Enfermedades mitocondriales
Por Marta García Arauzo y Miriam González Ferrero, Biología sanitaria UAH
Acaba de comenzar febrero, el mes de concienciación de las enfermedades raras. Aunque las enfermedades raras son aquellas con baja prevalencia; se estima que alrededor de un 7% de la población mundial se ve afectada por alguna, lo que resulta ser un gran número de personas. De hecho, cada 30 minutos nace un niño que para los 10 años habrá desarrollado una enfermedad mitocondrial. Las enfermedades mitocondriales son un grupo de trastornos asociados a fallos en el sistema de fosforilación oxidativa. Parte de los componentes de este sistema se codifican en el DNA mitocondrial, y mutaciones en él producirán patologías con síntomas bien definidos.
Cada 30 minutos nace un niño que para los 10 años habrá desarrollado una enfermedad mitocondrial.
Si quieres profundizar sobre las causas y conocer las enfermedades mitocondriales más importantes, ¡sigue leyendo!
Las mitocondrias son orgánulos citoplasmáticos con la función principal de generar energía en forma de ATP, mediante el proceso de fosforilación oxidativa en condiciones aerobias. Esto se realiza gracias a los complejos enzimáticos I-V de la cadena de transporte de electrones y a dos portadores de electrones (coenzima Q y citocromo c).

Otra función de las mitocondrias es contribuir a la apoptosis o muerte celular programada. Además, presentan funciones específicas de tipo celular como son el metabolismo del colesterol, la oxidación β de los ácidos grasos, la biosíntesis de esteroides sexuales y del hemo, y la termogénesis, entre otras.
Estos orgánulos presentan su propio sistema genético con toda la maquinaria necesaria para su expresión, es decir, replica, transcribe y traduce la información genética que contiene. Por ello se piensa que las mitocondrias tienen un origen endosimbiótico, donde una bacteria fue fagocitada por una célula eucariota ancestral.

El ácido desoxirribonucleico mitocondrial (mtDNA) es una molécula bicatenaria, circular, con un peso molecular de 11 000 000 daltons aproximadamente, sin intrones, se replica a partir de un solo origen y es muy pequeño. El genoma mitocondrial del ser humano se encuentra completamente secuenciado, está compuesto por 16 569 pares de bases que corresponden a un pequeño número de genes. Éstos se encuentran distribuidos entre la cadena pesada H (5 168 726 daltons) y la cadena ligera L (5 060 609 daltons).

La iniciación de la transcripción puede darse en ambas cadenas y se producirá un RNA precursor policistrónico, que será procesado para originar 13 mRNA individuales y 24 productos individuales de tRNA y rRNA.
El 93% de este ácido desoxirribonucleico comprende región codificante; el resto sería de DNA no codificante, que se piensa que actúa controlando la iniciación de la replicación y de la transcripción.
El DNA mitocondrial tiene información para 37 genes que codifican: 13 proteínas que forman parte de subunidades de 4 de los 5 complejos multienzimáticos del sistema de fosforilación oxidativa (sistema Oxphos), 22 ácidos ribonucleicos de transferencia (tRNAs) capaces de leer todo el código genético y 2 ácidos ribonucleicos ribosomales (rRNAs) componentes de los ribosomas específicos mitocondriales. Estos tRNAs y rRNAs se encargan de traducir las 13 proteínas codificadas por el mtDNA.

