ApoE4: La kriptonita de Thor
Por: Aitor Gil Iglesias, Milagros González Flores y Álvaro Mellado Cuesta
1.Introducción
Seguramente todos nosotros hemos leído que el actor Chris Hemsworth, más conocido como Thor en su papel dentro de la saga de Marvel, ha decidido retirarse del cine temporalmente al haber descubierto que tiene muchas posibilidades de desarrollar Alzheimer. Este hallazgo fue conocido por el actor cuando se encontraba rodando el documental “Limitless” para National Geographic al realizarse una prueba genética por la cual trataba de estudiar cómo su físico se vería afectado con el paso de los años. Sin embargo, estos estudios, que en un primer momento trataron de analizar el estado físico del actor, acabaron revelando la posibilidad de desarrollar Alzheimer al presentar dos copias del gen ApoE4 que hace que su riesgo de padecerla sea entre 8 y 10 veces más. Ahora bien, ¿qué es el Alzheimer y por qué es tan importante este gen en su desarrollo?

2.-¿Qué es el Alzheimer?
El Alzheimer (EA) es una enfermedad neurodegenerativa lentamente progresiva que se caracteriza por la pérdida de memoria de manera gradual. En ella también se describen otros síntomas como demencia irreversible y afectación global del resto de las funciones cognitivas (pensar, razonar…) que conllevan a un deterioro progresivo y que afectan al funcionamiento laboral y social.
Cabe destacar que la progresión de la enfermedad a nivel cerebral es la misma en todos los pacientes. Comienza en el neocórtex basal temporal, avanza hacia el hipocampo y se extiende por todo el neocórtex.
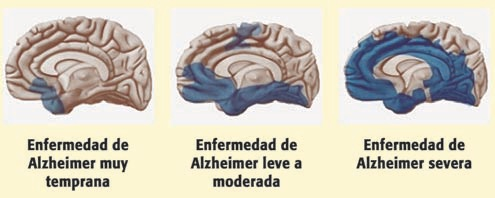
La mayoría de los casos aparecen a partir de los 70 años (“late onset”) , aunque puede haber una aparición temprana en menores de 60 (“early onset”). Afecta a alrededor de 50 millones de personas en el mundo y el principal factor de riesgo es la edad.
La early onset es autosómica dominante y hereditaria y, aunque la mayoría suceden como consecuencia de mutaciones esporádicas sobre el gen de la apolipoproteína (APOE, cromosoma 19), otros casos se deben a mutaciones en los genes que codifican para la proteína precursora amiloide (APP, cromosoma 21, por ello, las personas con Síndrome de Down, trisomía del 21, son más propensos a desarrollar la enfermedad de Alzheimer), para la presenilina 1 (PSEN1, cromosoma 14) y para la presenilina 2 (PSEN2, cromosoma 1).
No obstante, algunos otros factores de riesgo que podrían estar implicados son las enfermedades vasculares, infecciones, factores ambientales y lesiones encefálicas.

3.Etiopatología
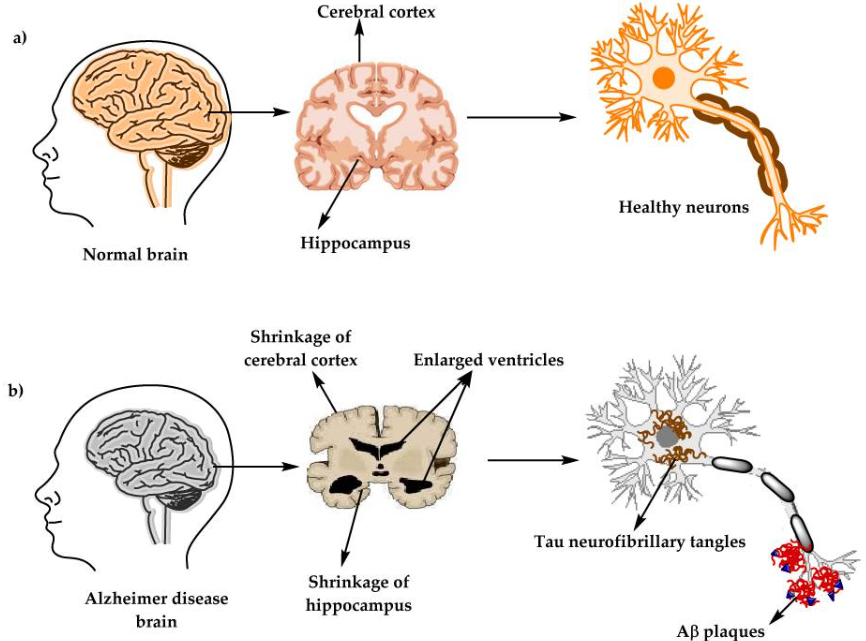
Bioquímicamente, esta enfermedad se produce como consecuencia de dos elementos neuropatológicos: las placas seniles o placas Aβ y los ovillos neurofibrilares (NFT), esenciales para entender cómo se desarrolla el Alzheimer.
