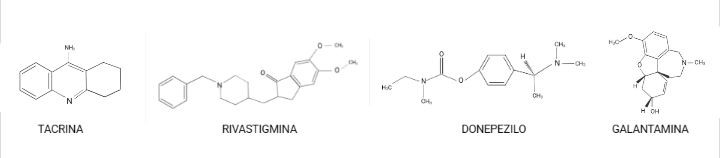
Como por ejemplo: tacrina, donepezilo, rivastigmina y galantamina son los principales tratamientos contra el Alzheimer (leve a moderada). Aunque poseen distintas propiedades farmacológicas, todos ellos inhiben la acción de la AChE.
Estos evitan la degradación del neurotransmisor (acetilcolina); de forma que favorecen un aumento de los niveles de ACh en la hendidura sináptica y facilitan los efectos de los neurotransmisores tanto nicotínico como muscarínico, lo que en ambos casos contribuye a mejorar la cognición. Sin embargo, en estudios realizados a largo plazo, se ha observado un deterioro progresivo de los pacientes, lo que nos indica que estos fármacos nos proporcionan una mejora de los síntomas de la enfermedad solamente durante un periodo de tiempo, y que, por tanto, no actúan sobre los mecanismos patogénicos de la enfermedad. (Golimstok et al., 2006)
ALZHEIMER EN LA ACTUALIDAD
La enfermedad causada por el mal corte de esta proteína es una de las principales causas de demencia en nuestro país, así como en el mundo. Según datos del SEN (“Sociedad Española de Neurociencia”), hoy en día en España unas 800.000 personas sufren la enfermedad, incrementando cada año aproximadamente en 40.000 nuevos casos. No obstante, se estima que entre el 30 y el 40% de los casos totales están aún sin diagnosticar.
En el año 2015, alrededor de 47 millones de personas sufrían demencia en el mundo, y conforme a las proyecciones de población, si las cifras actuales se mantienen, en 2050 la cifra podría llegar a 130 millones de personas. En el caso de España, entre un 3 y un 4% de la población de entre 75 y 79 años sufren Alzheimer, aumentando a 34% en mayores de 85. En mayores de 65, la SEN estima que alrededor de un 15 % de la población sufre deterioro cognitivo leve, que puede deberse a la enfermedad en el 50% de los años aproximadamente.
A continuación, se exponen citas del Dr. Juan Fortea, Coordinador del grupo de Estudio de Conducta y Demencias de la Sociedad Española de Neurología (SEN):
“La enfermedad de Alzheimer es la primera causa de demencia neurodegenerativa en el mundo y supone un problema sanitario de primer orden. Además, debido a que es una enfermedad cuya prevalencia aumenta exponencialmente a partir de los 65 años, ante el progresivo envejecimiento de la población española, urge el desarrollo de políticas sanitarias destinadas a garantizar el adecuado diagnóstico y acceso a los tratamientos presentes y futuros en nuestro país, así como la puesta en marcha de registros nacionales que permitan precisar la verdadera prevalencia e incidencia del Alzheimer”.
Dr. Juan Fortea, Coordinador del grupo de Estudio de Conducta y Demencias de la Sociedad Española de Neurología (SEN).
“Se estima que la mitad de los casos de la enfermedad de Alzheimer se puede atribuir a nueve factores de riesgo potencialmente modificables: diabetes mellitus, hipertensión arterial en edad media de vida, obesidad e edad media de vida, tabaquismo, inactividad física, depresión, actividad cognitiva o bajo nivel educativo, la hipoacusia y el aislamiento social, por lo que una reducción de entre un 10 y un 15% en dichos factores de riesgo podrían potencialmente prevenir entre 1 y 3 millones de casos de Alzheimer en el mundo”.
Dr. Juan Fortea, Coordinador del grupo de Estudio de Conducta y Demencias de la Sociedad Española de Neurología (SEN).
Referencias
Burley, S. K., Berman, H. M., Kleywegt, G. J., Markley, J. L., Nakamura, H., & Velankar, S. (2017). Protein Data Bank (PDB): the single global macromolecular structure archive. Protein Crystallography, 627-641.