El resto de polipéptidos que componen estos complejos, así como el complejo II completo, están codificados en el DNA nuclear. Por tanto, la biogénesis del sistema Oxphos requiere una regulación y expresión coordinada entre los genes del mtDNA y el DNA del núcleo celular.
Las subunidades que están codificadas en el DNA nuclear se sintetizan en el citosol, y deben ser transportadas a la mitocondria. Para ello, es imprescindible la proteína de choque térmico chaperonina, que despliega estas subunidades proteicas para que puedan atravesar la doble membrana mitocondrial en forma de largas cadenas. En la matriz mitocondrial, estas subunidades se vuelven a plegar para poder ensamblarse con los demás componentes que forman la cadena de transporte electrónico.
La genética del ácido desoxirribonucleico mitocondrial es muy peculiar, y presenta 5 aspectos fundamentales: poliplasmia, segregación mitótica, efecto umbral, herencia materna y una alta velocidad de mutación.
POLIPLASMIA
Cada mitocondria contiene entre 2 y 10 moléculas de mtDNA, y cada célula contiene múltiples mitocondrias. Por lo que hay un alto número de copias de mtDNA en cada célula (1 000 – 10 000), dependiendo del tejido. Al conjunto de mitocondrias de una célula se le denomina condrioma.
Normalmente, todas las mitocondrias poseen el mismo genoma, lo que se conoce como homoplasmia. Sin embargo, puede producirse una mutación en el mtDNA que afecte solo a algunas mitocondrias, así en una misma célula se encuentran mitocondrias con mtDNA mutado y mitocondrias con mtDNA normal; a esto se le denomina heteroplasmia.
También pueden existir células que en todas sus mitocondrias contengan mtDNA mutado, y la célula se encuentra en homoplasmia pero con mtDNA alterado.
SEGREGACIÓN MITÓTICA
El fenotipo de una línea celular puede variar durante la división celular, ya que las mitocondrias se distribuyen al azar entre las células hijas. Por tanto, si en una célula coexisten mtDNA normal y mtDNA mutado (heteroplasmia), tras varias divisiones celulares, se distribuyen distintas proporciones de genoma mitocondrial mutado y nativo en las células hijas. Así, se originarán tres genotipos diferentes: homoplasmia de mtDNA normal, heteroplasmia y homoplasmia de mtDNA mutado.

EFECTO UMBRAL
El fenotipo de una célula heteroplásmica depende del porcentaje de mtDNA mutado que presente. Si el número de moléculas de mtDNA alterado es relativamente bajo, se producirá una complementación con las moléculas de mtDNA normal y no habrá manifestación del defecto genético.
Cuando el mtDNA mutado sobrepasa un umbral determinado, sí se manifestará un fenotipo patógeno. De este modo, la producción de ATP está por debajo de los mínimos necesarios para el funcionamiento normal de los tejidos, ya que las proteínas codificadas en el mtDNA serán defectuosas, y esto origina una enfermedad en la persona.
El número de moléculas de mtDNA, es decir, el umbral, será distinto en cada órgano y tejido. Esto dependerá de la cantidad de energía requerida para su funcionamiento. Por ello, los tejidos que con mayor frecuencia se ven afectados son la visión, el sistema nervioso central, el corazón, el músculo esquelético, los islotes pancreáticos, el riñón y el hígado.
HERENCIA MATERNA
El mtDNA se hereda con un patrón vertical, no mendeliano, exclusivamente por vía materna. La madre transmite su genoma mitocondrial a todos sus hijos, pero solamente las hijas lo transmitirán a todos los miembros en la siguiente generación, y así sucesivamente.

