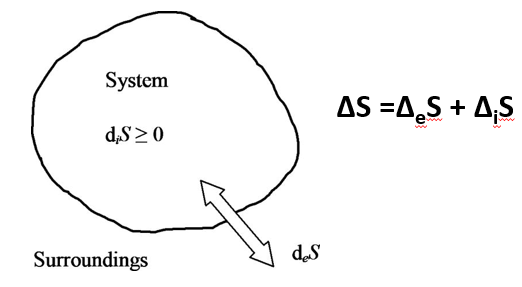
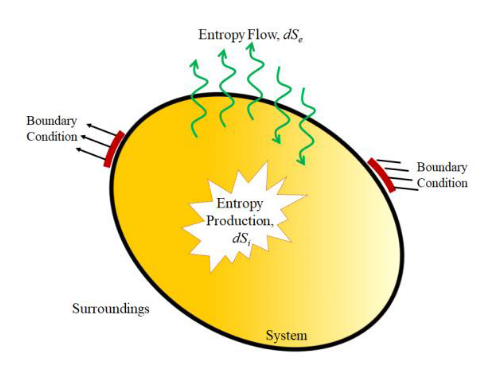
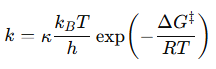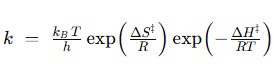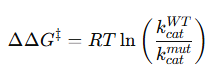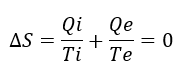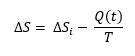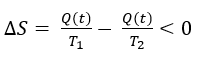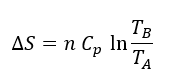La evolución es un hecho, no un relato. Y las moléculas nos hablan de ello.
escrito por C. Menor-Salvan | 17 diciembre, 2024
C. Menor-Salván 12/2024
Creacionismo y evolucionismo no son creencias o hipótesis alternativas; ni siquiera son conceptos establecidos con las mismas reglas. Son los magisterios no solapados tal como explicaba Stephen Jay-Gould. Así, un supuesto debate o confrontación entre supuestos «científicos» creacionistas y «creyentes en la teoría de la evolución» no tiene sentido.
En primer lugar, ¿de qué hablamos cuando hablamos de «teoría»? La evolución es un fenómeno natural. Por lo tanto, es observable. Sobre un fenómeno natural, los científicos construimos un marco teórico que permite explicar las observaciones y realizar predicciones; éste se actualiza al ir afinando nuestra capacidad para realizar observaciones y experimentos. Pero el hecho ocurre estemos nosotros o no para observarlo y construir hipótesis, modelos o teorías para explicarnoslo.
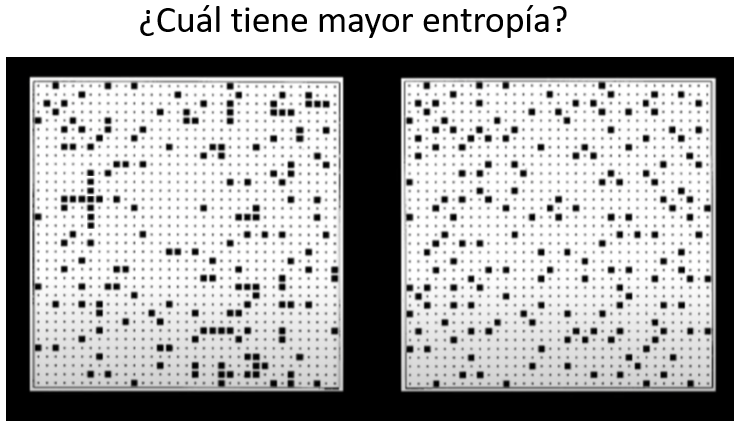
En todas las teorías hay puntos de especial dificultad. En el marco teórico sobre la evolución tenemos, por ejemplo, la cuestión del origen de la vida, donde observamos que las moléculas de la vida, y sus antecesoras, también siguen reglas de selección, adaptación y supervivencia que aún estamos entendiendo.
No creemos en la evolución; la evolución es un hecho observable
Usando un ejemplo quizá mas fácil de entender: la teoría que explica el funcionamiento de las enzimas también tiene puntos oscuros y aún hay discusión científica en torno a ello. Pero nadie pone en duda que las enzimas existen y su acción es un hecho, sea cual sea la teoría que construyamos para explicarlo. Este marco teórico ha ido cambiando, desde el obsoleto modelo de llave-cerradura hasta modelos como Circe o el de estabilización del estado de transición.
Los científicos no «creemos» en la evolución, del mismo modo que no «creemos» en la enzimas. Son fenómenos reales que tratamos de comprender y explicar. Decir «creo en la evolución» es, simplemente, absurdo.
Como tal hecho observable, las ideas en torno a la evolución no son algo nuevo. Según el historiador romano Diógenes Laercio, Anaxágoras de Clazomene enseñaba que
«los seres vivos se formaron de la humedad, el calor y sustancias terrosas; despues, se propagaron por generación unos de otros».
Esta idea evolutiva rudimentaria, lanzada por Anaximandro de Mileto, el maestro-abuelo de Anaxágoras, hace 2500 años, creaba una disonancia cognitiva con el creacionismo, lo que llevó a San Hipólito de Roma a recogerla en sus «Refutaciones de todas la Herejías«. Gracias a ello, tenemos testimonio de las ideas de la escuela de Anaximandro.
Aún hoy, la evolución es considerada pecaminosa por muchos grupos religiosos. Yo mismo he sido testigo de alguna manifestación en contra de la evolución en EEUU, recibida con bastante humor por parte de los científicos y estudiantes que estábamos en el campus, hay que decir.
Observando la evolución sin salir de tu barrio
La evolución no sólo es observable, sino que la llevamos utilizando siglos en nuestro beneficio. Basta comparar las variedades de lechuga cultivada que podemos encontrar en el supermercado, con su pariente silvestre más cercano y, posiblemente, su ancestro: la amarga e indigesta, aunque comestible lechuga silvestre (Lactuca serriola).
La lechuga silvestre. Antepasado de la lechuga cultivada. Foto: Olga Pokotilo/PlantNet
Todo el proceso de domesticación de plantas y animales llevado a cabo por los humanos durante milenios debería probar, en sí mismo, que la evolución es un hecho. Y no es exclusivo de los humanos. En la evolución de la vida terrestre, unas especies han ejercido presión selectiva sobre otras, condicionando su evolución en una compleja red.
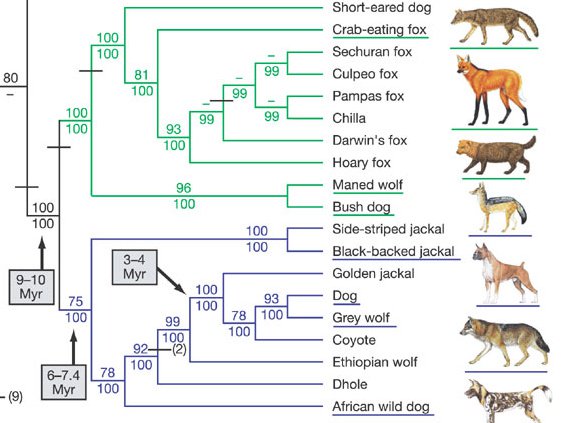
El perro original, que surgió de un ancestro común con el lobo gris actual, se ha diversificado un complejísimo arbol filogenético con todas las razas de perro doméstico. Un ejemplo de evolución divergente impulsada por la presión de selección ejercida por humanos. ¿cual es la frontera entre raza y nueva especie?
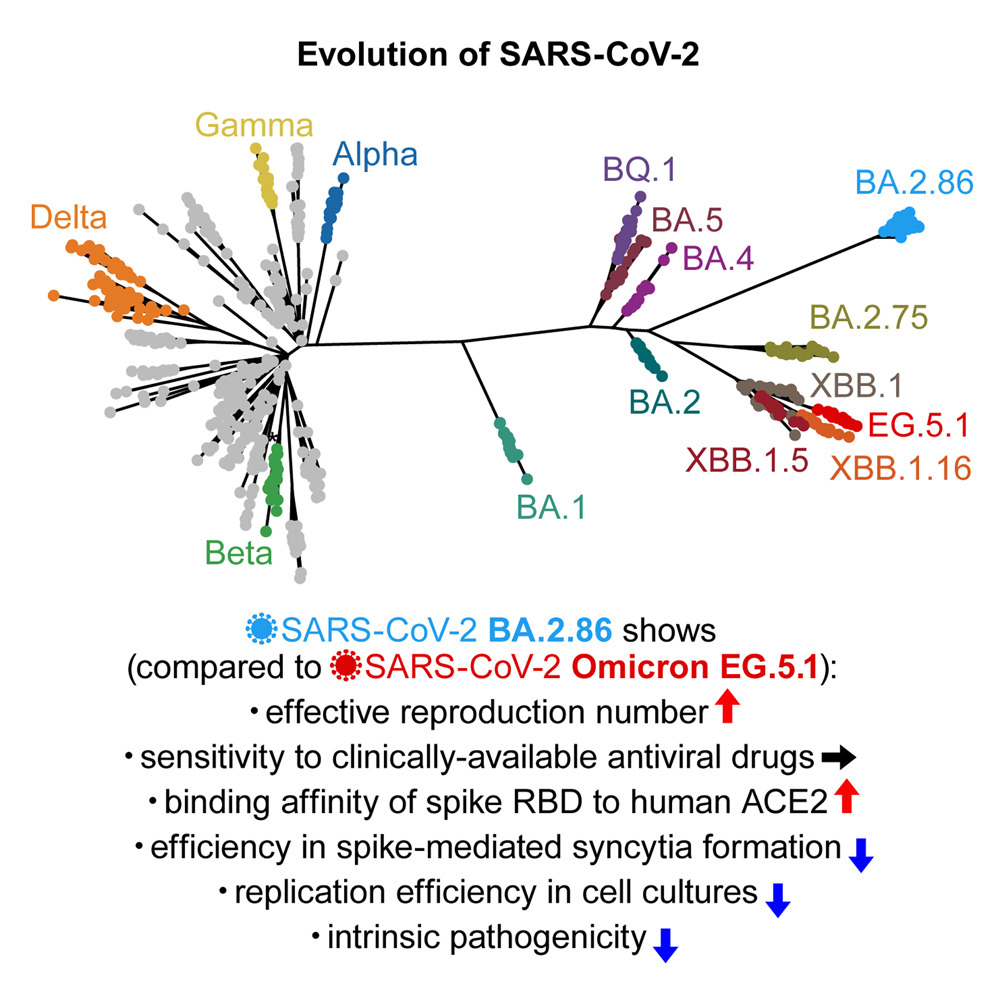
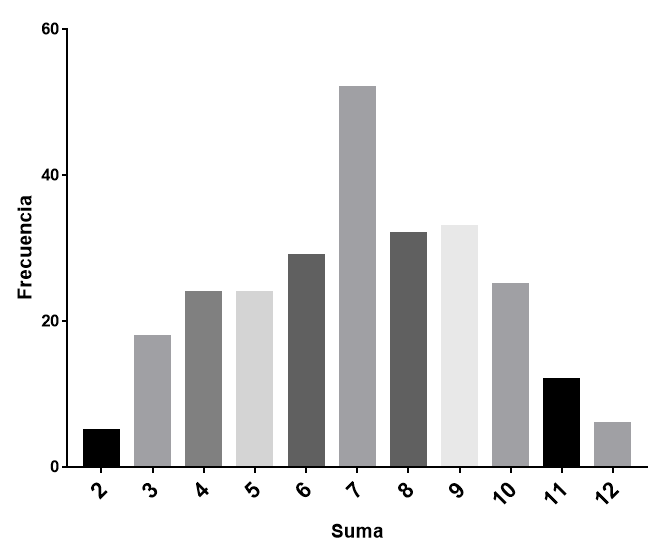
Tenemos un hecho evolutivo aún mas reciente: la pandemia de SARS-CoV-2. Nunca antes se había seguido la evolución de una especie viral tan de cerca y en tiempo real. Desde el inicio de la pandemia, gracias a la velocidad de replicación de los virus, fueron surgiendo variantes, creándose un complejo árbol filogenético. Durante el proceso, muchas variantes se extinguieron y otras han sobrevivido. Actualmente, tanto el virus como la enfermedad COVID son diferentes a los que existían en marzo de 2020. Este proceso de diversificación se basa en pequeños cambios en las estructuras moleculares que ocurren, inevitablemente, durante la replicación. Estos cambios, a veces, son silenciosos, si se mantiene la funcionalidad estructural; pero, a veces, llevan a las variantes a su desaparición, si la funcionalidad de las estructuras disminuye en las condiciones de ese momento, o a su prevalencia, si le aportan una funcionalidad ventajosa en las condiciones del momento. Con el tiempo, puede llegar a surgir una nueva especie.
Arbol filogenético del SARS-CoV-2. Evolución en vivo. Fuente: DOI 10.1016/j.chom.2024.01.001Dinámica de poblaciones de variantes virales. Estas curvas guardan una gran semejanza con las curvas de poblaciones de moléculas en procesos químicos como reacciones metabólicas o evolución química.
Ante todas las evidencias en torno a la evolución de la vida terrestre, la idea del creacionismo y el diseño inteligente se adaptan y, lejos de ser la herejía que añadía San Hipólito, la evolución se vuelve compatible con la idea religiosa de la creación. Así, en el diálogo público, el creacionismo se muestra con frecuencia como un _Deus Ex Machina_ que rellena convenientemente puntos oscuros en nuestra comprensión sobre la vida y el universo, dejando resueltas cuestiones espinosas, como el origen de la vida o del universo. Ello se combina a veces con una visión incorrecta sobre la evolución, cayendo en la trampa teleológica de que conduce hacia la inteligencia como máxima expresión de un proceso perfeccionador, puesto en marcha a partir de unas constantes físicas ajustadas cuidadosamente por un «diseñador» divino con este fin (el principio antrópico fuerte).
Sin embargo, la evolución biológica no es un proceso de mejora dirigido hacia un destino final. Su base es la preservación de estructuras supramoleculares funcionales, que sostienen la continuidad de la vida y que surgieron también mediante un proceso de evolución prebiótica.
La biología molecular es un tratado sobre la evolución
La biología molecular nos ofrece muchos ejemplos para ilustrar la evolución. Dos de los más interesantes son el ribosoma, cuya estructura relata la evolución de la vida desde su origen, y las polimerasas de DNA y RNA, que conectan todos los organismos y cuentan la historia evolutiva de los virus. El problema es que explicar brevemente para el público los complejos detalles moleculares de la evolución no es trivial. Voy a intentarlo, sin embargo, con un ejemplo sencillo (y simplificado), relacionado con un suceso que, desgraciadamente, se repite todos los inviernos: la toxicidad del monóxido de carbono.