3.1.-¿Qué son las placas seniles?
Estas consisten en acumulaciones extracelulares de la proteína beta-amiloide (Aβ) anómala que en principio no es tóxica pero que se agrega con otras proteínas beta-amiloide anómalas hasta formar oligómeros tóxicos que precipitan y se depositan como placas amiloides insolubles y que son realmente tóxicas.
Su síntesis tiene lugar por medio de la proteína precursora amiloidea (APP) cuyo gen se encuentra localizado en el cromosoma 21. Esta, es una proteína transmembrana que contiene una región amino-terminal extracelular larga y una región carboxilo terminal intracelular más corta que se sintetiza en el retículo endoplasmático, se glicosila en el aparato de Golgi y por medio de vesículas se inserta en la membrana celular.
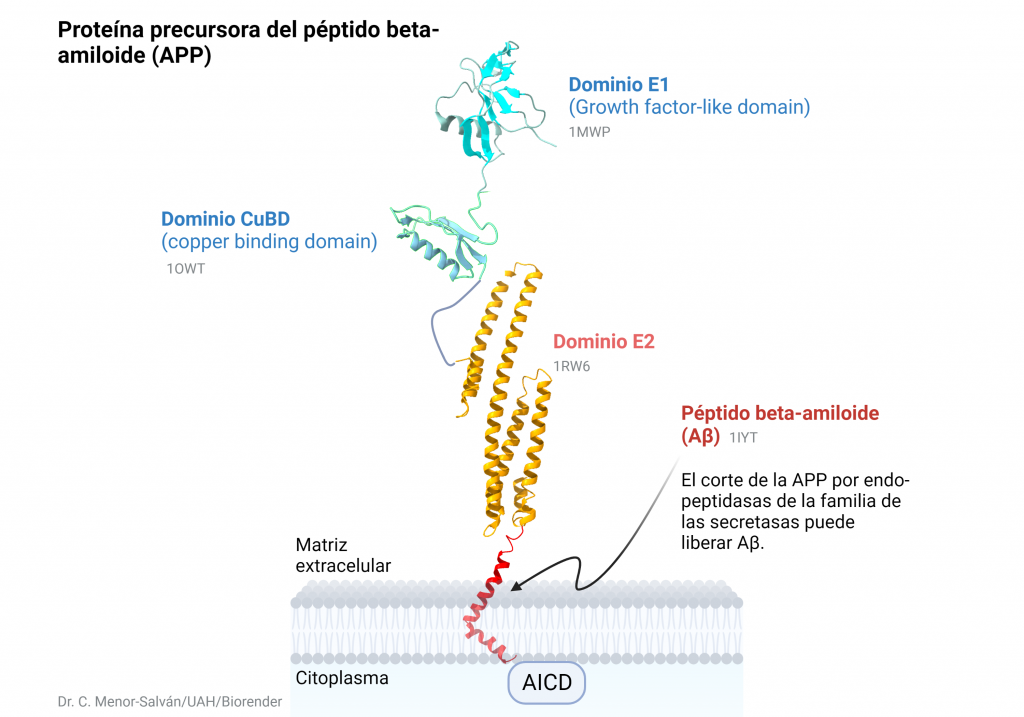
Una vez en la membrana celular, esta proteína puede ser procesada fisiológicamente por medio de dos vías:
- Vía no amiloidogénica (90% de los casos): por medio de esta vía, la proteína APP es escindida por la enzima α-secretasa en un fragmento extracelular soluble neuroprotector (SAPPα, Soluble APPα) compuesto por 665 aminoácidos. Posteriormente la región no escindida que ha quedado insertada en la membrana, es procesada por la enzima γ-secretasa, quien se encarga de liberar la región C-terminal para posteriormente poder ser degradada. De esta manera, se libera al medio extracelular un péptido de 36-40 aminoácidos denominado P3, que no es neuroprotector, pero tampoco patógeno. El fragmento restante C-terminal se desprende hacia el citosol y se denomina AICD (Amyloid Intracellular Domain), que se degrada enseguida. Este proceso es constitutivo (APP tiene una vida media de 1h) y fundamental para el funcionamiento neuronal.