Esto es debido al gran número de moléculas de mtDNA que contienen los óvulos (entre 100 000 y 200 000 copias), en comparación con unos pocos cientos que contienen los espermatozoides. Además, las mitocondrias que pueden entrar en el óvulo fecundado se eliminan por un proceso complejo en el que participa el sistema proteasoma ubiquitina.
En conclusión, las enfermedades vinculadas a mutaciones en el mtDNA son heredadas de manera exclusiva por línea materna.
ALTA VELOCIDAD DE MUTACIÓN
A diferencia de la recombinación de pares homólogos que tiene lugar en el núcleo, las moléculas de mtDNA no sufren recombinación, por lo que las mutaciones representan la única posibilidad de diversificación genética del mtDNA.
La tasa de mutación espontánea del mtDNA es de unas 10 a 20 veces mayor que en el DNA nuclear. Esto puede deberse a que en la mitocondria se producen continuamente especies reactivas de oxígeno (ROS), como consecuencia de la oxidación final de los compuestos carbonados. Por lo que el mtDNA es particularmente susceptible al daño oxidativo debido a este íntimo contacto con los radicales libres. Éstos dañarán este DNA que no está protegido por histonas y, además, presenta muy pocos mecanismos de reparación.
Algunas de las variaciones de la secuencia serán polimorfismos silentes que no tienen el potencial de un efecto patógeno, mientras que otras variaciones podrán ser mutaciones patógenas.
Las mitocondrias disfuncionales pueden ser eliminadas por digestión lisosomal o, también, las células que posean mitocondrias disfuncionales pueden sufrir apoptosis.
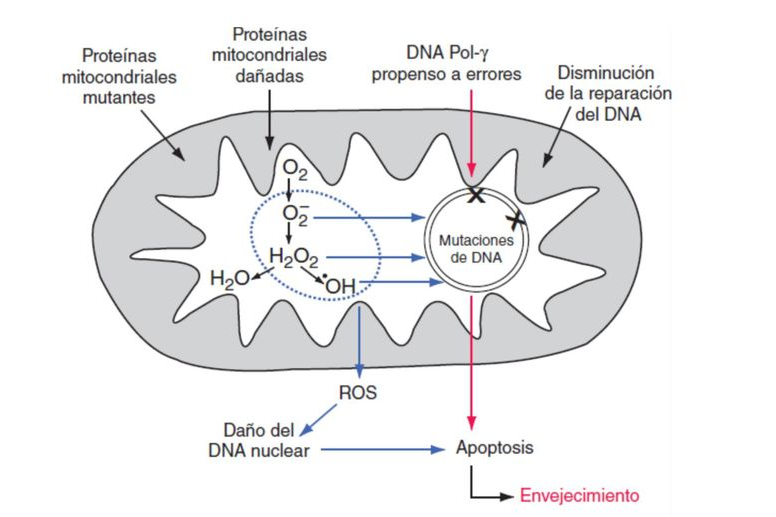
En relación al envejecimiento, la acumulación de las mutaciones en el mtDNA en células somáticas puede contribuir a enfermedades relacionadas con la edad, como el síndrome metabólico y diabetes, cáncer, trastornos cardiovasculares y degenerativos del sistema nervioso central (Alzheimer y Parkinson). Esto es debido a que estas mutaciones contribuyen a la declinación de la fosforilación oxidativa, y así disminución de la capacidad respiratoria de los tejidos. Por tanto, las mitocondrias se deterioran a lo largo de la vida, como consecuencia de la acumulación de mutaciones.
A su vez, la acumulación de mutaciones en el mtDNA producirá un sistema Oxphos ineficaz, con la posibilidad de generación excesiva de ROS, lo que hace que se cree un “círculo vicioso” de daño acumulativo en el genoma mitocondrial.
No obstante, dichas mutaciones somáticas del mtDNA no son transmitidas a la descendencia, por lo que el efecto hereditario de la mutagénesis del mtDNA requiere considerar aparte los acontecimientos en la línea germinal femenina.
Otras utilidades del genoma mitocondrial
Las variaciones en la secuencia de mtDNA entre diferentes individuos han resultado muy útiles para obtener un mayor conocimiento en otras áreas no relacionadas directamente con la clínica, como pueden ser estudios antropológicos, etnológicos y forenses.
En medicina forense, la genética mitocondrial se aplica para identificar personas a partir de restos humanos con abundante mtDNA, como cabello, huesos o dientes.
En el ámbito antropológico, el análisis comparativo del mtDNA entre distintos grupos étnicos del mundo permite establecer la manera en la que se desarrollaron las migraciones de grupos humanos a lo largo del planeta.
ENFERMEDADES DEL DNA MITOCONDRIAL
El término enfermedad mitocondrial agrupa a todas las afecciones relacionadas con alteraciones en el metabolismo oxidativo mitocondrial. El denominador común de estas patologías es la deficiencia en la biosíntesis de ATP, moneda energética de nuestro organismo.
El origen de estas enfermedades reside en mutaciones del mtDNA (mutaciones puntuales, deleciones o duplicaciones), y en alteraciones de la señalización genómica entre el DNA nuclear y el mitocondrial. Si la disfunción mitocondrial ocurre en la cadena respiratoria, estaremos hablando de citopatías mitocondriales.
Pinchando aquí puedes ver las enfermedades producidas por las mutaciones en los distintos genes relacionados con la cadena mitocondrial. Facilitado por la AEPMI (asociación de enfermos de patología mitocondrial).
Los déficits enzimáticos de la cadena respiratoria pueden ser únicos (cuando afectan a un solo complejo de la cadena respiratoria), o múltiples (afectan a varios). Las mutaciones del mtDNA suelen ser causadas por déficits múltiples, afectando a varios tejidos y causando la heterogeneidad de cuadros clínicos. De hecho; el mal funcionamiento de tres o más sistemas es característica principal del diagnóstico de las enfermedades mitocondriales.
Desde que en la década de 1960 se observaron por primera vez acúmulos musculares de mitocondrias en individuos con intolerancia al ejercicio, posteriormente se han ido relacionando las disfunciones mitocondriales con numerosas enfermedades: trastornos neurodegenerativos, cardíacos, enfermedades neurometabólicas, cáncer, obesidad, etc. Al limitarse el nivel de ATP, suelen verse más afectados los tejidos con alta necesidad energética, como cerebro, músculo, hígado, corazón, riñones y SNC. Entre las manifestaciones clínicas más frecuentes se encuentran la sordera, ceguera, diabetes, ataxia, debilidad muscular, disfunciones hepáticas y pancreáticas y fallos en el corazón, riñones e hígado.
Esta gran variedad de manifestaciones clínicas supone un reto para el diagnóstico de enfermedad mitocondrial. Cuando en un paciente se presentan uno o más de estos síntomas, la sospecha clínica debe ser ratificada por pruebas metabólicas específicas de disfunción mitocondrial, y finalmente, tras realizar pruebas de confirmación morfológicas, histoenzimáticas, bioquímicas o genéticas, sus resultados nos pueden conducir al diagnóstico de disfunción mitocondrial. En la siguiente imagen puedes consultar el protocolo a seguir desde la sospecha médica.