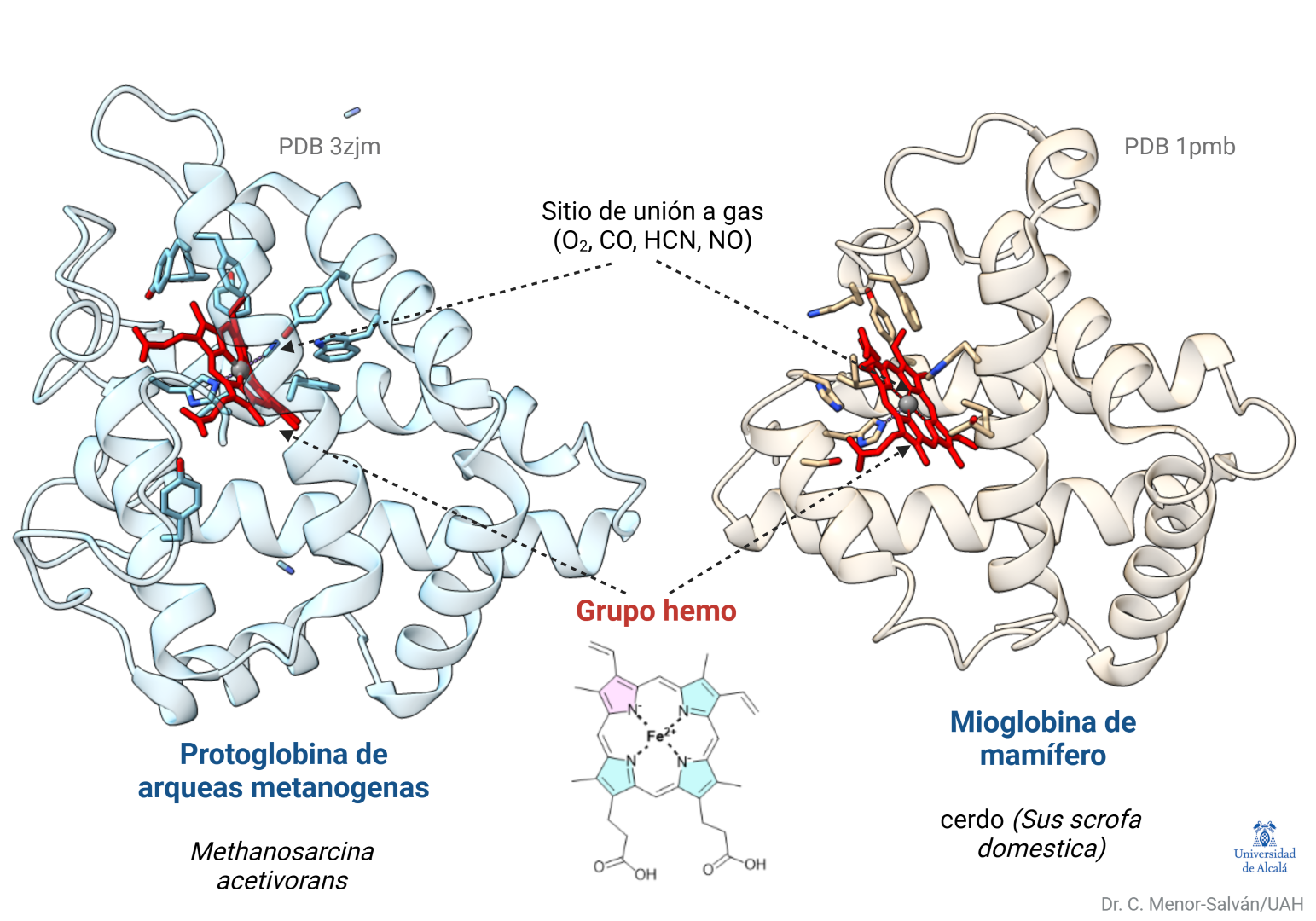
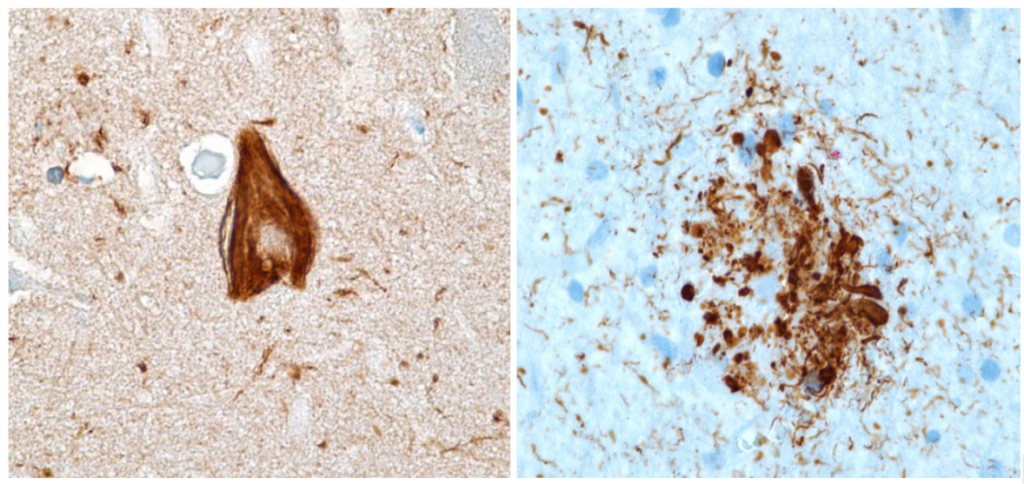
Estructuras de la protoglobina de arqueobacterias y la mioglobina de cerdo, mostrando el grupo hemo en su posición. Ambas proteínas, conectadas evolutivamente, tienen alta similitud estructural y gran afinidad por el monóxido de carbono.
Cuando la vida estaba en sus inicios y la atmósfera no contenía oxígeno, unas arqueobacterias productoras de metano ya estaban dotadas de la proteína protoglobina. Hoy día podemos encontrarlas en lugares tales como sistemas hidrotermales, aguas residuales, algunas minas, donde forman ecosistemas con otros procariotas que usan minerales (sulfuros metálicos) para obtener energía, o en fondos de lagos y mares, donde contribuyen a generar metano atmosférico.
Estas arqueas usan un sistema ancestral para metabolizar monóxido de carbono y usarlo como fuente de energía y carbono. En los inicios de la vida, posiblemente la protoglobina era un sensor de monóxido de carbono y, tal vez, de cianuro, otro componente quizá presente en aquel ambiente. Ambos gases se unen fuertemente al grupo hemo de la protoglobina.
Nosotros tenemos mioglobina y hemoglobina, que almacenan y transportan oxígeno. Estas proteínas evolucionaron a partir de aquellas globinas ancestrales de las arqueas, de quienes heredamos muchas estructuras moleculares, pues somos el resultado de una unión entre arqueas y bacterias que ocurrió hace unos 2000 millones de años.
Hace unos 600 millones de años, comenzó la evolución de la hemoglobina, que posibilitó el transporte de oxígeno desde el ambiente a los órganos, favoreciendo la evolución de los animales. Una consecuencia de este proceso de evolución es que el monóxido de carbono es muy tóxico para nosotros.
Esta toxicidad se debe a que nuestras mioglobina y hemoglobina tienen mucha afinidad por el monóxido de carbono, como la protoglobina arqueal; quizá, es un recuerdo de aquel lejano ancestro, que vivía en un ambiente sin oxígeno, alimentándose del monóxido de carbono. La mioglobina y hemoglobina no son el resultado de un «diseño inteligente», sino que resultan de la evolución de algo que surgió en un ambiente sin oxígeno y cuya función original se convirtió en un problema millones de años después.
Y no es el único problema. La vida nació en un ambiente sin oxígeno; cuando este empezó a aumentar en la atmósfera, tuvo lugar una de las primeras grandes extinciones. Los supervivientes desarrollaron adaptaciones moleculares que «parcheaban» los problemas que surgieron. En un ambiente sin oxígeno, el hierro reducido de la protoglobina era estable; en el ambiente con oxígeno, el hierro de la mioglobina y hemoglobina se oxida, por lo que tuvo que evolucionar un sistema antioxidante.
Otro de estos parches es una modificación en nuestras globinas que reduce ligeramente su afinidad por el monóxido de carbono. Ello reduce lo suficiente la toxicidad del gas como para que los fumadores puedan dar las gracias por ello. Sin esa modificación, los animales no soportaríamos un poco de humo.
La histidina 64, presente en la hemoglobina pero no en la protoglobina, estabiliza la unión del oxígeno al grupo hemo de la hemoglobina, que se produce formando un ángulo, mientras que obstaculiza ligeramente la unión del monóxido de carbono. Aunque el CO sigue teniendo mucha más afinidad por la hemoglobina que el O2, este pequeño cambio aportó una ligera ventaja a los animales.
Este ejemplo ilustra una característica básica de la evolución: el mantenimiento y adaptación de estructuras funcionales, no la mejora hacia un diseño óptimo. La mioglobina de mamífero y la protoglobina de arqueas sólo tienen en común un 13% de la secuencia de sus genes. Ese pequeño porcentaje permite que sus estructuras y función básica (la unión de estos gases) se hayan preservado y guardan una gran similitud estructural.
Las moléculas biológicas nos conectan a todos los organismos en un gran árbol cuya raíz está en el origen de la vida, hace más de 4000 millones de años. Y esto es un hecho, no un relato.
Tengo intolerancia a la lactosa: ¿debo comprar «mantequilla sin lactosa»?
escrito por C. Menor-Salvan | 17 diciembre, 2024
C. Menor-Salván. Mayo 2024.
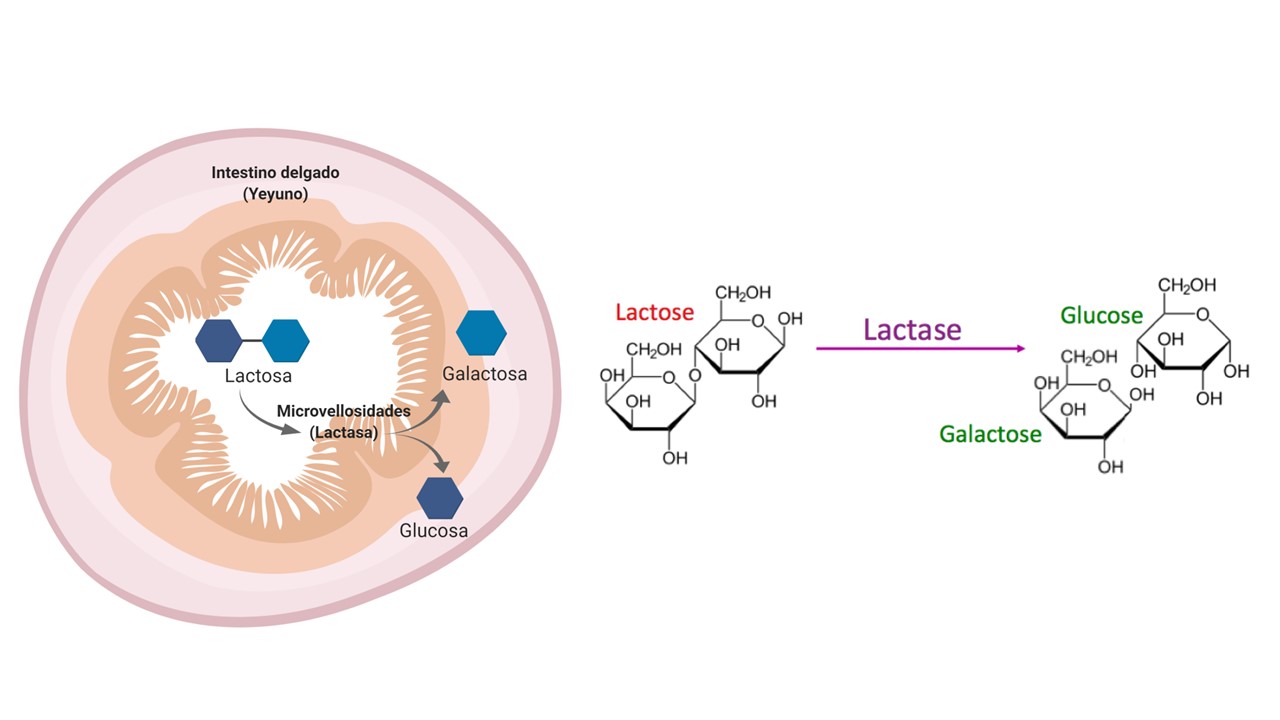
La lactosa es un azúcar natural presente en la leche de los mamíferos en una cantidad considerable. La leche humana contiene aproximadamente un 7-8 % de lactosa, y la leche de vaca comercial tiene una proporción variable entre el 3 y el 5 %.
Es un disacárido, es decir, un azúcar formado por la unión química de otros dos azúcares más simples (monosacáridos). En este caso, esos dos azúcares son glucosa y galactosa. Todos estamos muy familiarizados con el aspecto de los disacáridos: el azúcar común o sacarosa es un disacárido formado por glucosa y fructosa. En los disacáridos es muy importante, tanto la composición como la geometría con la que se unen los dos azúcares simples. Esto cambia sus propiedades; por ejemplo, la lactosa tiene aproximadamente un 20% del dulzor de la sacarosa, debido a que, por su estructura, no interacciona tan bien con los receptores de la lengua. De ahí que la leche no nos sepa tan dulce.
La lactosa es fundamental en las primeras fases del desarrollo, durante la lactancia, debido a que es una fuente de galactosa, necesaria, entre otras cosas, para el correcto desarrollo del sistema nervioso e inmunológico del niño, debido a que se incorpora estructuralmente en moléculas esenciales como los cerebrósidos y es necesaria para formar la mielina. Es importante también como prebiótico, y ayuda a crear y mantener un ecosistema microbiológico sano en el intestino, además de ayudar a la absorción de calcio y oligoelementos.
La lactosa requiere, para su absorción, una enzima, la lactasa, que rompe la molécula en sus dos componentes, para que puedan ser absorbidos:
La lactasa, presente en el intestino delgado, rompe la molécula de lactosa, permitiendo la asimilación de la glucosa y la galactosa.
Y aquí tenemos el problema: los bebés lactantes tienen una actividad lactasa elevada, pero ésta disminuye al terminar la lactancia, reduciéndose la absorción de lactosa. Gran parte de los humanos mantienen un cierto nivel de lactasa durante toda su vida, gracias a una mutación que hizo posible que podamos seguir tomando leche y productos lácteos durante la edad adulta. Pero muchos humanos tienen una actividad lactasa muy baja, lo que provoca intolerancia a la lactosa: ésta no se rompe y absorbe, quedándose en el tracto intestinal y produciendo malestar, hinchazón y gases, al llegar al intestino grueso, donde se convierte en un festín para las bacterias, e incluso diarreas, debido al desequilibrio osmótico que provoca. Aproximadamente el 30% de la población en España tiene intolerancia a la lactosa, con efectos que pueden ser desde casi inapreciables o leves, hasta moderados o intensos, haciendo imposible para la persona en este caso tomar leche. Es importante no confundir la intolerancia a la lactosa con la alergia o la intolerancia a las proteínas de la leche, algo completamente distinto.
¿Tiene lactosa la mantequilla?. Tests de lactosa
Vamos a realizar ahora un ensayo químico muy sencillo para detectar y determinar aproximadamente la lactosa en una muestra de leche y otra de mantequilla. Este test, en una versión un poco mas compleja, se ha utilizado para la determinación colorimétrica de la lactosa. Es el test del ácido pícrico. Se basa en la reacción entre la lactosa (u otros azúcares reductores) y el ácido pícrico, de color amarillo, en un medio alcalino. Según la cantidad de azúcar presente, el ácido pícrico se reduce a ácido picrámico, de color rojo, dando una coloración desde anaranjada a rojo intensa.
El primer paso es eliminar las proteínas de la leche. En este caso, hemos usado un poco de leche desnatada, y precipitamos las proteínas añadiendo la misma cantidad de una solución de ácido tricloroacético al 24%.
Izquierda, leche tras precipitar las proteínas. Derecha, muestra sin precipitar. Para el ensayo de azúcar tomamos el líquido claro tras centrifugar las proteínas.
Tomamos 1 ml del líquido claro sin proteínas que vemos en la foto, añadimos 1 ml de solución saturada de ácido pícrico y 1 ml de NaOH al 5%. Se calienta durante 2 minutos a 100º. En el caso de la mantequilla, el proceso es mas laborioso: extraemos una porción de mantequilla con agua, separamos la materia grasa y hacemos el ensayo con el extracto acuoso, una vez concentrado por liofilización, pues los azúcares están en baja proporción y son solubles en agua. Aquí tenéis el resultado:
En el tubo 1, la muestra de leche tiene un contenido alto de lactosa, como se ve comparado con el tubo 2. En el tubo 3 vemos el resultado con mantequilla de un conocido supermercado (segun etiqueta, sus azúcares totales son 0.4%) y en el 4 con agua. Como se ve, la mantequilla apenas contiene azúcar. El contenido de esta muestra, una vez calculado, nos sale en torno a 0.5%, por lo que, salvo que se tenga una intolerancia realmente severa, se puede consumir mantequilla y productos conteniendo mantequilla sin problema. Es lógico: la mantequilla se prepara con la fracción grasa; la lactosa es una molécula polar que no se disuelve en la grasa y se separa en la fracción acuosa durante la preparación de la mantequilla. Pero, ¿realmente contiene lactosa?
Etiqueta nutricional de la mantequilla que hemos usado en el ensayo. Según ella, los azúcares totales son el 0.4%, comparable al resultado de nuestro ensayo.