- Vía amiloidogénica (10% de los casos): por medio de esta vía, la enzima β-secretasa o BACE libera un fragmento N-terminal de la proteína APP (SAPPβ) que no es neuroprotector. Posteriormente, el péptido que queda en la membrana será procesado por la enzima γ-secretasa dando lugar a diferentes péptidos que presentan diferentes solubilidades: el péptido Aβ (42 aá) y el AICD, que tiene un tiempo de vida mayor, va al núcleo y favorece la transcripción de genes como la propia APP, GSK3 (Ser/Thr kinasa), BACE, NEP (nefrilisina) y LRP1 (lipoprotein receptor-related protein 1).
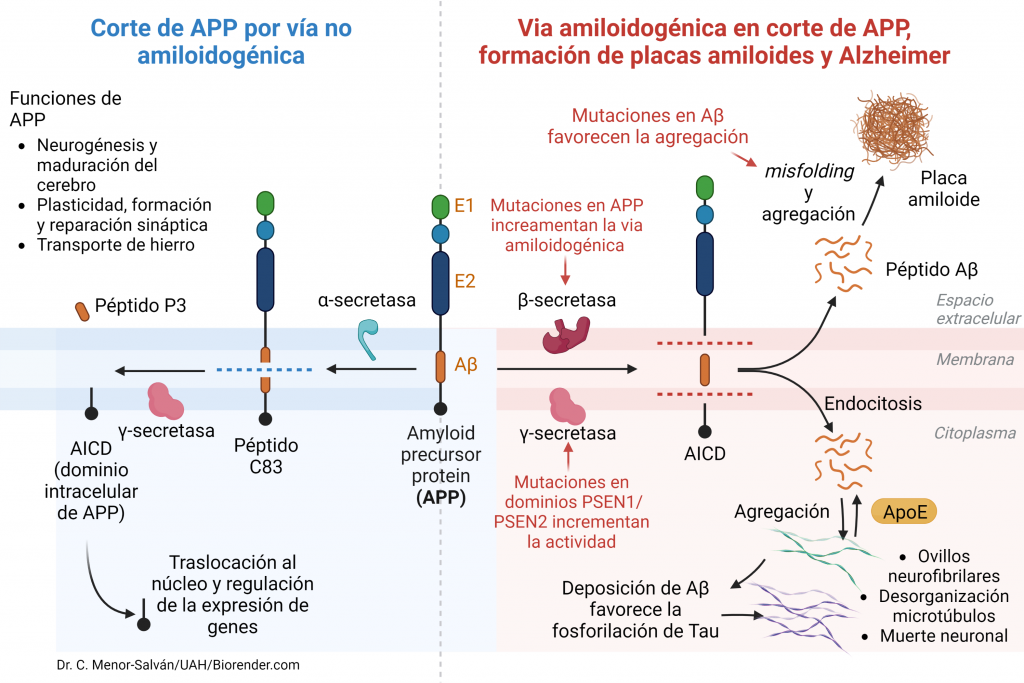
Los péptidos más comúnmente formados en la vía amiloidogénica son los péptidos Aβ40, los cuales presentan una estructura en forma de hélice alfa que puede ser fácilmente degradada. Sin embargo, en situaciones patológicas predomina la síntesis del péptido Aβ42 y Aβ43, los cuales son mucho más insolubles, tienden a formar láminas β y a interaccionar entre ellos formando fibrillas que pueden acumularse y formar placas extracelulares amiloides insolubles que se diseminarán por otras estructuras como el hipocampo, la amígdala y la corteza cerebral resultando realmente dañinas.

Código PDB: 5KK3
PDB DOI:10.2210/pdb5KK3/pdb
Ahora bien, ¿por qué es tan importante Aβ?
De manera normal, esta proteína se encarga de modular la transmisión del impulso eléctrico entre las neuronas, por lo que, un mal funcionamiento o no funcionamiento supondría la pérdida de la comunicación neuronal, es decir, una pérdida de sinapsis. Esto se debe a que puede alterar la actividad de quinasas, producir señales de estrés oxidativo, daños mitocondriales e incluso defectos en el transporte axonal que son los que finalmente conducen a una degeneración neuronal. Todo ello es lo que acabaría produciendo en estos pacientes una gran deficiencia cognitiva.