Existen síndromes clínicos específicos, bien definidos, de los que vamos a introducir los más representativos.
CPEO: Oftalmoplejía externa progresiva crónica
El signo principal de este síndrome es la blefaroptosis, también llamada ptosis palpebral o párpado caído, junto con paresia (debilidad) simétrica de los músculos extraoculares. También suele acompañarse de miopatía proximal de las extremidades, intolerancia al ejercicio y debilidad muscular.

El CPEO está causado por deleciones grandes y normalmente espontáneas del mtDNA. Se ha observado que muchas de las deleciones encontradas están delimitadas por repeticiones directas de 3 a 13 nucleótidos. Esto podría significar que la presencia de estas repeticiones conlleva a errores en la replicación, que serían los responsables de la deleción.
Algunos casos también ocurren por mutaciones puntuales, como la del gen del tRNA LeuUUR en A3243 (la A proviene de la base adenina y el número representa la posición del nucleótido 3,243). En un ínfimo porcentaje de individuos se ve afectada la región del DNA mitocondrial con genes que codifican para rRNA.
Este síndrome aparece antes de los primeros 20 años de vida.
Síndrome de Kearns-Sayre
Es una enfermedad variante de la CPEO más grave y peculiar que también aparece antes de los 20 años de edad. Además de oftalmoplejía progresiva crónica y retinitis pigmentaria atípica se suma uno de los siguientes problemas: bloqueo cardíaco, síndrome cerebeloso (alteración funcional del cerebelo) e hiperproteinorraquia, que consiste en una cantidad de proteína superior a 100 mg/L en LCR. Al conjunto de estos tres problemas multisistémicos se denomina la tríada clásica.
La siguiente imagen es útil para explicar la retinopatía pigmentaria:

Imagen y pie de foto tomados del artículo de referencia [11].
MERRF o síndrome de epilepsia mioclónica con fibras rojo-rasgadas
Las manifestaciones clínicas de este síndrome son epilepsia mioclónica o mioclonías, convulsiones generalizadas y miopatía con presencia de fibras rojo-rasgadas.
Con estos síntomas, realizar un electroencefalograma es muy interesante para el diagnóstico, pues registra las mioclonías y puede determinar su origen.
Las mioclonías son movimientos musculares involuntarios originados por una actividad anormal de la corteza sensomotora. Las fibras rojas forman unidades motoras tónicas, de contracción lenta. Son resistentes a la fatiga y de ellas dependerá el correcto mantenimiento del equilibrio y la postura y los movimientos de poca intensidad. Por eso, la disfuncionalidad de las fibras rojas conlleva errores en este tipo de ejercicios.