Azúcar bajo el microscopio
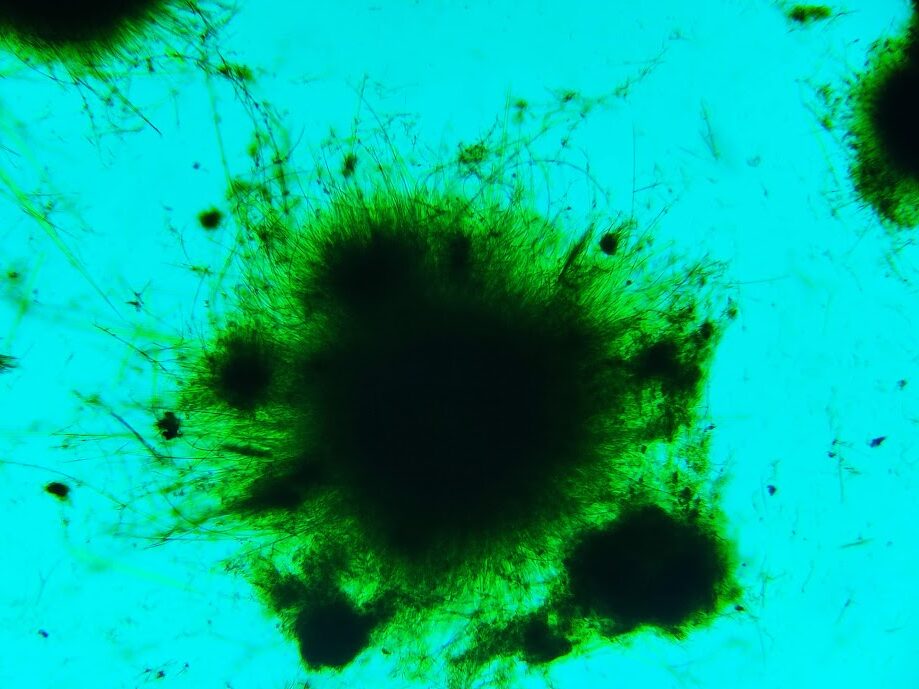
El test del ácido pícrico es muy sencillo y eficaz para detectar lactosa, pero no es específico, por lo que estamos viendo realmente los azúcares reductores totales. Otros azúcares y sustancias reductoras tienen capacidad para reducir el ácido pícrico, como la glucosa o la fructosa. ¿podemos comprobar, de modo simple, que la muestra que hemos usado tenga realmente lactosa?. Hay un test que, con ayuda de un microscopio biológico normal, puede resolverlo: los azúcares reaccionan con la fenilhidracina, dando lugar a un compuesto insoluble llamado fenilosazona. Las fenilosazonas son diferentes para cada azúcar y cristalizan, formando agregados cristalinos distintos, que pueden diferenciarse fácilmente con el microscopio (si se han hecho en las mismas condiciones), excepto en el caso de azúcares que forman la misma fenilosazona, como el caso de la glucosa y la fructosa.
Si hacemos el test con nuestra muestra de leche vemos este resultado:
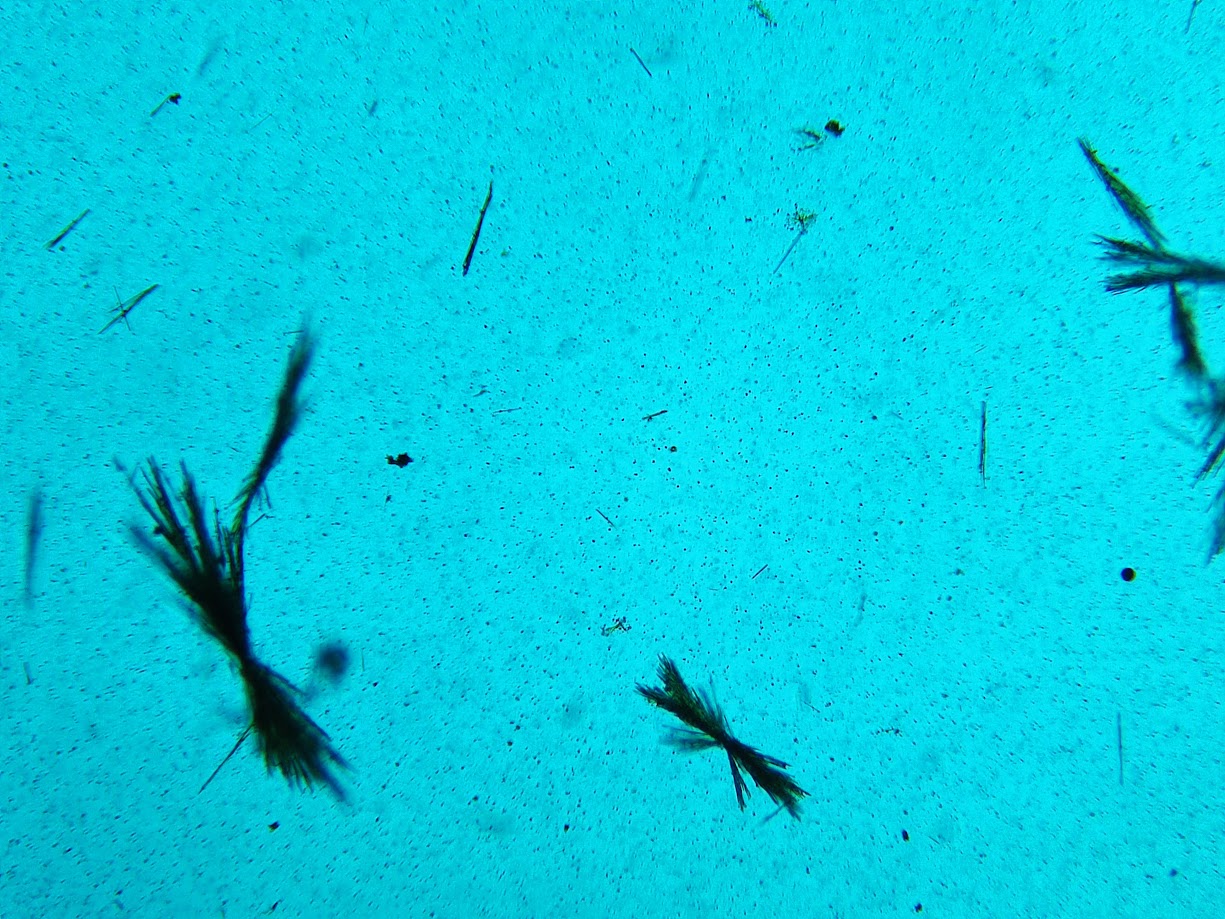
¿A qué azúcar diríais que corresponde?. Correcto, es lactosa. Si hacemos el test despues de extraer y concentrar las trazas de azúcar que contiene la mantequilla y la analizamos observamos esto:
La proporción de azúcar es bajísima y formada principalmente por glucosa/fructosa y otras. De hecho, no hemos podido detectar lactosa y la proporción de azúcar real es incluso inferior a la que observamos con el test del ácido pícrico, que podemos considerar nuestro máximo, ya que da una proporción aparente algo mayor de la real en concentraciones muy bajas. También depende de la marca, pero, en general, las mantequillas tienen una proporción real de azúcares reductoresusualmente inferior al 0.1%. Durante la elaboración de la mantequilla se incorporan, además, microorganismos como bacterias lácticas, que metabolizan la poca lactosa que llevaba durante la elaboración, y fermentan las grasas; es el proceso de maduración, que proporciona el olor y sabor característico de la mantequilla. El escaso contenido en azúcares totales depende de cómo se ha preparado cada mantequilla en particular (en muchos casos, por ejemplo, se bate con leche para rebajar el contenido graso), pero casi siempre va a ser inferior al 0.5%.
Mantequilla ‘sin lactosa’, ¿un producto innecesario fruto del temor o rechazo a los lácteos con lactosa? ¿para que los intolerantes a la lactosa que no saben bioquímica puedan usar mantequilla con tranquilidad? Mantequilla tradicional de Soria. «contiene cantidades insignificantes de azúcares». De hecho, la lactosa es indetectable aquí.
En conclusión: La mantequilla contiene una proporción muy baja de azúcares (de 10 a 100 veces inferior a la leche entera) y un contenido en lactosa usualmente inferior al 0.1%, comparable a la de la leche ‘sin lactosa’. Un vaso de leche de 200 cc nos va a proporcionar aproximadamente entre 10 y 15 gramos de lactosa, lo cual es una cantidad considerable para una persona con intolerancia a la lactosa; pero, suponiendo el improbable caso de que la mantequilla llegara a contenter un 0.4% de lactosa, la típica pastilla de mantequilla de 10 gramos con la que suelen acompañar la tostada en el desayuno, nos proporcionaría tan solo 40 miligramos de lactosa. Por ello y dada la cantidad de mantequilla que se suele consumir, no es necesario comprar mantequilla «sin lactosa», ni es necesario pedirle al camarero que nos cambie la pastillita o tarrinita de mantequilla por una ‘sin lactosa’.
Una mutación en la glucosa-6-fosfato dehidrogenasa y su influencia en la constante de Michaelis-Menten y la eficiencia catalítica de la enzima.
escrito por C. Menor-Salvan | 17 diciembre, 2024
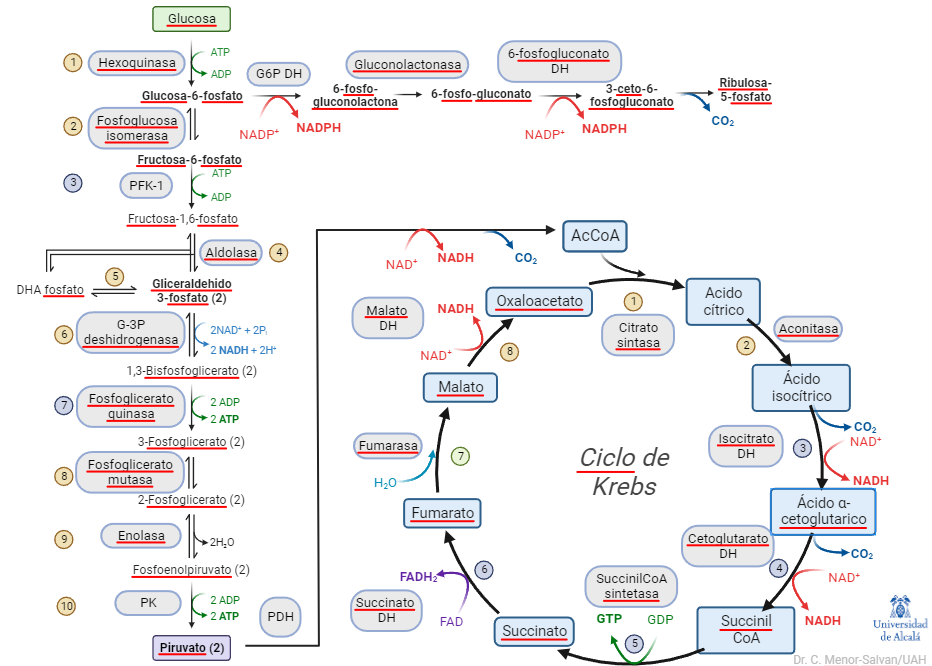
La glucosa-6-fosfato dehidrogenasa (G6PDH) es una enzima fundamental en el metabolismo central. Conecta la glucolisis con el metabolismo de los azúcares en el ciclo de las pentosas fosfato, a través de la oxidación de glucosa-6-fosfato a 6-fosfogluconolactona por NADP+. La reacción es una fuente de NADPH, que es un cofactor crucial en algunos procesos bioquímicos, como la recuperación del glutation, un pequeño péptido esencial para evitar daños por estrés oxidativo.
Esquema simplificado del metabolismo central. La reacción de la G6PDH (arriba, izquierda) conecta la glucolisis con el ciclo de las pentosas fosfato
La recuperación de glutation es especialmente significativa en los glóbulos rojos, en los que esta reacción es su única fuente de NADPH. Si sus niveles se ven comprometidos, los glóbulos rojos se ven afectados por daño oxidativo y puede producirse anemia hemolítica
Esto puede tener lugar por deficiencia de la enzima G6PDH, que es, precisamente, una de las enzimopatías más comunes, afectando a casi 500 millones de personas en todo el mundo, principalmente en Africa, Peninsula Arábica y Sudeste Asiático. Se han descrito 230 mutaciones con relevancia clínica, principalmente hombres, al estar ligadas al cromosoma X. Las más graves producen anemia hemolítica y las menos severas producen hemolisis asociadas a tóxicos que producen estrés oxidativo o algunas toxinas, como las lectinas presentes en las judías.
La razón de esa distribución es que, curiosamente, la deficiencia de esta enzima confiere resistencia contra la malaria producida por el parásito Plasmodium falciparum. Entonces, en las zonas donde la enfermedad es endémica, las mutaciones de G6PDH proporcionan una ventaja evolutiva, prevaleciendo gradualmente sobra la enzima más activa, ya que el parásito requiere de ésta para su ciclo vital.
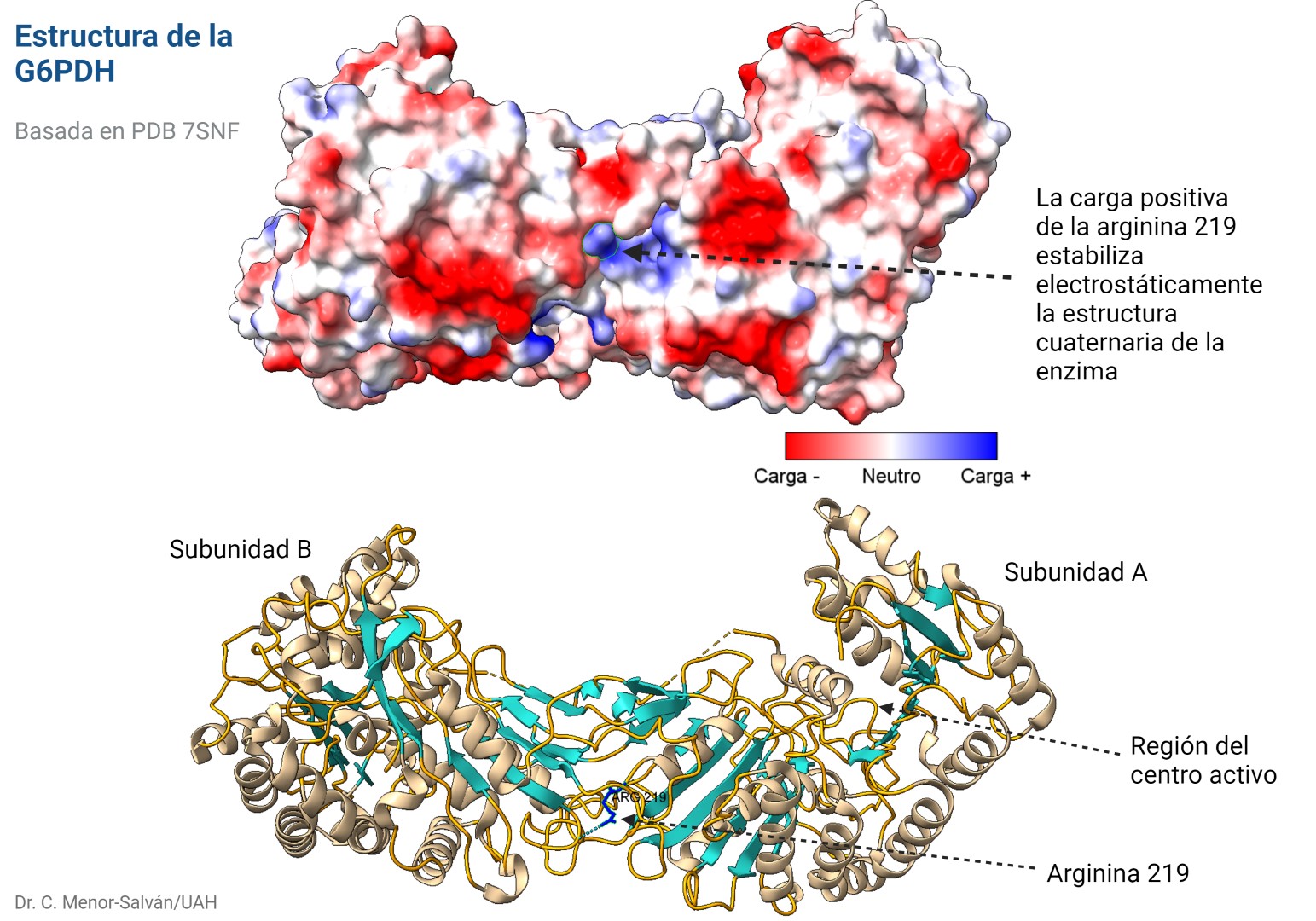
Mutación en la arginina 219
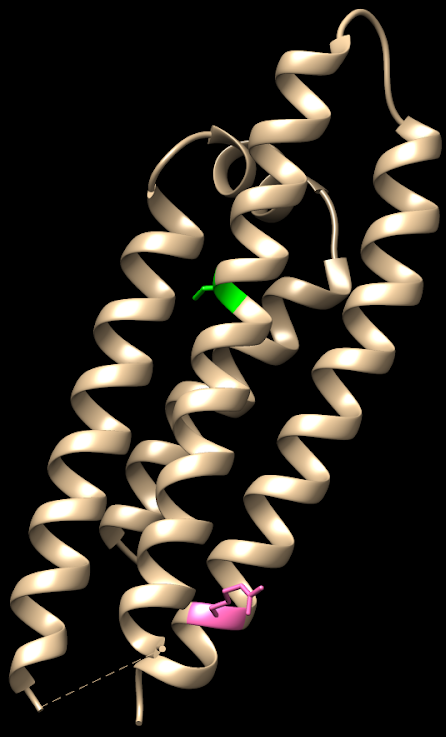
En el trabajo publicado en Nature Communications por Zgheib et al., se describe un caso muy interesante desde el punto de vista de la bioquímica estructural. Un joven paciente de 15 años sufrió hemolisis y pancitopenia tras una infección viral. Los autores estudiaron el caso y, tras ver en su historia que un tío suyo tenía deficiencia de G6PDH, llevaron a cabo el estudio bioquímico de la enzima.