Además, Aβ en bajas concentraciones es quelante de metales, mejora la memoria y el aprendizaje, es antioxidante, interviene en el mantenimiento de la barrera hematoencefálica etc.
Esta acumulación de Aβ anómala en condiciones patológicas, en las que no solo se incluye el Alzheimer, es como consecuencia de las mutaciones en algunos genes como APP, PSEN1 (proteína que se encarga de activar el complejo de la γ-secretasa) y PSEN2 (poco frecuentes), los cuales, afectan al catabolismo y anabolismo de Aβ y como consecuencia producen una rápida progresión de neurodegeneración.
Concretamente, dichas mutaciones afectan al procesamiento alternativo de APP, y como consecuencia se generan fundamentalmente proteínas Aβ truncadas, Aβ42 y Aβ43. Estas presentan residuos poco polares que tienden a apilarse en paralelo induciendo a un plegamiento incorrecto de las láminas beta las cuales, son muy insolubles y al no ser reconocidas por los sistemas de degradación acaban precipitando para formar fibrillas amiloides que generan una gran neurotoxicidad.
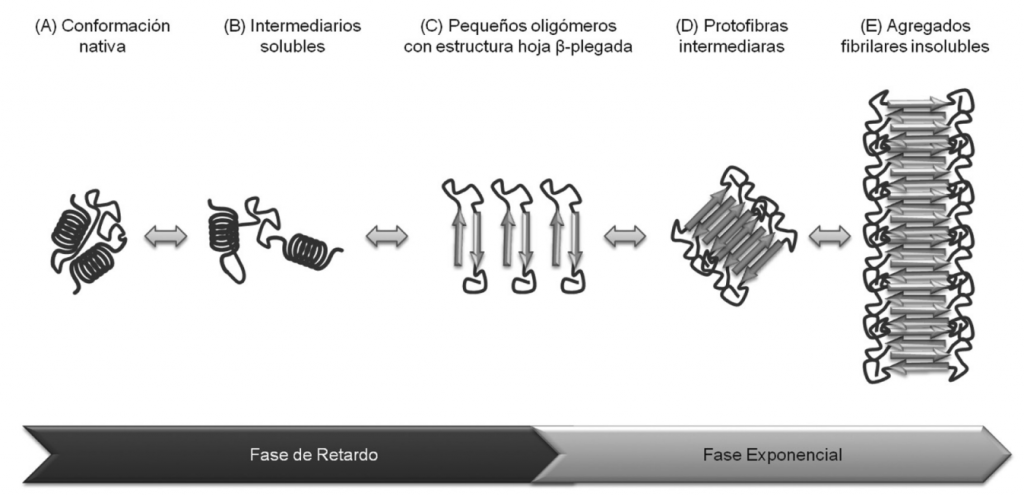
3.2-¿Qué son los ovillos neurofibrilares (NFT)?
Son lesiones de tipo intracelular formadas por filamentos helicoidales emparejados (PHF) que se producen como consecuencia de la hiperfosforilación de las proteínas Tau y que puede deberse a la aparición de proteínas Aβ, al estrés oxidativo o a mediadores inflamatorios. La formación de estos ovillos producen muerte neuronal por apoptosis.
Estas proteínas se encuentran de manera mayoritaria en neuronas y células de la Glía y son esenciales para promover el ensamblaje de los microtúbulos neuronales así como para mantener la integridad estructural neuronal y por tanto para permitir el crecimiento de los axones y el transporte axonal. Concretamente, la proteína Tau se encarga de facilitar la polimerización de la tubulina para permitir que se formen los microtúbulos.
Código PDB: 2MZ7.
PDB DOI: 10.2210/pdb2MZ7/pdb
El gen que codifica para la proteína Tau, localizado en el gen 17, contiene 16 exones y en el cerebro adulto sufre diferentes tipos de empalme alternativo en los exones 2, 3 y 10 originando finalmente 6 isoformas de Tau. Estas isoformas contienen entre 3-4 repeticiones peptídicas de 31-32 residuos en la región carboxilo terminal que se corresponde con el dominio de unión a los microtúbulos. Así mismo, el estado de fosforilación del gen varía durante el desarrollo y es lo que conduce a un cambio en el empalme del ARN.