Imagen y pie de foto tomados del artículo de referencia [12].
En algunos pacientes aparecen síntomas clínicos adicionales como demencia, sordera neurosensorial, neuropatía sensitiva, atrofia óptica, fallo respiratorio y cardiomiopatía.
Es un síndrome de curso progresivo que aparece tanto en la infancia como en edad adulta. Como es de herencia materna, podemos estudiarlo mediante pedigríes.
MERRF se asocia principalmente a mutaciones puntuales del gen del tRNA Lys en la posición A8344G (transición de adenina por guanina en la posición 8,344) o, de forma minoritaria, en T8356C. La mutación A8344G altera específicamente el bucle TᴪC del tRNA Lys, lo cual reduce la síntesis proteica celular. Todas las mutaciones son en forma heteroplásmica. Los jóvenes con un 95% de heteroplasmia pueden desarrollar este síndrome, mientras que los adultos de entre 60-70 años o más podrán hacerlo con solo un 60% del DNA mutado. La razón de que haya personas que nacen con fenotipo normal y desarrollan MERRF en la edad adulta puede ser que, con la edad, la capacidad oxidativa de la mitocondria se reduce, promoviendo la aparición de la enfermedad.

Extraído del artículo de referencia [8].
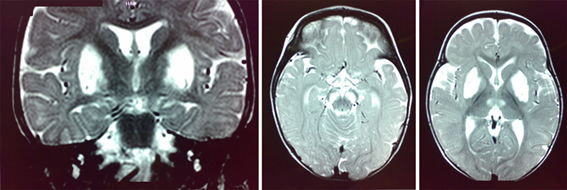
MELAS: Síndrome de encefalomiopatía mitocondrial con acidosis láctica y episodios de accidentes cerebro-vasculares
Con solo leer el nombre, ya nos hacemos una idea de los síntomas de la enfremedad. Básicamente, el acrónimo MELAS nos explica que este síndrome consiste en encefalomiopatía mitocondrial, acidosis láctica e ictus cerebrales semejantes a apoplejías. Estos ictus son reversibles y pueden ser visualizados a través de resonancia magnética o PET. Los recién nacidos con este síndrome son sanos, sin embargo, durante los primeros años de vida dejan de crecer súbitamente y se empiezan a presentar los síntomas comentados. Estas manifestaciones suelen acompañarse de convulsiones generalizadas, dolor de cabeza, sordera, demencia y, a veces, de fibras rojo-rasgadas.

La fila inferior muestra la presencia de fibras rojo-rasgadas en diafragma (izquierda) y corazón (derecha) teñidos con tinción de Gomori.
Imagen tomada del artículo de referencia [14].
MELAS, al igual que MERRF, se hereda por vía materna. Esta enfermedad ha sido asociada fundamentalmente con mutaciones en el gen del tRNA LeuUUR del mtDNA. Alrededor del 80% de los casos son causados por una transición en A3243G, pero se han encontrado otras con menor incidencia, como A3271G, también en el tRNA LeuUUR y alguna en genes codificantes de proteínas, todas en forma heteroplásmica.
Si lo recuerdas, la transición A3243G también la hemos visto en CPEO (oftalmología progresiva externa crónica), y además también aparecen sordera, miopatía o diabetes; es decir, esta mutación tiene efectos fenotípicos muy variables.
Al ser dañado el tRNA, la síntesis de proteínas mitocondriales se verá comprometida.
LHON: Neuropatía óptica de Leber
Se caracteriza por la pérdida de la visión bilateral, indolora, aguda o subaguda, debido a la atrofia del nervio óptico. Aparece en la segunda o tercera década de la vida de hombres y mujeres, más frecuentemente en los primeros. A parte de la afectación de la visión, también puede aparecer neuropatía retrobulbar y periférica, síndrome piramidal y cerebeloso, el fondo de ojo con edema, tortuosidad de los vasos retinianos e incluso trastornos en la conducción cardiaca.