Descubrieron una nueva mutación en la enzima, en la que una arginina en la posición 219 se había sustituido por una glicina. Esta mutación puede estar asociada a un cambio de un simple nucleótido, pues los codones de glicina y arginina sólo se diferencian en su primer nucleótido, siendo G para arginina y C para glicina.
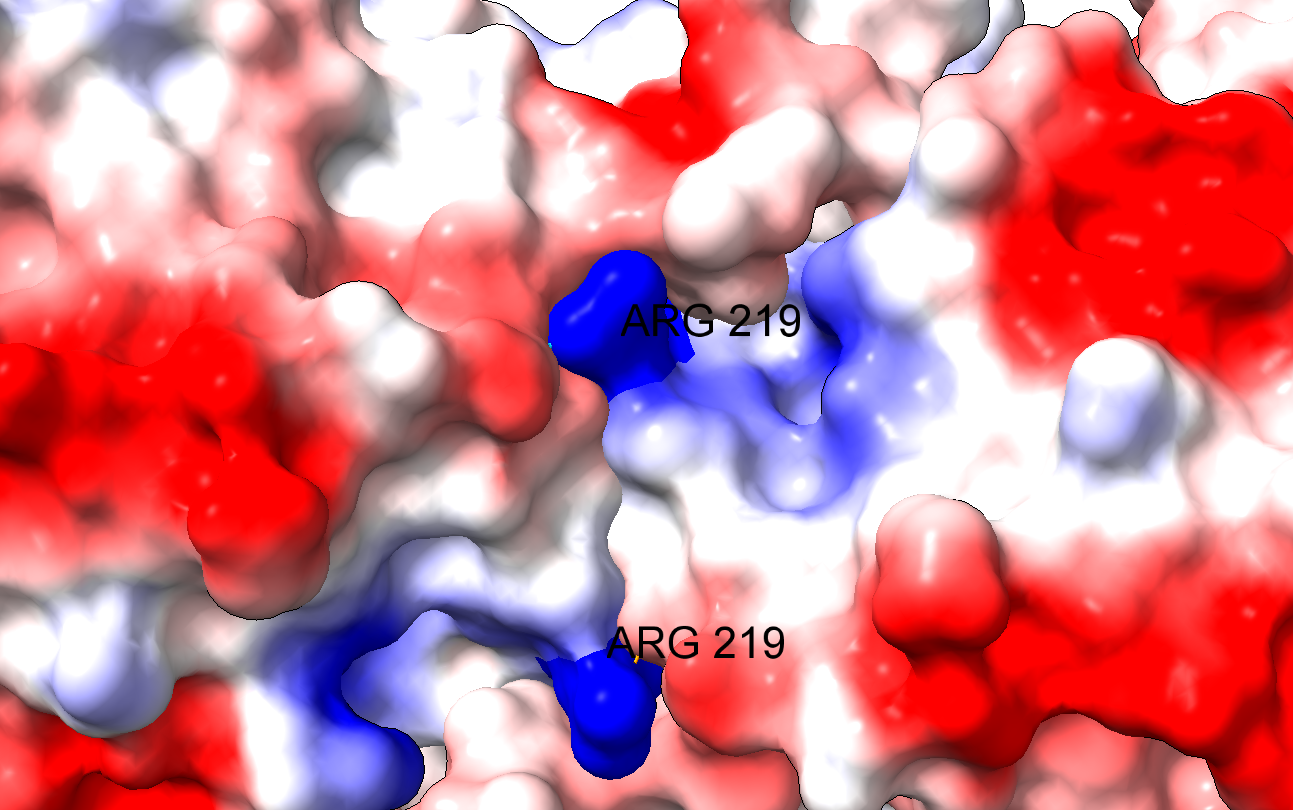
El problema es que el cambio de un aminoácido con carga como la arginina por un aminoácido neutro de pequeño tamaño como la glicina puede tener importantes implicaciones en la estructura de la proteína. En efecto, la arginina 219 estabiliza la estructura proteica, que es un dímero, a través de un puente de hidrógeno y de la carga positiva, por interacción electrostática. Al cambiar por la glicina, se pierden esas interacciones electrostáticas en una posición clave, desestabilizándose la estructura de la enzima y dificultando su acción catalítica. Los autores comprueban que, en efecto, la enzima mutante es mucho más sensible a la desnaturalización, debido a la desestabilización estructural. La enzima mutante pierde actividad a temperatura superior a 44ºC, mientras que la enzima WT (wild type, la enzima funcional normal) pierde actividad a temperaturas superiores a 50ºC.
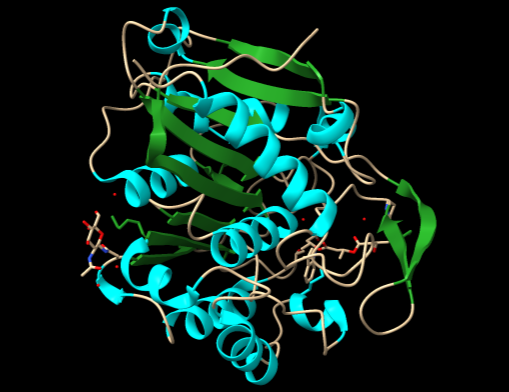
Estructura de la glucosa-6-fosfato dehidrogenasa. Arriba, superficie electrostática de la molécula, un dímero estabilizado por la carga positiva de la arginina 219. Abajo, modelo de cintas que muestra la posición de la arginina y su puente de hidrógeno con la otra subunidad, y la posición del centro activo de la enzima. Figura realizada con ChimeraX.
Es interesante que la arginina 219 no está en el centro activo de la enzima. Así, podemos explicar los datos experimentales de la actividad de la enzima que obtienen los autores:
Km=49.5 µM para el sustrato (glucosa 6 fosfato) en la enzima WT
Km=46.1 µM para el sustrato en la enzima con la mutación
Kcat=326 s-1 para la enzima WT
Kcat=6 s-1 para la enzima mutante.
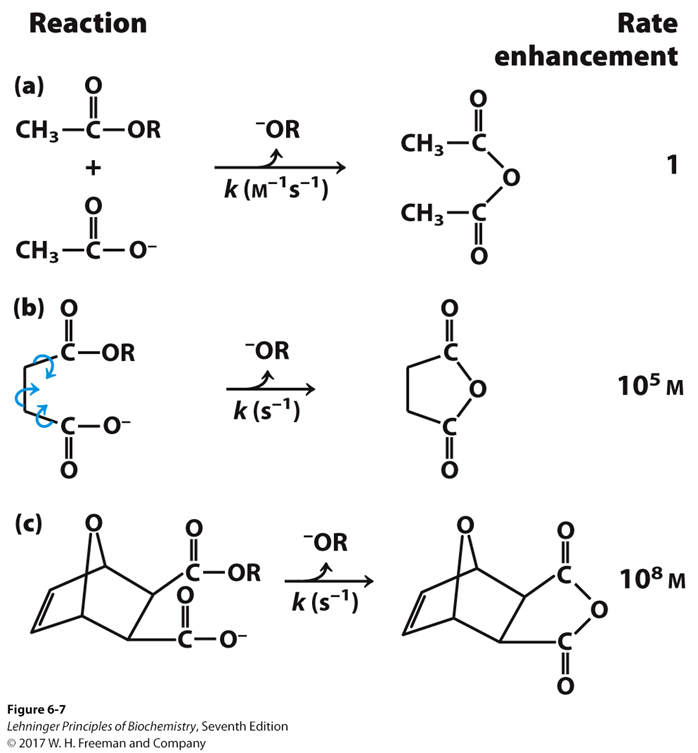
¿que nos dicen estos resultados?. Que la enzima mutante muestra una constante de Michaelis-Menten similar a la WT para el sustrato glucosa-6-fosfato, por lo que no ha perdido afinidad por el sustrato; no es sorprendente, pues la mutación no afecta al centro activo y la unión sustrato-enzima no se ha visto afectada. Sin embargo, la enzima mutante ha perdido eficiencia catalítica. Recordemos que el Kcat o numero de turnover son los moles de sustrato que son transformados por mol de enzima y por segundo. Así, su actividad se ve enormemente reducida: la enzima WT es 54 veces más activa.
Detalle de los ‘anclajes’ electrostáticos mediante las argininas 219 de las dos cadenas del homodímero de G6PDH. La mutación a glicina hace que se pierdan las uniones electrostáticas, desestabilizando la estructura.
Este caso ilustra bien la diferencia de significado entre las dos constantes fundamentales de la actividad enzimática y cómo la discusión teórica que realizamos en la clase de Bioquímica Estructural tiene gran relevancia práctica.
¿Qué está ocurriendo a nivel termodinámico?
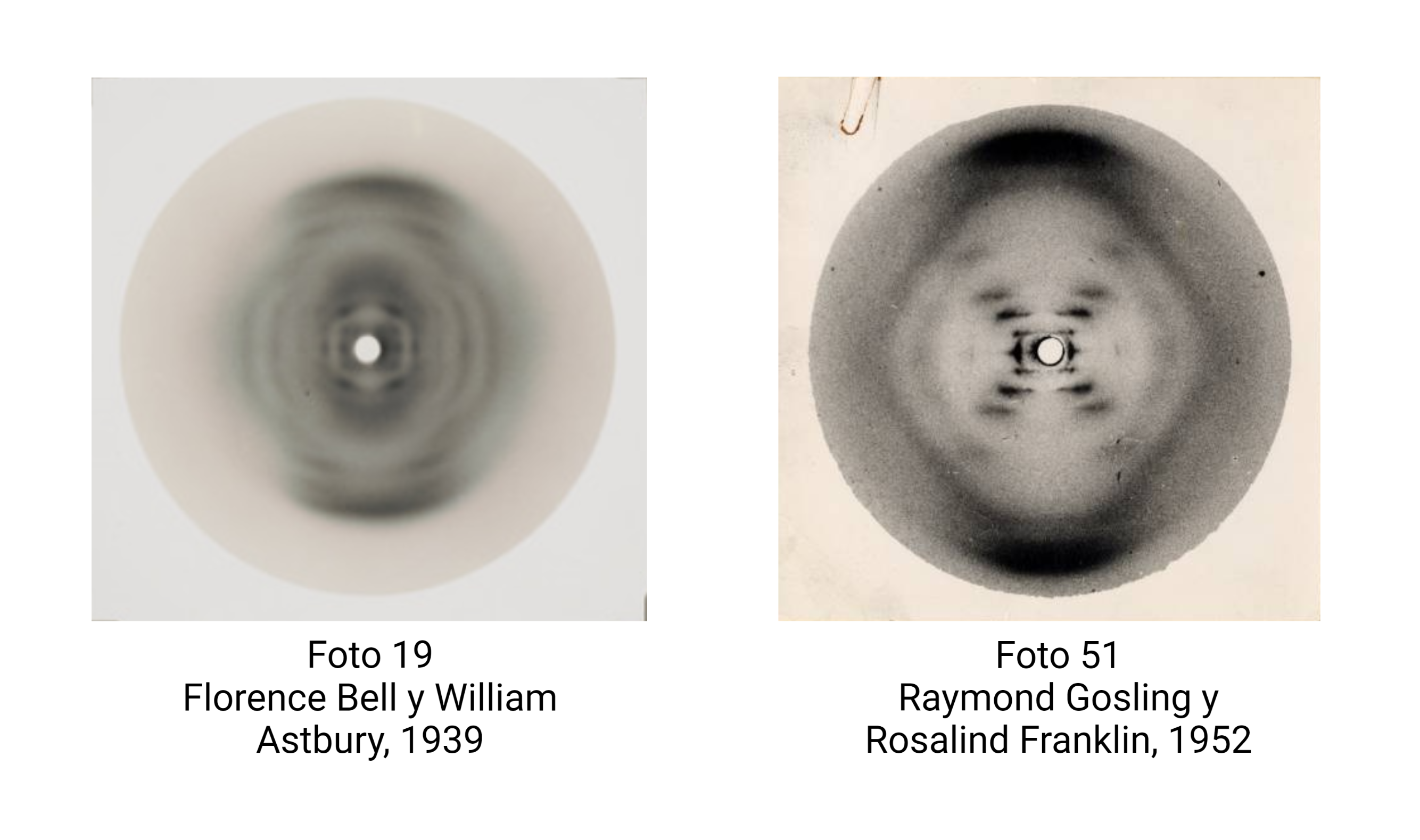
El hecho clave es que la mutación R219G varía poco la Km, pero reduuce drásticamente la Kcat. ¿hay una relación con la ΔG‡, variación de energía libre hacia el estado de transición?.
Podemos aproximar Km a la afinidad por el sustrato, es decir, a la estabilidad del complejo ES, mientras que Kcat se relaciona directamente con la barrera de activación que tiene que superar el sistema para alcanzar el estado de transición. Sabemos, según la teoría de Eyring, que ΔG‡ es inversamente proporcional a la velocidad de la reacción. Es decir, cuanta más alta sea la energía libre de transición, menor será la velocidad y la eficacia de la enzima:
donde k es la constante de velocidad. Esta ecuación la podemos separar en sus componentes entrópica y entálpica:
En las reacciones enzimáticas, la enzima acelera la reacción al disminuir la energía libre de activación necesaria para alcanzar dicho estado de transición, lo que consigue, bien por control entrópico, logrando que el paso del complejo ES al estado de transición sea favorable, ΔS‡>0 o próximo a cero, o bien por control entálpico, logrando que ΔH‡ se reduzca o sea negativo.
En este contexto, sabemos que kcat WT/kcat mut es 54.3, es decir, que la enzima mutante es 54 veces más «lenta». Para evaluar la diferencia entre las energías libres de activación, podemos simplificar a:
dado que
ΔΔG‡ = ΔGmut‡ − ΔGWT‡
y que podemos sustituir k por kcat si ésta refleja el paso limitante asociado a cruzar la barrera de activación.
De los datos, obtenemos que ΔΔG‡ = 2.4 kcal/mol. Es decir, que la mutación eleva la barrera impuesta por la energía de activación respecto a la enzima silvestre. Esto implica que el estado de transición es menos accesible, provocando la caída de velocidad. Como se ve, un cambio pequeño en la energía de activación tiene una consecuencia importante en la actividad de la enzima.
Haciendo un cálculo similar para la energía libre de unión obtenemos que ΔΔGB es -0.04 kcal/mol, un valor muy pequeño. La unión de sustrato es algo más favorable en el mutante, pero muy pequeño y no sabemos si esta diferencia puede ser simple error experimental, así que vamos a considerar que la mutación R219G no altera apreciablemente la energía libre de unión del sustrato o la estabilidad del complejo ES.
La reducción de Kcat sugiere que la Arg219 desempeña un papel clave en la estabilización del estado de transición. Dada la posición de la mutación, critica para la estabilidad del dímero de G6PD, podemos explicar el efecto de dos maneras:
Arg219 es esencial para estabilizar el dímero, y es éste la forma activa de la proteína. Ello favorece que la enzima mutante esté como una mezcla de monómero y dímero, mientras que la WT es sólo el dímero activo. Sabemos que la Kcat son los moles de sustrato convertidos en producto por mol de enzima y unidad de tiempo. Si disminuimos la concentración de enzima activa, aunque la concentración de enzima total se mantenga, tendremos que
Kcat aparente = factiva x Kcat
Es decir, que la enzima mutante tendrá una Kcat aparente que es la fracción de enzima activa por la Kcat de la enzima activa. Si despejamos obtenemos que factiva = 1.8%. ¡la mayor parte de la enzima estaría desactivada!