Tau en el embrión aparece fosforilada, pero en adultos está muy poco fosforilada. Se puede fosforilar por más de 80 sitios Ser/Thr y 5 de Tyr, gracias a GSK-3, PKA, la cuales son Ser/Thrk o Fyn (TyrK). En la enfermedad de Alzheimer, está hiperfosforilada.
Además, cuando Tau está fosforilada cambia de localización y, en lugar de localizarse preferentemente en los axones, se dirige hacia las dendritas y se lleva a Fyn secuestrada consigo. Fyn fosforila a receptores de glutamato (Glu) (receptores NMDA sobre todo) lo que producirá una muerte neuronal por sobreexcitación.
De esta manera, y dado la importancia de la fosforilación en este gen, se ha podido observar que en los procesos de neurodegeneración Tau sufre una fosforilación anormal irreversible que impide su función normal y su agregación. Como consecuencia, no puede unirse correctamente a los microtúbulos y todo ello junto con la formación de las fibrillas por su agregación, impiden que la neurona pueda transmitir señales eléctricas. Además todo ello favorece un mal transporte axonal de nutrientes, lo cual contribuye a la degeneración sináptica y a la muerte neuronal.
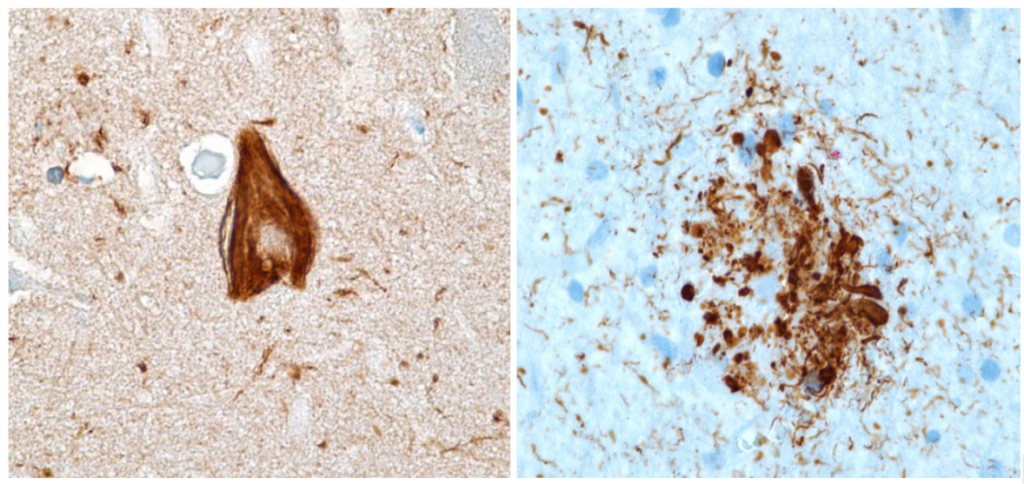
Además, parece ser que, una vez Tau está fosforilada y ha perdido su estructura, se empieza a transmitir de unas neuronas a otras, induciendo el cambio conformacional patológico en proteínas Tau normales por contacto (como los priones). Gracias a ello, la enfermedad puede propagarse y afectar a diferentes estructuras.
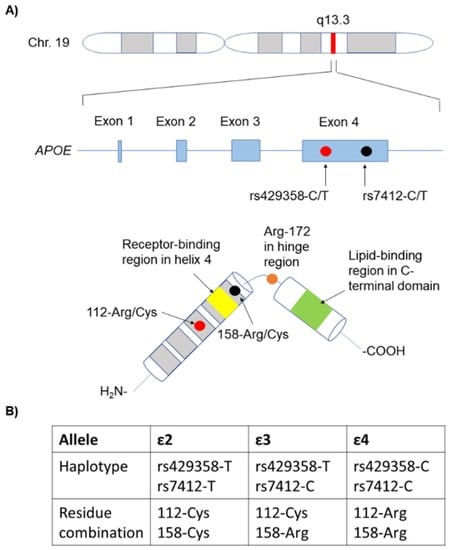
4.-¿Qué es la ApoE?
La ApoE es una proteína de 299 aminoácidos, que está dentro del grupo de las apolipoproteínas, las cuales están implicadas en el metabolismo de los lípidos (forman las lipoproteínas), siendo la ApoE la única que encontramos en el cerebro.
En la ApoE hay varios dominios de unión a destacar, como por ejemplo el de unión a lípidos, al receptor, a heparina o la región bisagra. Todos ellos con gran importancia en las funciones de la ApoE como veremos a continuación.