El síndrome de Lohn fue la primera enfermedad humana de herencia materna que fue asociada con una mutación puntual del mtDNA. Estas mutaciones se pueden encontrar de forma homo y heteroplásmica y se han descrito en gran número. Esto es relevante pues las manifestaciones clínicas dependen de la mutación existente. Las tres mutaciones primarias o patogénicas verdaderas están en genes que codifican para proteínas constituyentes del complejo I de la cadena respiratoria. Son G3460A, T14484C y G11778A, esta última asociada al 50% de los casos y responsable de la forma más severa de la enfermedad.
MILS: Síndrome de Leigh de herencia materna
Consiste en encefalomielopatía necrosante subaguda; es decir, lesiones necróticas cerebrales en tálamo, ganglios basales, tronco cerebral y médula espinal. La esperanza de vida es muy limitada y suele aparecer durante el primer año de vida.
Se puede acompañar de alteraciones respiratorias y de la deglución (por estar afectado el tronco cerebral), además de parálisis oculomotoras, atrofia óptica, leucodistrofia, movimientos involuntarios o hiperproteinorraquia (disminuyendo así la conducción nerviosa). Además, las habilidades psicomotoras adquiridas tienden a empeorar.
El síndrome de Leigh está causado por la transversión de T por G en el gen de la subunidad 6 de la ATPasa, T8993G.
Hasta aquí hemos comentado las enfermedades mitocondriales más representativas, pero cabe destacar que, en numerosas ocasiones, no es posible catalogar dentro de los síndromes específicos a todos los pacientes diagnosticados con enfermedad mitocondrial. Esto se debe fundamentalmente a la enorme diversidad en las manifestaciones clínicas que cada paciente desarrolla. Además, el fenotipo clínico de un paciente mito evoluciona a lo largo del tiempo, desarrollando otros síntomas y signos diferentes a los que presentaba cuando se realizó el diagnóstico.
En cuanto al tratamiento, no existen medidas curativas que reviertan la disfunción mitocondrial, sino que se seguirá un tratamiento farmacológico y nutricional destinado a enlentecer el proceso de degeneración natural. También es importante la terapia psicológica y de mantenimiento para lograr la mayor calidad de vida de los pacientes.
Como hemos visto, casi la totalidad de estas enfermedades se manifiestan en las primeras dos décadas de vida. Por esto, es importante que, especialmente los niños, se enfrenten de manera positiva y esperanzadora al diagnóstico recibido. Una herramienta útil para ellos puede ser el siguiente cuento redactado por la Obra Social del Hospital Sant Joan de Déu, que explica la enfermedad de una forma amena y lúdica.
Es muy importante que todos los enfermos de estas patologías se sientan acompañados y comprendidos. Para ello, resulta esencial la concienciación poblacional de estas enfermedades, de cara no solo a conseguir financiación para seguir investigando, sino también para acercar el conocimiento de las mismas a todas las personas, aunque no les afecte directamente. Existen varias asociaciones que ayudan a conseguir esto, como la Asociación de Enfermos de Patologías Mitocondriales (AEPMI).
Además, la página web de una fundación australiana contiene numerosos testimonios de personas afectadas por enfermedades mitocondriales, lo cual es otra forma muy útil de que un mayor número de personas conozcan estas patologías.
También se realizan numerosas campañas como carreras solidarias #RunForMito, recogida de tapones o dispositivos tecnológicos en desuso, venta de productos solidarios, o campañas de iluminación como #LightUpForMito, que se realizó con razón de la Semana mundial de la concienciación de enfermedades mitocondriales, celebrada en septiembre, y a la que se sumaron numerosas asociaciones y ayuntamientos, consiguiendo promover la sensibilización muchas personas y dando voz a todas las familias afectadas.