De ahí que su eficiencia baje: aunque la enzima total es constante y tiene capacidad para unir el sustrato (y por ello Km se mantiene igual), la mayor parte de la enzima es incapaz de llevar a cabo la reacción.
Por otro lado, la mutación puede haber alterado la organización estructural. Al perderse el puente salino que forma la arginina, aumenta la entropía conformacional (aumenta la flexibilidad y la «libertad» de la enzima para adoptar diversas conformaciones), penalizando el término TΔS‡. Con la mutación, el «salto» al estado de transición implica que tiene que restringir más la «libertad» de la estructura, lo que hace que ΔS‡, que antes era favorable, se haga más pequeño, o incluso negativo, elevando la energía libre de activación. Esto encaja con la pérdida de actividad a temperatura más baja en la enzima mutante.
Curiosamente, este caso también nos explica por qué el modelo llave-cerradura no es posible, ya que implica una alta estabilización del complejo ES unida a un alto coste entrópico para obtener el estado de transición.
Como se ve, pequeños cambios, como una mutación en un aminoácido o una pequeña variación en las energías del proceso tienen importantes consecuencias fisiológicas y médicas.
Referencias
Zgheib, O. et al. (2023) ‘Substitution of arginine 219 by glycine compromises stability, dimerization, and catalytic activity in a G6PD mutant’, Communications Biology. Springer US, 6(1), p. 1245. doi: 10.1038/s42003-023-05599-z.
El error de Pauling y la carrera por la estructura del ADN
escrito por C. Menor-Salvan | 17 diciembre, 2024
C. Menor-Salván. Ver. 2.5. Abril 2023
El descubrimiento de la estructura del ADN fue uno de los logros científicos destacados del siglo XX. En él participaron además de los conocidos Watson y Crick y Rosalind Franklin, una serie de científicos relevantes, cuyo nombre apenas se recuerda; sin sus contribuciones, no podemos entender la historia completa. Esta aventura nos enseña además que, en la ciencia, aunque se hagan famosos los «goleadores», el conocimiento se construye de modo colectivo y los errores pueden ser tan importantes como los aciertos. Delicioso es el fruto que surge tras el amargor del error y la ignorancia, y de ellos es desde donde se construye la ciencia. Además, los aspectos mundanos pueden ser tan relevantes como los técnicos.
El ADN se descubrió en el siglo XIX, pero se tardó más de medio siglo en revelar su estructura
No hay que confundir el descubrimiento de la estructura del ADN, con el descubrimiento del ADN en sí. Se atribuye el descubrimiento del ADN al químico suizo Friedrich Miescher, entre 1860 y 1874, siendo Phoebus Levene quien dio, en 1909, su descripción química precisa. Miescher propuso, en 1874, que, de alguna manera la «nucleína» (nombre que dió al ADN) era la «causa específica de la fertilización».
Levene acuñó el término ‘ácido nucleico’. Propuso la ‘teoría del tetranucleótido’, sugiriendo que el ADN estaba compuesto por cuatro bases, un azúcar y fosfato. Sin embargo, en aquel momento, aún no existía la tecnología necesaria para entender la arquitectura de la molécula.
Aspecto real del DNA puro. Esta muestra se obtuvo de timo de vaca, la fuente usual de DNA para su estudio. De hecho, en la época del descubrimiento de su estructura, no se le llamaba DNA sino «ácido timonucleico». Imagen: C. Menor-Salván/UAH
Pasó casi medio siglo hasta que se determinó su estructura. Una vez disponible el arsenal técnico adecuado, no había nadie mejor para lograrlo que el genial químico estadounidense Linus Pauling (1901-1994), quien estuvo a las puertas de conseguirlo antes de que Watson, Crick, Wilkins y Franklin publicaran su famoso artículo triple de abril de 1953.
Pauling fue uno de los científicos más relevantes del siglo XX. Recibió el premio Nobel en Química en 1954 por su contribución al conocimiento de los enlaces químicos. Para esa fecha había realizado tantos descubrimientos importantes en numerosos campos de la Química y la Biología, que, cuando le llamaron para comunicarle la concesión del premio, pidió a su interlocutor que le leyese la comunicación, pues no tenía claro por cuál de sus trabajos recibía el premio.
Pauling había resuelto otro gran problema: la estructura de las proteínas. Con Robert Corey y Herman Branson, publicaron en 1951 las estructuras secundarias que ahora ilustran los libros de texto. Este importante trabajo era ya suficiente para que tuviera fama imperecedera en el mundo de la Ciencia, pero Pauling era ambicioso y estaba obsesionado con resolver todas las estructuras de macromoléculas biológicas. Así, era el favorito en la carrera por el ADN. Él mismo estaba convencido de ello.
Astbury, Bell y la triple hélice de Pauling
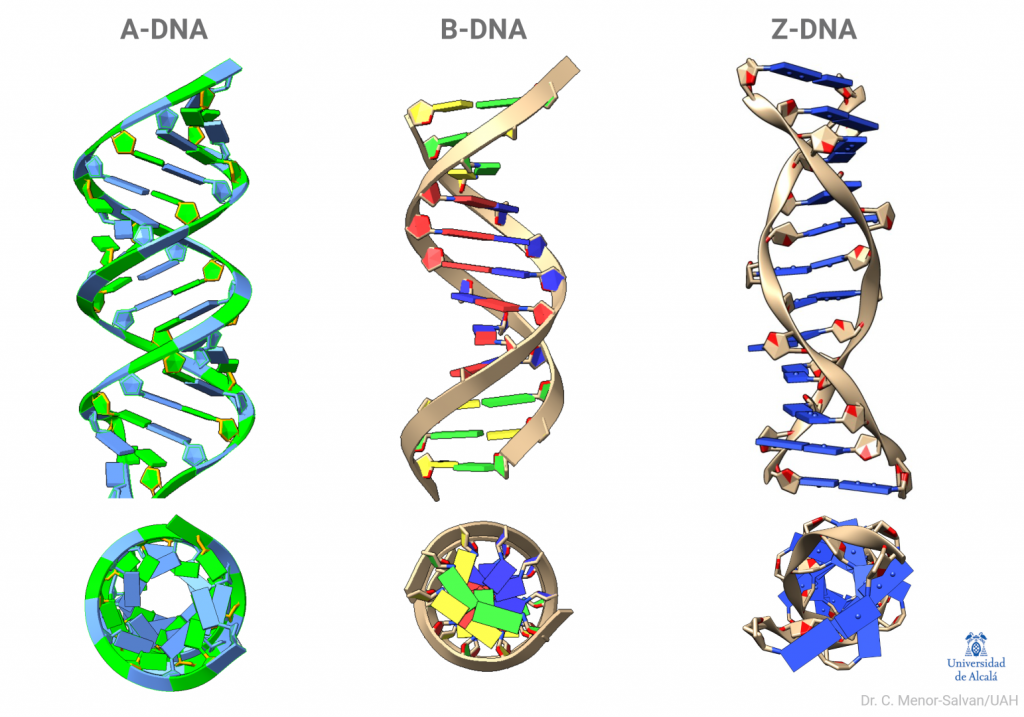
Una brillante química británica, Florence Bell, presentó, en 1939, su tesis sobre la estructura del ADN y las proteínas, bajo la dirección de William Astbury. En ella, Bell y Astbury presentaron, por primera vez en la Historia, imágenes de difracción de rayos X de ADN. Esta compleja técnica, que se aplica en campos desde la Bioquímica Estructural hasta la Mineralogía, es esencial para resolver las estructuras macromoleculares así como las estructuras de los sólidos cristalinos. Y ahí está el problema: EL ADN es muy difícil de cristalizar y tiene una peculiaridad clave, que descubrió Rosalind Franklin: puede cambiar de forma.
Portada de la tesis doctoral de Florence Bell. Su lectura es muy interesante, está redactada con un tono honesto y cálido, y constituye un hermoso documento sobre el nacimiento de la Biología Molecular. Después, con el inicio de la Segunda Guerra Mundial, todo quedó en un impasse; entre tanto, Bell se casó con un oficial norteamericano y emigró a EEUU tras la guerra. A pesar de los esfuerzos de Astbury, Bell no quiso continuar su carrera científica, aunque trabajó como química en EEUU.
Bell logró obtener un esbozo de la estructura del ADN, formado por largas fibras con las bases colocadas en paralelo y unidas, a modo de cuentas de collar, por el fosfato; a partir de ello, no pudo avanzar más. Su tesis relata los intentos para obtener una muestra de ADN lo suficientemente cristalina, que terminaban en imágenes de difracción borrosas. El ADN se resistía a revelar su estructura. Había algo que Bell ignoraba: el ADN no tiene una única forma. Al preparar el ADN puro, se formaba una mezcla de lo que ahora conocemos como ADN B y ADN A, que hacían muy difícil interpretar los datos. Además, todavía tenía que mejorar la tecnología para la obtención de éstos.
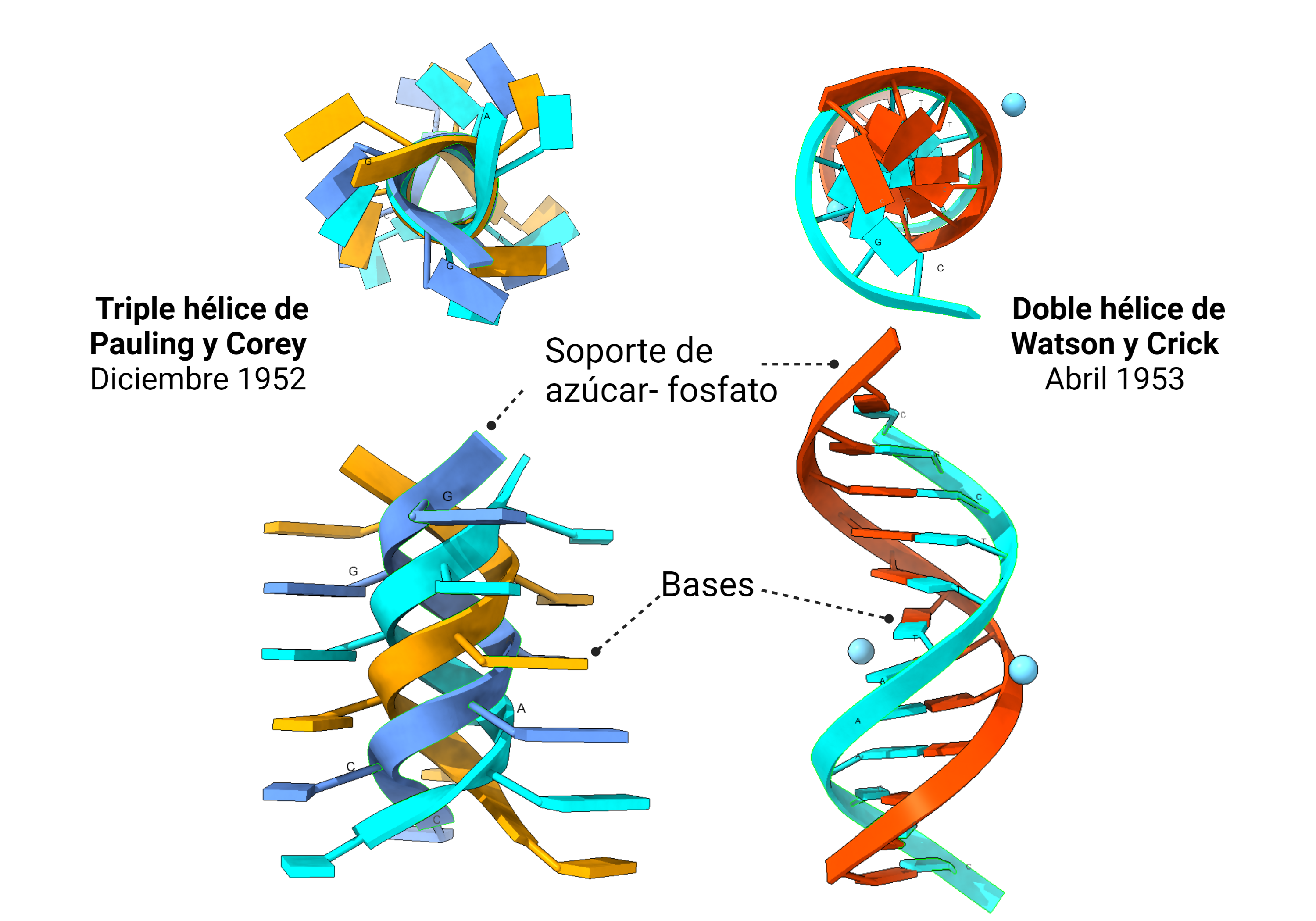
Fue Rosalind Franklin, 15 años después, quien logró lo que Bell no pudo: obtener una forma única de ADN cristalino, en su forma canónica ADN B, con el que hizo posible la famosa foto 51, que aclaró definitivamente la estructura.
Florence Bell propuso en su tesis de 1939 este esbozo de la estructura de ADN, en el que los nucleótidos, unidos por enlaces fosfodiester 3′-5′, formaban largas cadenas con las bases apiladas. Si Bell hubiera conseguido mejores datos de DRX, seguramente ella misma, o con Pauling, no habrían tenido ningún problema en determinar la estructura. Imagen: F. Bell, 1938
Aun así, Bell proporcionó datos importantes, gracias a una imagen particularmente clara, la imagen 19, tales como la distancia entre las bases y que los nucleótidos formaban largas cadenas.
Pauling estudió los resultados de Bell y Astbury, tomando como hipótesis que las bases del ADN se orientan al exterior, para enlazarse con otras moléculas y que los fosfatos se organizan en un patrón similar al de algunos minerales. También tuvo en cuenta otro dato fundamental: el biofísico Robley Williams había logrado, en 1952, obtener una imagen en 3D de una molécula de ADN con un microscopio electrónico. Pauling observó que ese pequeño cilindro podría ser una trenza helicoidal.
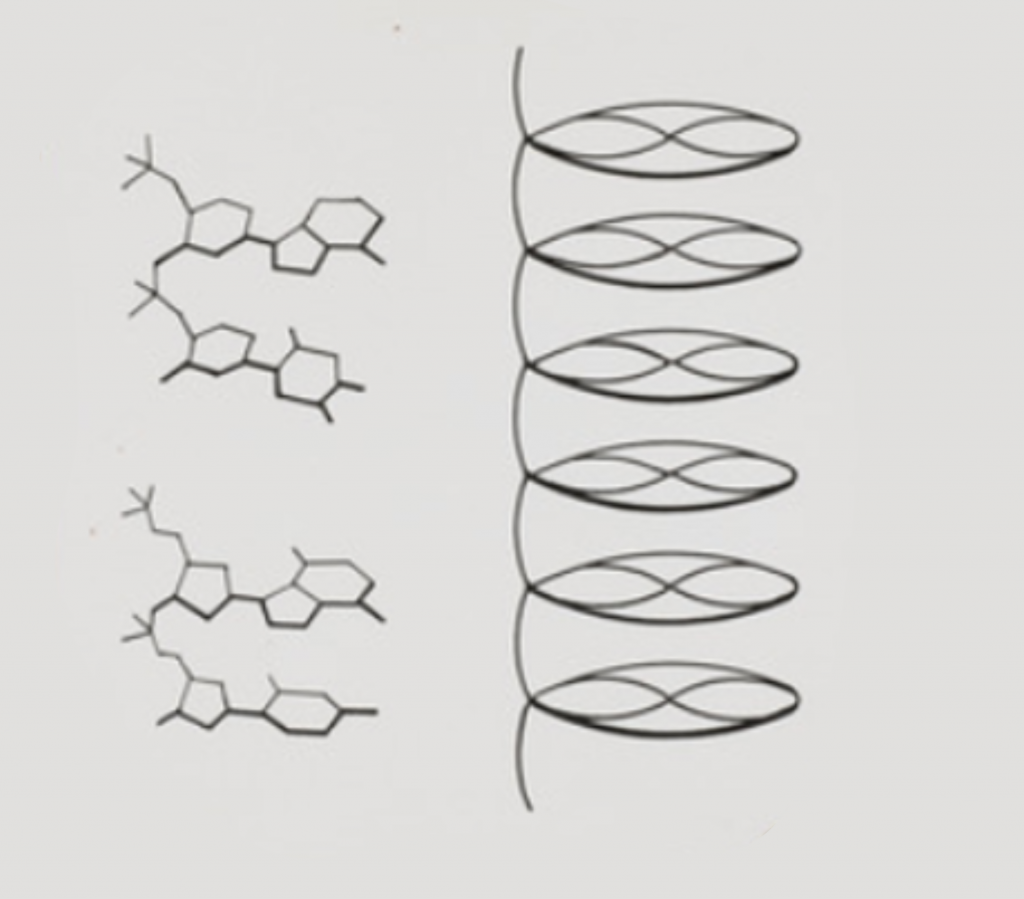
Con estas ideas, propuso un modelo en triple hélice, en un artículo enviado, no sin prisas, el 31 de diciembre de 1952. En él, publicado en febrero de 1953, Pauling sugiere que su modelo es todavía algo preliminar y requería refinado. Su colaborador Corey le avisó de que había problemas, como que el modelo no encajaba del todo bien, no se podían incluir iones de sodio, a pesar de que el ADN formaba una sal sódica, y estaba el problema de la repulsión de las cargas del fosfato; Pauling reconoció que su modelo estaba algo «apretado». Pero la prisa estaba justificada: sabía que, en Gran Bretaña, Maurice Wilkins y su equipo estaban obteniendo imágenes de difracción del ADN y que unos entonces desconocidos James Watson y Francis Crick estaban trabajando en un modelo de ADN.