Sin embargo, el interés reciente hacia la ApoE no es por su papel en el metabolismo de los lípidos, sino por su relación con el péptido Aβ y la proteína Tau, ambos implicados en el inicio y avance de la enfermedad de Alzheimer.
4.1- Relación ApoE – péptido Aβ
La ApoE es sintetizada por astrocitos y la microglía, debido a la alta importancia de los lípidos para el desarrollo y mantenimiento de las neuronas, pero, ¿cuál es su función dentro del metabolismo de lípidos? ¿y que nexo tiene esto con el péptido Aβ y la enfermedad de Alzheimer?
FUNCIÓN DE LA ApoE
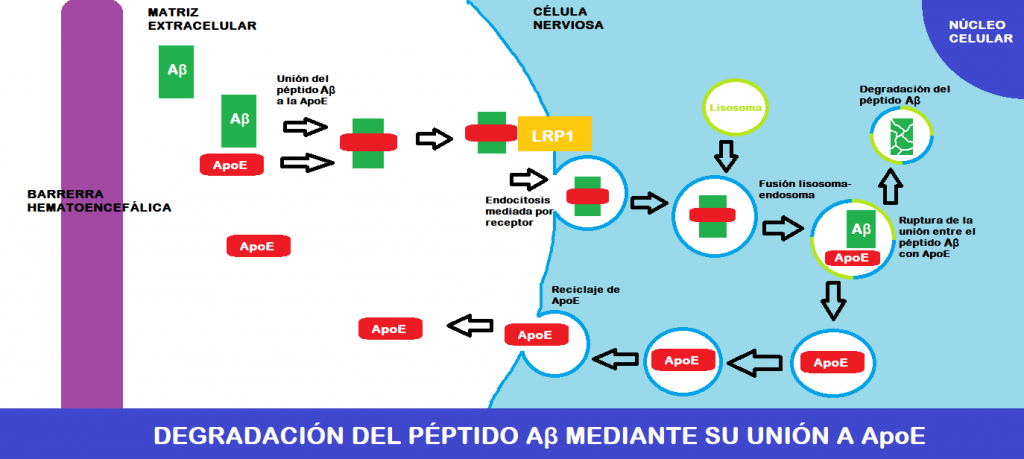
La ApoE se encuentra circulando por la matriz extracelular nerviosa, y es ahí dónde se une al colesterol, que proviene del interior de las células nerviosas (sale a través de transportadores de la familia ABC), y ambos forman lipoproteína discoidal. Esta se une a recetores celulares para lipoproteínas (LRP1 principalmente) y entra en las células por endocitosis mediada por receptor. Ya en el interior, esta lipoproteína entra en el metabolismo de los lípidos.
Sin embargo, lo más interesante de esto es que el péptido Aβ es capaz de unirse a la ApoE circulante (a su dominio de unión a heparina concretamente), formando una molécula que es capaz de entrar en las células al igual que la lipoproteína que hemos mencionado antes (endocitosis mediada por LRP1).
Una vez dentro, el endosoma donde está la molécula ApoE-péptido Aβ se fusiona con lisosomas, y esto hace que por aumento del pH y acción de proteasas lisosomales ApoE y el péptido Aβ se separen. El destino de cada uno es bien distinto:
- ApoE queda en una vesícula que se fusionará con la membrana plasmática, permitiendo que salga al exterior y se recicle.
- El péptido Aβ es sustrato para proteasas lisosomales, por lo que acaba siendo degradado.
Pero yendo más allá, lo que más ha despertado el interés científico de todo esto y lo que está relacionado con el caso concreto de Chris Hemsworth son las distintas isoformas de ApoE que aparecen en las poblaciones, cada una de ellas con un papel distinto en la predisposición a sufrir Alzheimer.
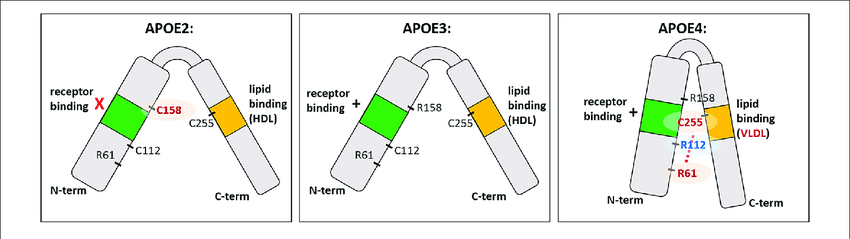
4.2- Isoformas de ApoE y su importancia en el Alzheimer
Hay 3 isoformas de la ApoE, las cuales sólo se diferencian en los residuos 112 y 158:
- ApoE2: posee cisteínas (en 112 y 158). Se la considera «protectora» frente a la enfermedad de Alzherimer. Se cree que su eliminación del péptido Aβ es más eficaz.