Bibliografía
- Solano, A., Playán, A., López-Pérez, M. J. and Montoya, J. (2001) ‘Enfermedades genéticas del ADN mitocondrial humano’, Salud Pública de México, 43(2), pp. 151–161. doi: 10.1590/s0036-36342001000200010.
- Dra. Aglais Arredondo Falagán, I Lic. Gleymis Venet Cadet,I Dra. Dra. Olivia Román Guerra I y MsC. Eglis Yanet Ramírez Delgado. (2012) Bases moleculares de las enfermedades mitocondriales, Medisan, 16(5), pp. 795–805.
- Barrera-Ramírez, C. F., Barragán-Campos, H. M., Sánchez-Guerrero, J., García-Ramos, G., Vega-Boada, F. and Estañol, B. (1999) ‘El otro genoma: El concepto clínico de las citopatías mitocondriales o enfermedades de la fosforilación oxidativa’, Revista de Investigación Clínica, 51(2), pp. 121–134.
- Boveris, A., Costa, L. and Junqueira, V. (2000) ‘Envejecimiento mitocondrial. La teoría del envejecimiento mitocondrial por la acción continua de los radicales libres del oxígeno y del nitrógeno’, Ciencia e Investigación, pp. 13–23. Available at: http://revistasinvestigacion.unmsm.edu.pe/index.php/farma/article/view/4393/4457.
- Jesus, E. P., Carmen, G. L., Oscar, B. B. and Manuel, C. G. (2014) ‘Protocolos Diagnóstico Terapeúticos de la AEP: Neurología Pediátrica’, Revista de Neurología, 58(6), p. 288. doi: 10.33588/rn.5806.2013478.
- Feillet, F., Schmitt, E., Gherardi, R. and Bonnemains, C. (2014) ‘Enfermedades mitocondriales’, EMC-Pediatría, 49(2), pp. 1–12. doi: 10.1016/s1245-1789(14)67271-1.
- Campos, Y., Pineda, M., García Silva MT., Montoya J., Antoni L. Andreu (2014) ‘Enfermedades mitocondriales’, Revista de Neurología, 58(06), p. 288. doi: 10.33588/rn.5806.2013478.
- Montoya, J. (2000) ‘Diseases of the mitochondrial DNA’, Revista de Neurología, 31(4), pp. 324–333. doi: 10.33588/rn.3104.2000236.
- Sydow, G., Inde, K. and Lindermark, H. (2017) ‘Guía Biopsicosocial; Conoce la neuropatía óptica hereditaria de Leber – NOHL’, Asociación Atrofia del Nervio Óptico de Leber, I. Available at: https://enfermedades-raras.org/index.php?option=com_content&view=article&id=10484.
- Calderón-Garcidueñas, A. L., Perez-Loria, O., Alberto-Sagastegui, J. and Farias- Garcia, R. (2000) ‘Oftalmoplejia progresiva externa secundaria a miopatía mitocondrial. Presentación de un caso y revisión de la literatura’, Gaceta Médica de México, 136(3), pp. 267–271.
- Delgado Pelayo Sari (2007) Retinosis Pigmentaria. Preguntas y Respuestas, Revista Médica MD.
- Miranda Nava, G. and Ortega Ponce Fabiola, E. E. (2010) ‘Epilepsia mioclónica con fibras rojas rasgadas’, Revista Mexicana de Neurociencia, 11(3), pp. 243–245.
- Gutiérrez, A., Saldaña Martínez, A., García Ramírez, R., Rayo Mares, D., Carreras, M., López Pérez, M. J., Montiel Sosa, J. F. (2009). Síndrome de Leigh causado por la mutación G14459A del ADN mitocondrial en una familia mexicana. Revista de neurología.(Ed. impr.), pp. 248-250.
- Conforto, A. B., Yamamoto, F. I., Oba-Shinjo, S. M., Pinto, J. G. C., Hoshino, M., Scaff, M. and Marie, S. K. N. (2007) ‘Screening for melas mutations in young patients with stroke of undetermined origin’, Arquivos de Neuro-Psiquiatria, 65(2 B), pp. 371–376. doi: 10.1590/S0004-282X2007000300001.
- Donaire de la Torre, C. (2015). Enfermedades mitocondriales: una forma aplicada de enseñar genética. Trabajo Fin de Máster. Universidad de Jaén.
- Skorecki K, Behar D. (2019). Dna mitocondrial y enfermedades y rasgos hereditarios. In: Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J. eds. Harrison. Principios de Medicina Interna, 19e. McGraw-Hill. https://accessmedicina.mhmedical.com/content.aspx?bookid=1717§ionid=114913378