Sin embargo, el modelo de Pauling resultó ser erróneo.
Comparación del modelo erróneo de Pauling (izquierda) y el DNA B canónico de Watson y Crick (derecha). La diferencia fundamental es que Pauling preparó un modelo en el que las bases se orientan hacia el exterior. Pauling pensaba que las bases formarían puentes de hidrógeno con otras moléculas, como proteínas. Sin embargo, aunque encajaba más o menos bien con los datos de Bell y Astbury, el modelo tenía problemas muy profundos, como ignorar las cargas de los fosfatos. Imagen: C. Menor-Salván
Pares de bases y la precaución de Wilkins
El modelo en triple hélice encajaba mas o menos bien con los datos de difracción de rayos X de los que disponía Pauling. Es más, el primer modelo de ADN que idearon Watson y Crick, en 1951, era una triple hélice similar a la de Pauling. La diferencia es que ellos contaron con Rosalind Franklin. Cuando mostraron a Franklin su modelo, ella les dio razones convincentes por las que el modelo no era válido. Franklin ya había observado la importancia del agua y de los iones en la estructura (ella estaba trabajando con la sal sódica del ADN) y de cómo debían distribuirse los fosfatos. Ellos la escucharon y volvieron a los cálculos de un nuevo modelo. En ese momento, Franklin y Wilkins, aunque no estaban a favor de determinar un modelo aún, también pensaban que se trataba de una triple hélice. Tendría que llegar la foto 51, obtenida por Franklin, a despejar las dudas: la molécula de ADN era una doble hélice.
Comparación de la foto de DRX de ADN favorita de Bell, la foto 19 (izquierda) con la histórica foto 51 de Franklin y su doctorando Gosling. Franklin realizó un descubrimiento clave: el ADN se presentaba en dos formas, la forma A y la forma B (actualmente sabemos que hay alguna más), mutuamente intercambiables por deshidratación y rehidratación y con diferente grado de coordinación con el agua. Aislar las formas permitió obtener imágenes de DRX claras, algo que Bell no logró, al obtener mezclas de las dos estructuras.
Las claves científicas del error de la triple hélice de Pauling las explica Rosalind Franklin en su magnífico artículo de 1953, en el que muestra que tenía clara la estructura: los datos de Pauling no eran lo suficientemente resolutivos; además, una hélice con los fosfatos en el interior y las bases en el exterior no encaja, ni con las propiedades del ADN, ni con la química del fosfato; y, no menos importante, Pauling no tuvo en cuenta un hallazgo previo fundamental al que Franklin si da crédito.
En 1947, un joven bioquímico, Michael Creeth, propuso un modelo de ADN formado por dos cadenas unidas por puentes de hidrógeno entre sus bases. Los mentores de Creeth eran Gulland y Jordan, quienes habían demostrado el apareamiento entre bases del ADN. Esta observación, para Franklin, tal como ella misma cuenta en su artículo de 1953, es fundamental. Considerando los pares de bases y los datos de difracción, la famosa doble hélice emergía como la estructura correcta.
Modelo propuesto por Michael Creeth para la estructura del ADN, tal como figura expuesto en el National Centre for Macromolecular Hydrodynamic, en Nottingham (Inglaterra). Actualmente, Bell y Creeth, así como sus mentores, son grandes olvidados en el descubrimiento del ADN.
Consciente de que los datos de Bell y Astbury no tenían resolución suficiente, Pauling escribió a Wilkins, solicitando ver los suyos. Wilkins, que no deseaba que el gran Pauling tomara el control de la investigación, se negó. Suele decirse que Pauling no tuvo oportunidad de reunirse con ellos directamente, debido a que el gobierno de EEUU le denegó la renovación del pasaporte para viajar a Gran Bretaña en 1951. Lo cierto es que el impacto de esta anécdota no fue grande, pues, finalmente, Pauling visitó Inglaterra durante un mes en 1952, coincidiendo con el trabajo clave de Franklin y, durante el cual, lamentablemente, no prestó atención al ADN.
Mientras Pauling, pensando que Wilkins seguiría negándose a colaborar, se centraba en sus trabajos con las proteínas, Franklin y su doctorando Gosling obtenían las imágenes históricas que confirmaron la estructura del ADN. Franklin no tenía inconvenientes para mostrar sus resultados y, si Pauling hubiera hablado con ella directamente, la historia del ADN sería distinta. Es más, ocurrió algo extraño: Franklin mostró sus imágenes a Corey, mano derecha de Pauling. Corey sugirió a Pauling que el modelo de triple hélice no encajaba bien y que había varios problemas. Pero él insistió, pensando que, bueno, ya resolverían los problemas pendientes. No visitar a Franklin y no escuchar a Corey fue un error histórico.
No fue el único error de Pauling. Watson y Crick tuvieron en cuenta otro dato clave: el bioquímico austriaco Erwin Chargaff les explicó que las bases del ADN siguen una proporción muy sencilla, que hoy conocemos como reglas de Chargaff. Estas apoyaban la idea de que las dos cadenas estarían unidas por las bases. Pauling y Chargaff se conocieron en 1947 durante un crucero de vuelta a EEUU y hablaron de ello; pero, considerándole un tipo «molesto y desagradable», Pauling despreció sus observaciones.
El modelo erróneo de Pauling fue el impulso definitivo que llevó a Watson y Crick a publicar, sólo dos meses después, su doble hélice. Cuando el artículo de Pauling apareció en febrero de 1953, los británicos estaban sorprendidos: era un modelo ingenuo, erróneo, similar al que ellos concibieron en 1951 y que Franklin les hizo desechar, y que, como los mismos Watson y Crick comentaron, incluso violaba las reglas químicas básicas del fosfato, expuestas por el propio Pauling en su famoso libro de texto de Química General. Eufóricos por la oportunidad que el error de Pauling les brindaba, era el momento perfecto para arrebatarle la gloria de resolver la estructura del ADN; se apresuraron, entonces, a publicar su modelo antes de que Pauling tuviera tiempo de darse cuenta de su fallo y rehacer su modelo, a la vista de los nuevos datos de difracción de rayos X.
Página del famoso artículo de Watson y Crick, en la que figura su modelo de doble hélice y agradecen específicamente a Wilkins y Franklin por sus contribuciones. Watson y Crick eran teóricos y dependían de los datos experimentales. La foto que se ve en la parte inferior no es de Franklin, sino de Wilkins.
Así, en abril de 1953, tras un acuerdo con Wilkins y Franklin, se publicaron tres artículos: uno por Watson y Crick, otro por Wilkins y el tercero por Franklin y Gosling, detallando la estructura del ADN y los datos esenciales que la sostienen. Sorprende que mucha gente piense que aquel mes se publicó un sólo artículo, firmado por Watson, Crick y Wilkins, y que Franklin fue ignorada completamente. Como ocurre con El Quijote, que todo el mundo lo conoce, pero casi nadie lo ha leído. Sin embargo, la lectura de los artículos aclara cosas, como que, en efecto, no fue un artículo, sino tres, cada uno preparado por los líderes del descubrimiento: Watson y Crick eran los teóricos que desarrollaron el modelo, y Wilkins y Franklin los experimentalistas que lograron preparar el ADN puro y obtener los datos de su estructura. A priori no parece mal arreglo. Con ojos actuales, cada uno de ellos tendría su paper histórico en Nature como IP (Los problemas de índole personal que llevaron a que Wilkins y Franklin publicaran por separado, o por qué publicaron tres papers separados en lugar de uno todos juntos, no son objeto de esta entrada, y es un tema que se discute mucho, aunque para mi forma parte del gossip científico, no de la ciencia en sí)
Aceptando la derrota
Pauling aceptó sus errores con elegancia y, en la Conferencia Solvay de ese mismo abril de 1953, expresó su apoyo al modelo de Watson y Crick:
«Aunque solo han pasado dos meses desde que el profesor Corey y yo publicamos nuestra estructura propuesta para el ácido nucleico, debemos admitir que probablemente esté equivocada; Aunque se podría hacer algo de refinamiento, creo que es muy probable que la estructura de Watson-Crick sea esencialmente correcta»
En 1988, durante una conversación informal en un congreso, Pauling recapitulaba:
«Supongo que siempre pensé que la estructura del ADN era mía para resolver y, por lo tanto, no la perseguí con suficiente agresividad».
Definitivamente, no escuchar las señales de que su modelo no era correcto y su ambición, que le llevó a estar convencido de que era el único que podría resolverla, no le ayudaron.
Anexo 1: Cómo ayudó la foto 51 a revelar la estructura
La técnica de difracción de rayos X (DRX) es y ha sido esencial para la Biología. Es una técnica compleja, costosa y que requiere de una larga formación específica, por lo que los cristalógrafos, como lo fueron Rosalind Franklin, Florence Bell o el propio Pauling, expertos en DRX, son muy valiosos para la investigación y su labor no siempre es suficientemente reconocida. Prácticamente todas las estructuras que podemos encontrar en el Protein Data Bank, por ejemplo, han sido resueltas mediante DRX.
Para obtener un buen patrón de difracción, o difractograma, es necesario disponer de una muestra muy pura del compuesto que se está analizando, uniforme y cristalina (es decir, todas las moléculas de la muestra están ordenadas). Esto fue muy difícil de conseguir con el ADN, en especial por su tendencia a la transición entre ADN A y ADN B según su contenido en agua.
El DNA no tiene una única estructura. Son posibles múltiples conformaciones.
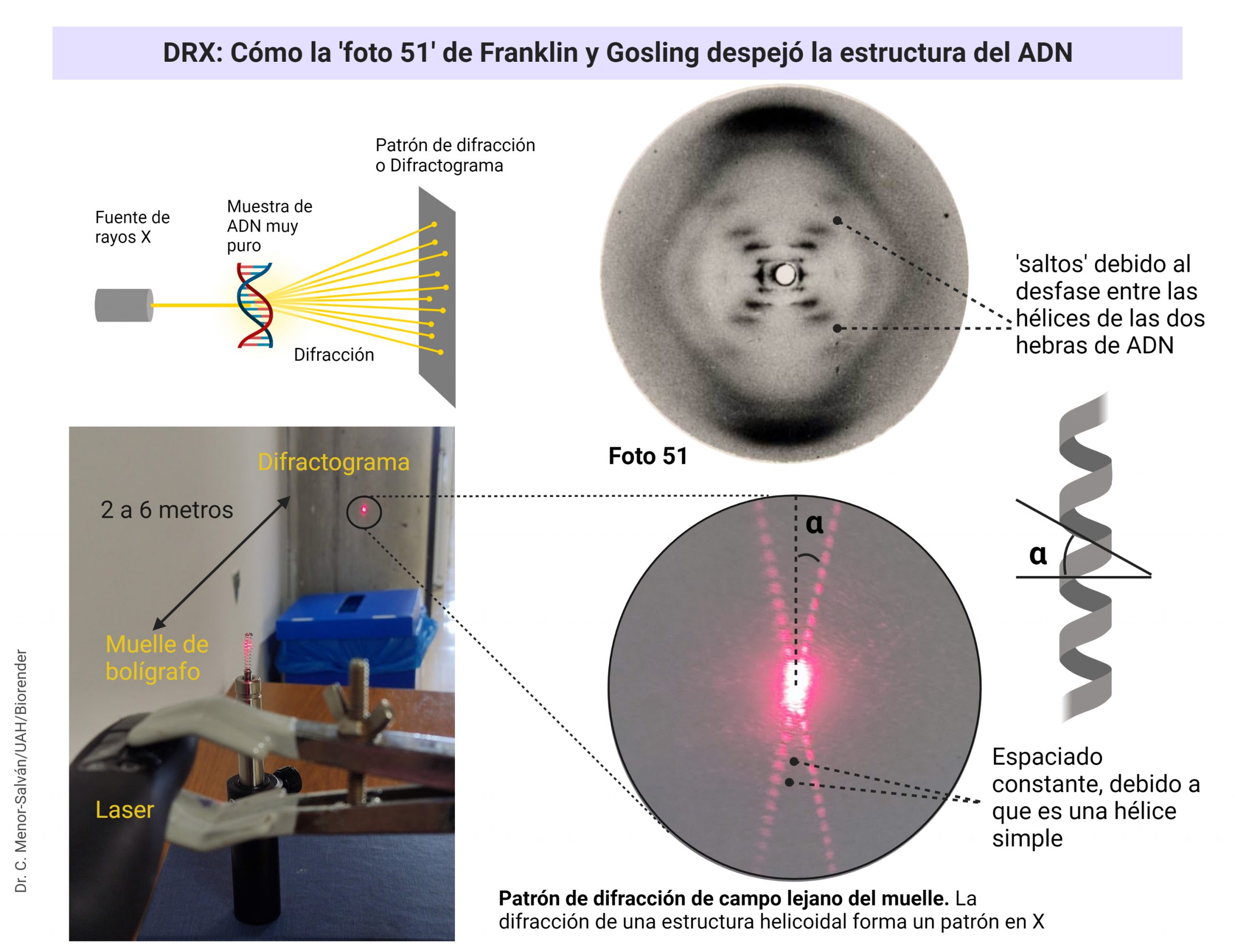
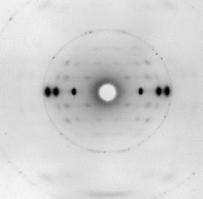
Rosalind Franklin y Raymond Gosling (a quien nadie recuerda, a pesar de haber trabajado en el laboratorio codo con codo con Franklin) consiguieron una imagen excepcional, como hemos visto: la famosa foto 51. Esta foto se reproduce en muchas ocasiones, pero ¿qué significa? ¿cómo se interpreta?