- ApoE3: posee cisteína (en 112) y arginina (en 158). Es la isoforma mayoritaria en la población y se considera que su papel es neutro frente a la enfermedad de Alzheimer.
- ApoE4: posee argininas (en 112 y 158). Su presencia aumenta la probabilidad de sufrir la enfermedad de Alzheimer hasta 4 veces si aparece en heterocigosis y hasta 10 veces si aparece en homocigosis.
PDB DOI: 10.2210/pdb1NFO/pdb
PDB DOI: 10.2210/pdb1NFN/pdb

PDB DOI: 10.2210/pdb1B68/pdb
Pero, ¿por qué ApoE4 aumenta tanto la probabilidad de desarrollar Alzheimer?, ¿en qué afecta el cambio de tan solo 2 aminoácidos a una proteína que posee 299? Ahora mismo te lo explicamos
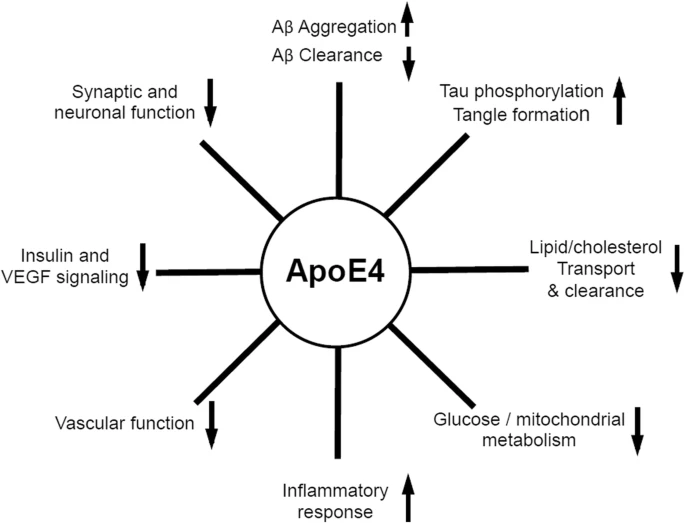
4.3- ApoE4
Su única diferencia estructural frente a la isoforma protectora es el cambio de dos cisteínas (aminoácidos polares sin carga), por dos argininas (aminoácidos polares con carga positiva). En principio parece un cambio insignificante, pero este pequeño cambio hace que la ApoE4 tenga una conformación algo más «cerrada» que el resto de ApoE, afectando más de lo que creemos en su función.
La conformación que adopta ApoE4 hace que se una más deficientemente a sus receptores, por lo que ApoE4 se encuentra durante más tiempo circulante por la matriz extracelular. Al disminuir la endocitosis mediada por receptor, disminuye la proteólisis intracelular y para compensarlo, aumenta la proteólisis extracelular.
Esta proteólisis favorece la acumulación del péptido Aβ, su unión con otros péptidos Aβ (formando oligómeros Aβ) y el inicio y desarrollo de la formación de placas amiloides y de la enfermedad de Alzheimer. De hecho se han descubierto restos de esta ApoE en las placas amiloides.
Pero esto no es todo, ya que recientemente se ha visto que ApoE también interactúa con la proteína Tau, una proteína muy importante en el desarrollo de la enfermedad de Alzheimer y cuya hiperfosforilación explica muchos de los signos clínicos de la enfermedad.
4.4- Relación ApoE – proteína Tau
Se ha visto que ApoE está relacionada con la acumulación de Tau y Tau fosforilada, especialmente ApoE4 es quien favorece en mayor medida está acumulación, la cual deriva en la formación de los ovillos neurofibrilares. Para explicar está relación de manera más detallada vamos a presentaros un experimento realizado con ratones en 2017.
En este experimento se quiso conocer más acerca del papel de ApoE en la neurodegeneración a través de Tau. Para ello se utilizaron ratones que sobreexpresaban Tau humana (ratones P301S) y las distintas isoformas de ApoE, creando distintos grupos de ratones:
- Tau-ApoE2 (TE2): expresaban Tau y ApoE2.
- Tau-ApoE3 (TE3): expresaban Tau y ApoE3.