Chen, Y., Liu, W., McPhie, D. L., Hassinger, L., & Neve, R. L. (2003). APP-BP1 mediates APP-induced apoptosis and DNA synthesis and is increased in Alzheimer’s disease brain. The Journal of cell biology, 163(1), 27-33.
Chen, Y., McPhie, D. L., Hirschberg, J., & Neve, R. L. (2000). The amyloid precursor protein-binding protein APP-BP1 drives the cell cycle through the SM checkpoint and causes apoptosis in neurons. Journal of Biological Chemistry, 275(12), 8929-8935
Cho, Y., Bae, H. G., Okun, E., Arumugam, T. V., & Jo, D. G. (2022). Physiology and pharmacology of amyloid precursor protein. Pharmacology & Therapeutics, 108122..
Crescenzi, Orlando et al. “Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain.” European journal of biochemistry vol. 269,22 (2002): 5642-8. doi:10.1046/j.1432-1033.2002.03271.x
Goodsell, D. Molecule of the month: Amyloid-beta precursor protein. 10.2210/rcsb_pdb/mom_2006_7
Golimstok, A. (2006). Tratamiento de la enfermedad de Alzheimer. Rev. Hosp. Ital. B. Aires Vol, 26(4), 132.
Hoe, H. S., & William Rebeck, G. (2008). Functional interactions of APP with the apoE receptor family. Journal of neurochemistry, 106(6), 2263-2271.
Lee S, Xue Y, Hu J, Wang Y, Liu X, Demeler B, Ha Y. The E2 domains of APP and APLP1 share a conserved mode of dimerization. Biochemistry. 2011 Jun 21;50(24):5453-64. doi: 10.1021/bi101846x. Epub 2011 May 26. PMID: 21574595; PMCID: PMC3120129.
Nalivaeva NN, Turner AJ. The amyloid precursor protein: a biochemical enigma in brain development, function and disease. FEBS Lett. 2013 Jun 27;587(13):2046-54. doi: 10.1016/j.febslet.2013.05.010. Epub 2013 May 16. PMID: 23684647
Pietrzik, C. U., Busse, T., Merriam, D. E., Weggen, S., & Koo, E. H. (2002). The cytoplasmic domain of the LDL receptor-related protein regulates multiple steps in APP processing. The EMBO journal, 21(21), 5691-5700.
Ramage, S. N., Anthony, I. C., Carnie, F. W., Busuttil, A., Robertson, R., & Bell, J. E. (2005). Hyperphosphorylated tau and amyloid precursor protein deposition is increased in the brains of young drug abusers. Neuropathology and applied neurobiology, 31(4), 439-448.
Santos-Espinosa, A., Manzanarez-Quin, C. G., Reyes-Díaz, R., Hernández-Mendoza, A., Vallejo-Cordoba, B., & González-Córdova, A. F. (2018). Ácido γ-aminobutírico (GABA) producido por bacterias àcido lácticas en alimentos fermentados. Interciencia, 43(3), 175-181.
Setó-Salvia, N., & Clarimón, J. (2010). Genética en la enfermedad de Alzheimer. Revista de Neurología, 50(6), 360-364.
Shah, R. S., Lee, H. G., Xiongwei, Z., Perry, G., Smith, M. A., & Castellani, R. J. (2008). Current approaches in the treatment of Alzheimer’s disease. Biomedicine & Pharmacotherapy, 62(4), 199-207.
Soba, P., Eggert, S., Wagner, K., Zentgraf, H., Siehl, K., Kreger, S., … & Beyreuther, K. (2005). Homo‐and heterodimerization of APP family members promotes intercellular adhesion. The EMBO journal, 24(20), 3624-3634.
Sosa, L. J., Cáceres, A., Dupraz, S., Oksdath, M., Quiroga, S., & Lorenzo, A. (2017). The physiological role of the amyloid precursor protein as an adhesion molecule in the developing nervous system. Journal of neurochemistry, 143(1), 11-29.
Stein, T. D., Montenigro, P. H., Alvarez, V. E., Xia, W., Crary, J. F., Tripodis, Y., … & McKee, A. C. (2015). Beta-amyloid deposition in chronic traumatic encephalopathy. Acta neuropathologica, 130(1), 21-34.
Vallés-Saiz, L. (2022). Nuevas funciones para la proteína tau en el Sistema Nervioso Central y tejidos periféricos.