Usando un simple muelle y un puntero laser para entender el patrón de difracción de una hélice
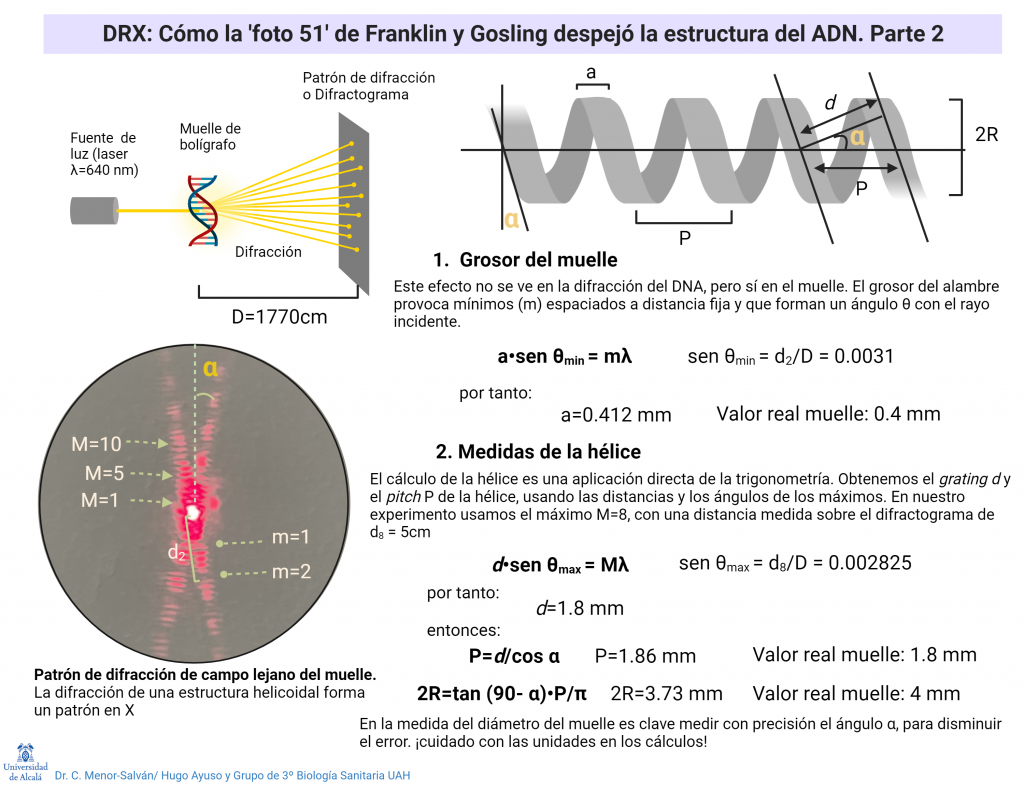
La interpretación completa es muy compleja, pero podemos entenderla de modo sencillo, en especial si usamos un experimento: si tomamos un muelle de boli o cualquier otro muelle pequeño y fino y lo iluminamos con un láser, que tenga el haz cuanto más grueso mejor (de modo que ilumine varias vueltas de hélice al mismo tiempo), el muelle va a difractar la luz laser. Como la diferencia entre la distancia de vueltas del muelle y la longitud de onda del laser es muy alta, el patrón de difracción que se produce es la difracción de campo lejano o difracción de Fraunhofer, que podemos ver al proyectar la luz a una distancia de entre 2 y 6 metros del muelle. El patrón de difracción nos permite medir con mucha precisión las medidas del muelle. Si cambiamos el muelle por moléculas ordenadas de un compuesto, y el láser por un haz fino de rayos X, muy penetrante y de longitud de onda muy pequeña, comparable a las dimensiones de la molécula, obtendremos un patrón de difracción de su estructura.
El patrón de difracción de una estructura en hélice da lugar a una «X» muy característica. El ángulo de la «X» y la distancia entre las manchas luminosas nos permite calcular las medidas de la hélice (ver mas abajo). Con el sencillo experimento del muelle y el puntero láser, podemos reproducir el experimento y los cálculos que realizaron Franklin y Gosling.
Si nos fijamos en las imágenes de Florence Bell, este patrón no está nada claro, debido a las dificultades que tuvo para preparar la muestra. Pauling supo que la molécula de ADN era helicoidal porque vio una imagen de microscopía electrónica.
Otro aspecto clave de la foto 51 es que revelaba claramente que el ADN no estaba formado por una hélice, sino por dos hélices unidas, con un desfase entre ellas. Este desfase daba lugar a los huecos que se pueden ver en la foto. Este dato confirmaba definitivamente que el ADN estaba formado por dos hebras unidas formando una hélice doble. Franklin pudo medir la molécula usando el patrón, obteniendo un ajuste muy bueno con el modelo teórico propuesto por Watson y Crick.
¿Qué habría ocurrido si el modelo de Creeth hubiera sido correcto? El patrón de difracción habría sido completamente distinto. Podemos modelizarlo también mediante la difracción de Fraunhofer de dos alambres paralelos muy juntos:
Esto nos da un patrón de difracción o difractograma lineal:
Como en el caso anterior, con el espaciado entre las manchas luminosas podemos medir la distancia entre los dos alambres. Comparemos este difractograma con el difractograma de rayos X real de un poliester, un polímero lineal formado por cadenas paralelas:
Y aquí vemos el patrón de difracción de un fragmento proteico formado por láminas beta paralelas:
Así queda demostrada la estructura de la molécula usando un método físico clave, la DRX. La verdad es que las cosas son como tienen que ser: si el DNA tuviera la estructura de Creeth, la Biología Molecular sería imposible, y por tanto la vida. Pero eso todavía no lo sabían en los años 1940-1950.
Anexo 2: Extracto de los artículos históricos de abril de 1953
Watson y Crick mencionan el modelo erróneo de PaulingÚltimo párrafo del artículo de Rosalind Franklin de abril de 1953, relatando sus observaciones y concluyendo que «la idea general no es incosistente con el modelo propuesto por Watson y Crick». Como experimentalista, apoya con sus datos el modelo teórico propuesto. Franklin, además, cita el trabajo clave de Gulland y sus colaboradores, descubridores del apareamiento de bases en el ADN. El modelo de Pauling fue el ‘punching ball‘ de los tres artículos. Aquí, Franklin discute por qué el modelo de Pauling es erróneo. El modelo de Pauling, a pesar de ello, fue un catalizador de la publicación de estos tres trabajos históricos. El resto del artículo de Franklin es un relato detallado de los datos e interpretación de los análisis por difracción de rayos X. Primer párrafo del trabajo de Franklin. Aquí relata algo esencial que, a veces, se pasa por alto: el descubrimiento de las formas B y A del ADN. Franklin reconoce aquí a otro científico realmente olvidado por la Historia del descubrimiento del ADN: el noruego Sven Furberg, quien, en 1948, estuvo también a punto de descubrir la estructura del ADN y propuso, por primera vez, que debía tener una estructura helicoidal. Pero Furberg tropezó en la misma piedra que Bell y Astbury…
Referencias
ASTBURY, W. T. & BELL, F. O. (1938) ‘X-Ray Study of Thymonucleic Acid’, Nature, 141(3573), pp. 747–748. doi: 10.1038/141747b0.
Dahm, R. (2008) ‘Discovering DNA: Friedrich Miescher and the early years of nucleic acid research’, Human Genetics, 122(6), pp. 565–581. doi: 10.1007/s00439-007-0433-0.
FRANKLIN, R. E. & GOSLING, R. G. (1953) ‘Molecular Configuration in Sodium Thymonucleate’, Nature, 171(4356), pp. 740–741. doi: 10.1038/171740a0.
Gann, A. & Witkowski, J. A. (2013) ‘DNA: Archives reveal Nobel nominations’, Nature, 496(7446), p. 434. doi: 10.1038/496434a.
Harding, S. E., Channell, G. & Phillips-Jones, M. K. (2018) ‘The discovery of hydrogen bonds in DNA and a re-evaluation of the 1948 Creeth two-chain model for its structure’, Biochemical Society Transactions, 46(5), pp. 1171–1182. doi: 10.1042/BST20180158.
Lake, J. (2001) ‘Why Pauling didn’t solve the structure of DNA’, Nature, 409(6820), pp. 558–558. doi: 10.1038/35054717.
Levene, P. a, Hydrolysis, A. & Rockejeller, T. (1917) ‘Yeast Nucleic’, J. Biol. Chem., 40, pp. 415–424.
Pauling, L. & Corey, R. B. (1953) ‘A Proposed Structure For The Nucleic Acids’, Proceedings of the National Academy of Sciences, 39(2), pp. 84–97. doi: 10.1073/pnas.39.2.84.
Pederson, T. (2020) ‘The double helix: “Photo 51” revisited’, The FASEB Journal, 34(2), pp. 1923–1927. doi: 10.1096/fj.202000119.
Ronwin, E. (1953) ‘The Phospho-Di-Anhydride Formula and Its Relation to the General Structure of the Nucleic Acids’, Science, 118(3071), pp. 560–561. doi: 10.1126/science.118.3071.560.
Schmidt, G. (1950) ‘NUCLEIC ACIDS, PURINES, AND PYRIMIDINES’, Annu. Rev. Biochem, 19, pp. 149–186.
Thompson, J. et al. (2018) ‘Rosalind Franklin’s X-ray photo of DNA as an undergraduate optical diffraction experiment’, American Journal of Physics, 86(2), pp. 95–104. doi: 10.1119/1.5020051.
WATSON, J. D. & CRICK, F. H. C. (1953) ‘Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid’, Nature, 171(4356), pp. 737–738. doi: 10.1038/171737a0.
Polihidroxibutirato Sintasa (PhaC), ¿el nuevo salvavidas ecológico?
escrito por celiycintia_1C | 17 diciembre, 2024
Realizado por: Cintia Merino Piñón y Celia Juara Díaz (1ºC Biología Sanitaria – UAH)
Ya por 1862, de la mano de Estados Unidos, encontramos los primeros tipos de plástico que se empezaron a emplear. Estos consisten básicamente en polímeros formados a partir de largas cadenas moleculares obtenidas del petróleo. Desde sus inicios, todo apuntaba al éxito que tendrían, y al cambio radical que significaría para el humano y la producción mundial, a causa de sus múltiples usos y rentabilidad económica.
Pese a todo, con el tiempo hemos podido observar que el uso excesivo y los problemas que acarrea su mala degradación, nos han llevado a consecuencias tales como: la liberación masiva de CO2, la acumulación de miles de toneladas de plástico en muchos puntos del planeta, etc. Desgraciadamente, todo apunta a que la situación se agravará con los años si seguimos así, al no disponer de una opción más sostenible.
Es en este punto, donde entra en juego la ciencia, presentando ante el mundo a las “bacterias capaces de sintetizar plástico biodegradable”, Cupriavidus necator, Pseudomonas putida, Bacillus bataviensis, Allochromatium vinosum… las posibles soluciones.
¿QUÉ ES LA POLIHIDROXIBUTIRATO SINTASA (PhaC)?
En el transcurso de la investigación, iniciada en la década de los 80, en busca de una alternativa sostenible al plástico tradicional, centraron su atención en los “polihidroxialcanoatos (PHA)”. Estos son biopoliésteres sintetizados de forma intracelular por algunos microorganismos, utilizados como reserva de carbono y energía, al igual que ocurre con el glucógeno en animales y hongos.
Cupriavidus necator, también conocida como “Ralstonia eutropha” (perteneciente a la familia de las Burkholdariaceae), es una bacteria muy versátil, localizada en suelos y aguas, con capacidad para convertirse en una solución muy efectiva al problema del plástico. Se trata de una bacteria flagelada de forma alargada, gram negativa, con libertad de movimiento. Dependiendo de las condiciones ambientales, presentaría un metabolismo heterótrofo, o autolitotrofo.
Fig.1: Bacterias de la especie Cupriavidus necator y sus visibles gránulos de plástico en el interior de la bacteria. Imagen obtenida de: https://www.researchgate.net/publication/231182767_Whey_Lactose_as_a_Raw_Material_for_Microbial_Production_of_Biodegradable_Polyesters DOI:10.5772/48737Fig.2: Cupriavidus necator con polihidroxibutirato (PHB) contenido en gránulos en su interior. Imagen obtenida de: https://www.cell.com/trends/biotechnology/fulltext/S0167-7799%2821%2900007-X DOI:https://doi.org/10.1016/j.tibtech.2021.01.001
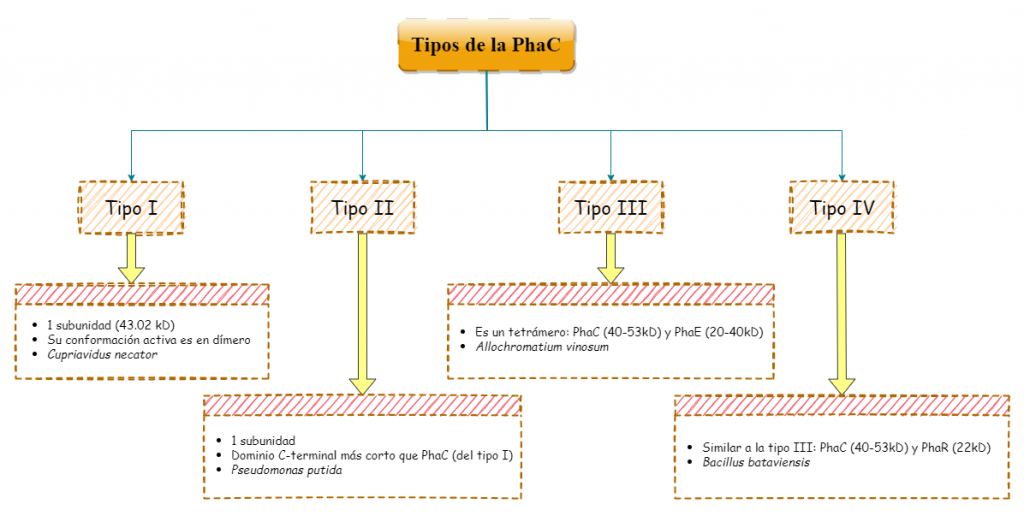
La síntesis de polihidroxibutirato (PHB), no sería posible sin la polihidroxibutirato sintasa (Pha C), responsable de la catálisis del proceso. Este grupo de enzimas presentes en muchos microorganismos sintetizadores de bioplásticos, se divide en 4 tipos distintos que difieren en su especificidad y la composición de sus subunidades (aunque todas ellas comparten el mismo mecanismo de acción y un centro activo similar al de las lipasas).
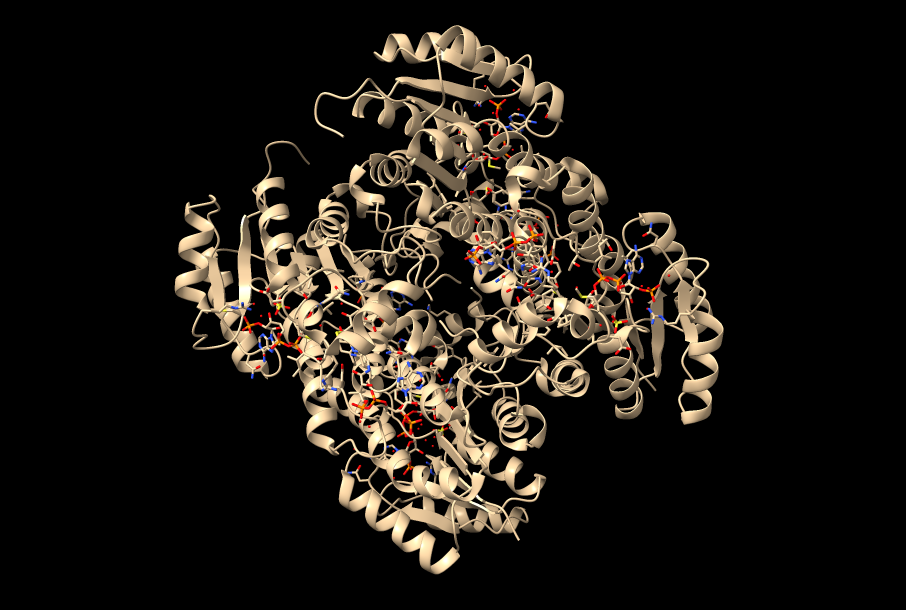
Fig.3: La enzima polihidroxibutirato sintasa (PhaC), responsable de la síntesis del plástico. Imagen creada con ChimeraX a partir de PDB 5t6o.