- Tau ApoE4 (TE4): expresaban Tau y ApoE4. Es el grupo de ratones en el que mayor neurodegeración se observó a los 9 meses.
- Sin Tau y con ApoE (KI): no se observó neurodegeneración a los 9 meses. Esto confirmaba que ApoE por sí sola no ocasiona neurodegeneración.
- Con Tau y sin ApoE (TEK0): se observó neurodegeneración a los 9 meses, pero en menor nivel que en cualquiera de los grupos con Tau y las distintas formas de ApoE.
Con esto se confirmó que ApoE (especialmente ApoE4) juega un papel importante en la neurodegeneración a través de Tau. Además en este experimento se observó que ApoE4 no influye en la síntesis de Tau, sino que la acumulación de Tau se debe a que ApoE4 afecta a su degradación por autofagia.
Por último se vio que cuando había patología de Tau (hiperfosforilación y acumulación), ApoE4 provoca neuroinflamación, promoviendo la activación de la microglía y la expresión de genes A1 en astrocitos.
4.5- Genes implicados
En la prueba genética que se realizó el actor Chris Hemsworth, se analizo el gen que da lugar a la síntesis de la ya mencionada ApoE. El gen de la ApoE está formado por 4 exones y se encuentra en el cromosoma 19.
Las mutaciones de mayor interés en este gen son las que afectan a los codones que dan lugar a los aminoácidos 112 y 158, y por tanto a las distintas isoformas ya vistas (ApoE2, ApoE3 o ApoE4) y su influencia en la probabilidad de desarrollar la enfermedad de Alzheimer.
5.Referencias
- Breijyeh, Z., & Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules (Basel, Switzerland), 25(24), 5789. https://doi.org/10.3390/molecules25245789
- Lane, C. A., Hardy, J., & Schott, J. M. (2018). Alzheimer’s disease. European journal of neurology, 25(1), 59–70. (2020) https://doi.org/10.1111/ene.13439
- Lin, Y. T., Seo, J., Gao, F., Feldman, H. M., Wen, H. L., Penney, J., Cam, H. P., Gjoneska, E., Raja, W. K., Cheng, J., Rueda, R., Kritskiy, O., Abdurrob, F., Peng, Z., Milo, B., Yu, C. J., Elmsaouri, S., Dey, D., Ko, T., Yankner, B. A., … Tsai, L. H. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron, 98(6), 1141–1154.e7. (2018) https://doi.org/10.1016/j.neuron.2018.05.008
- Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., … & Holtzman, D. M. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature, 549(7673), 523-527. (2017)
- Spinney, L. The forgetting gene: for decades, most researchers ignored the leading genetic risk factor for Alzheimer’s disease. That is set to change. Nature, 510(7503), 26-29. (2014) https://doi.org/10.1038/510026a
- Figura III: Lane, C. A., Hardy, J., & Schott, J. M. Alzheimer’s disease. European journal of neurology, 25(1), 59–70. (2018) https://doi.org/10.1111/ene.13439
- Figura IV: Breijyeh, Z., & Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules (Basel, Switzerland), 25(24), 5789. (2020)
- Figura VIII: Duran-Aniotz, C., Moreno-Gonzalez, I., & Morales, R. Agregados amiloides: rol en desórdenes de conformación proteica. Revista médica de Chile, 141(4), 495-505. (2013)
- Figura X: Luna-Muñoz, J., Zamudio, S., De La Cruz, F., Minjarez-Vega, B., & Mena, R. Acción protectora de la proteína tau en la enfermedad de Alzheimer. Revista Mexicana de Neurociencia, 13(3), 160-167. (2012)
- Figura XI: Castellani, R. J., Rolston, R. K., & Smith, M. A. Alzheimer disease. Disease-a-month : DM, 56(9), 484–546 (2010)
- Figura XIII: Fernandez, Celia & Hamby, Mary & McReynolds, Morgan & Ray, William. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Frontiers in Aging Neuroscience. (2011) 11. 10.3389/fnagi.2019.00014
- Figura XVII: Safieh, M., Korczyn, A. D., & Michaelson, D. M. ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC medicine, 17(1), 1-17. (2019)
- Figura XVIII: bondio, P., Sazzini, M., Garagnani, P., Boattini, A., Monti, D., Franceschi, C., Luiselli, D., et al. The Genetic Variability of APOE in Different Human Populations and Its Implications for Longevity. Genes, 10(3), 222. (2019)