Tipos I y III
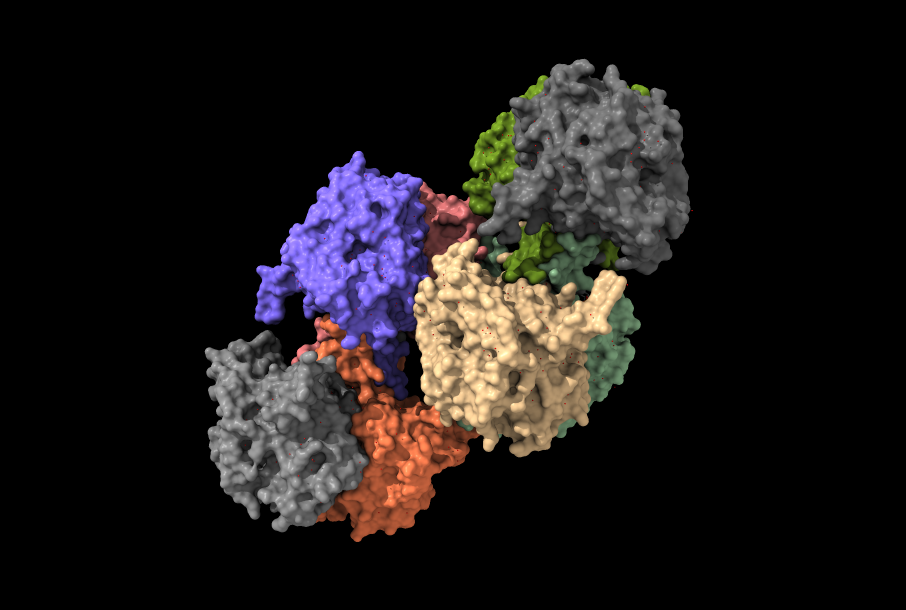
Los tipos I y III, los más estudiados hasta el momento, comparten esa especificidad por el HB como sustrato a la hora de sintetizar PHAs. El tipo I (Cupriavidus necator), se constituye únicamente de una cadena de polipéptidos de 589 residuos en total (una única subunidad, PhaC). Posee un dominio N-terminal (191 residuos) con funciones desconocidas por el momento, y un dominio C-terminal (398 residuos) encargado de la catálisis. Dicho dominio catalítico, se compone de una zona central de láminas-β, rodeada a ambos lados por α-hélices (aspecto que recuerda al centro activo de las lipasas, de ahí su parecido con esta enzima). No obstante, su conformación activa es un dímero, con sus dominios catalíticos enfrentados, que presenta una mayor actividad catalítica. El responsable de la dimerización, es un dominio helicoidal que se aleja del núcleo de la proteína, del que parte un bucle desordenado de 66 residuos que se integra a otra cadena polipeptídica, formando el dímero.
Por otra parte, el tipo III estaría compuesto de dos subunidades distintas formando un tetrámero: la PhaC (40-53 kD), responsable de la catálisis; y la PhaE (alrededor de 20-40 kD). Ejemplos de microorganismos que contienen este tipo de enzima son: Chromatium vinosumy Allochromatium vinosum.
Tipos II y IV
El tipo IV comparte una gran similitud con el tipo III: ambos son un dímero con dos subunidades distintas, aunque en lugar de PhaE como subunidad, este tipo tiene la PhaR (22kD). Esta variedad de polihidroxibutirato sintasa es propia de bacterias como: Bacillus cereuso B. bataviensis.
El tipo II está presente en muchos tipos de bacterias en la naturaleza, tales comoPseudomonas putida. Sería muy parecido al primer tipo, salvo que su dominio C-terminal presenta una secuencia más corta, y el sustrato sobre el que actúa cambia, existiendo variantes del tipo II con gran afinidad por el hidroxihexanoato (P. oleovorans).
Cabe destacar que a pesar de todas estas diferencias, todas las PhaC sintasas son enormemente similares entre ellas, presentando secuencias homólogas a las del primer tipo, en sus dominios catalíticos.
Fig.4: Diagrama a modo de resumen de los distintos tipos de PhaC y sus características.
Otras enzimas
Por otro lado, la PhaC no es la única enzima que participa en la formación del bioplástico. Existen otras enzimas que desempeñan sus funciones durante la biosíntesis para formar los monómeros que componen el polímero.
Centrándonos en la Cupriavidus necator, la beta ketotiolasa (PhaA), es la enzima que inicia todo el proceso, al ser la encargada de comenzar la síntesis del hidroxibutirato. Esta transferasa, consiste en un tetrámero, de subunidades dimerizadas.
Finalmente, la síntesis de los monómeros termina tras la intervención de la acetoacetil CoA reductasa (PhaB), una oxidorreductasa que emplea NADH para reducir el acetoacetil CoA, obteniéndose el hidroxibutirato. Desgraciadamente, con respecto a su estructura tridimensional, a día de hoy sigue sin disponerse de suficiente información de los dominios que participan en la actividad de la enzimática. Aunque se cree que su estructura primaría es muy similar a una enzima que participa en la síntesis de ácidos grasos, la FabG.
Fig.6: Enzima PhaB (acetoacetil-CoA reductasa). Imagen creada con ChimeraX a partir de PDB 3vzs.Fig.5: Enzima Pha A (beta-ketiolasa). Imagen creada con ChimeraX a partir de PDB 4o9c.
ESTRUCTURA Y FUNCIONAMIENTO
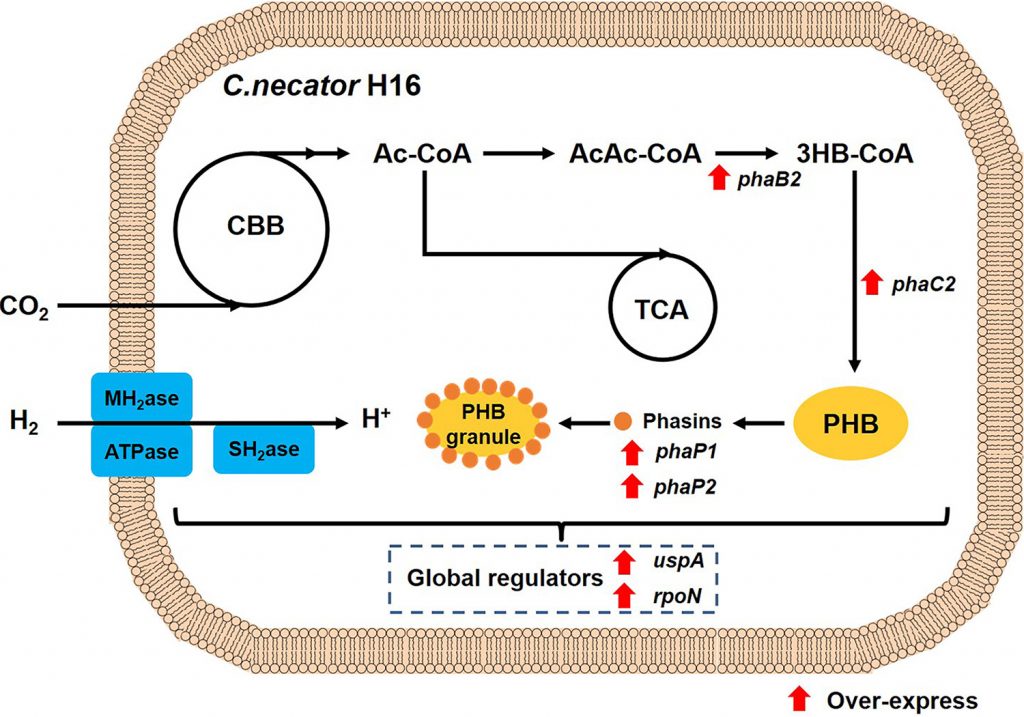
El polihidroxibutirato, sintetizado por la bacteriaCupriavidus necator, es un polihidroxialcanoato de cadena corta, formado por la adición sucesiva de “n” unidades de hidroxibutirato. La biosíntesis comienza con el acetil-CoA, que puede provenir del metabolismo de carbohidratos, ácidos grasos e incluso proteínas, aunque la ruta más estudiada es la que comienza con los carbohidratos. La molécula de acetil-CoA es transferida, gracias a la enzima PhaA, a otro acetil-CoA, formándose acetoacetil-CoA.
Fig.7: Proceso metabólico del PHB por la bacteria Cupriavidus necator. Imagen obtenida de: https://journals.asm.org/doi/10.1128/AEM.01458-21 DOI: https://doi.org/10.1128/AEM.01458-21Fig.8: Proceso metabólico para la síntesis de PHB y enzimas que lo llevan a cabo. Imagen obtenida de:https://pubs.acs.org/doi/10.1021/acs.chemrev.6b00804 DOI:https://doi.org/10.1021/acs.chemrev.6b00804
En el siguiente paso consecutivo, la enzima PhaB reduce la molécula a hidroxibutirato, empleando para ello NADH como cofactor. Una vez obtenido el HB, comienza la formación del PHB, catalizada por la enzima PhaC.
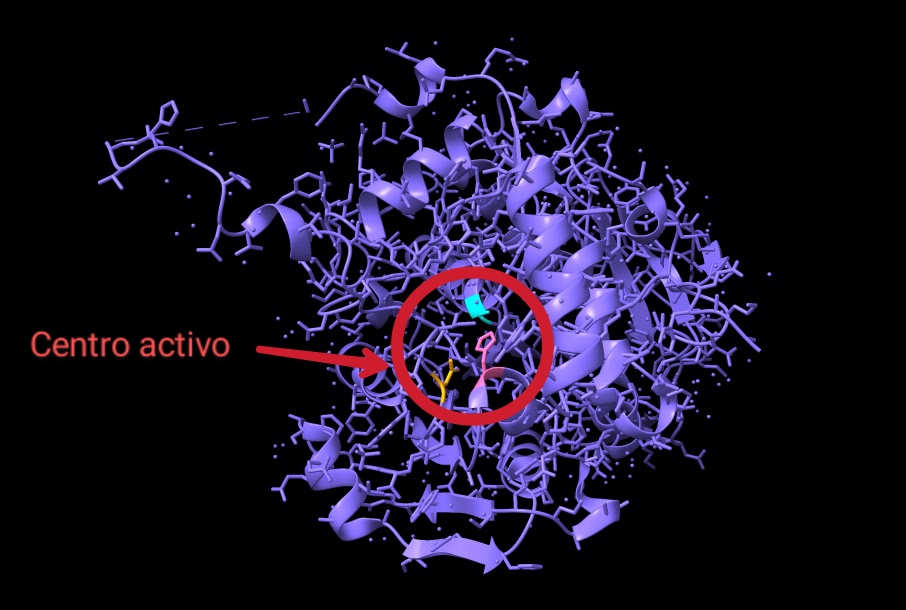
Enfocándonos, en el mecanismo de acción y la estructura de su dominio catalítico, diversas investigaciones bioquímicas llevaron a cabo la cristalización de esta enzima con el fin de estudiarlos. Con respecto al experimento, la estructura cristalizada presentaba una mutación: la cisteína del centro activo fue sustituida por alanina (Fig.10), otorgándole mayor estabilidad. En cuanto a la actividad enzimática de PhaC, se teorizaba que su funcionamiento era similar al de una lipasa, sintetizando el PHB de forma similar a los ácidos grasos. Ahora bien, actualmente el mecanismo de acción más aceptado consiste en un único sitio activo, porque la distancia que existe entre los dos centros activos del dímero (unos 33 Å) haría imposible el intercambio del bioplástico en proceso de ser sintetizado entre ambos.
Fig.9: Centro activo de PhaC (Cupriavidus necator) visto desde fuera. Imagen creada con ChimeraX a partir de PDB 5t6o.
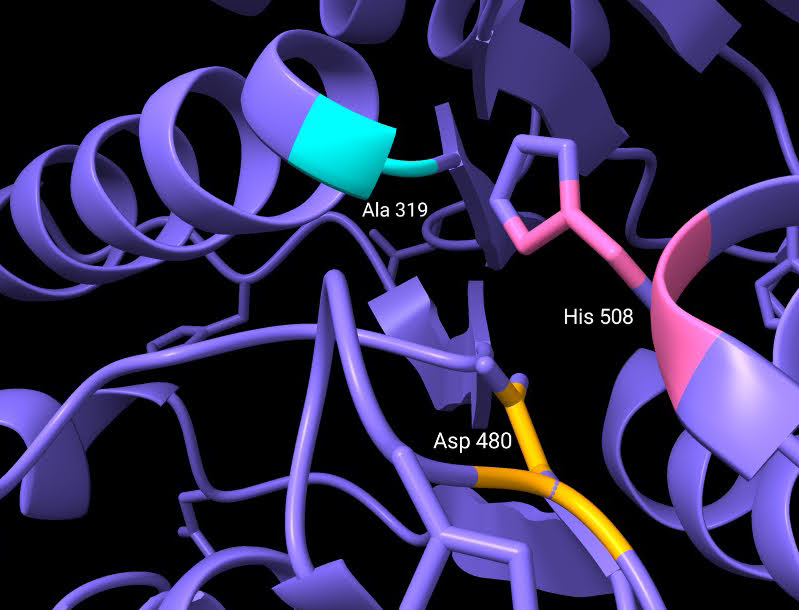
Los residuos que forman parte del centro activo de esta curiosa enzima son: Cys319, His508 y Asp480 (Fig.9). Estos se encuentran alojados en una cavidad situada a unos 10Å de la superficie proteica. La alanina del centro activo está localizada en lo que se conoce como “codo nucleófilo”, es decir, se halla entre una lámina beta y una alfa hélice. A su alrededor observamos un bucle, donde se ubica la histidina, separada por 3,5 Å de la alanina (cisteína en la enzima sin mutación) en la estructura cristalizada estudiada. Por último, el aspartato del centro activo se localiza justo detrás de la histidina, en otro bucle.
Fig.10: Centro activo de la enzima cristalizada, con Cys sustituida por Ala. Imagen creada con ChimeraX a partir de PDB 5t6o.
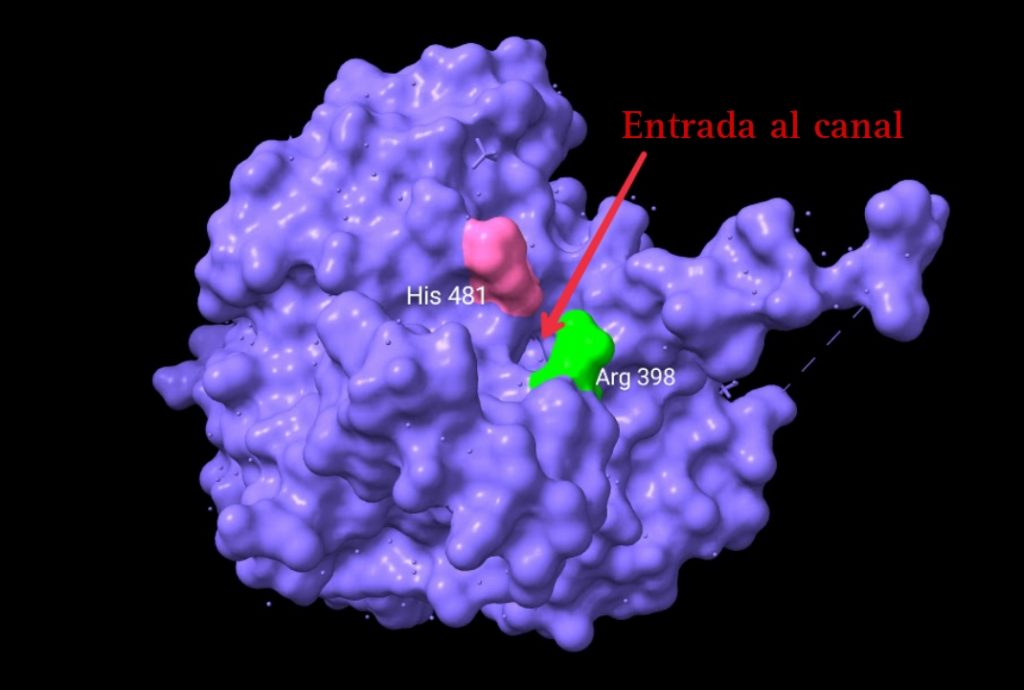
La entrada de los monómeros al centro activo de la enzima pudo observarse claramente gracias a un análisis esta estructura con un software, CAVER . Esta, consiste en un canal que accede directamente a una cavidad llena de agua en el interior de la proteína (contigua al centro activo) desde la superficie. Con unos 18 Å de longitud, este canal permite el acceso de la mitad de la molécula de HB al centro activo. Dos residuos de arginina, uno de cada monómero, están muy próximos a la apertura del canal, y su unión con el grupo CoA (del HB), es la causa del aumento de la actividad enzimática del dímero.
Fig.11: Ubicación de la entrada del canal propuesto.Imagen creada con ChimeraX a partir de PDB 5t6o.
La formación del polímero comienza con la histidina (His 508) del centro activo, que se encargaría de desprotonar el tiol de la cisteína (Cys 319), mutada en alanina, gracias a la escasa distancia que las separa. De esta forma, se puede producir un enlace covalente entre la cisteína desprotonada y el primer monómero de HB de la cadena, creando una unión estable entre residuo y monómero. Es entonces cuando entra en acción el residuo de aspartato (Asp 480) que, debido al puente de hidrógeno con la His 508, participa de forma indirecta en la desprotonación de los monómeros de la cadena que se van añadiendo. La adición de un segundo monómero al “plástico” naciente requiere la correcta orientación del HB (unido covalentemente a la Cys 319), que se produce gracias a la naturaleza del propio enlace que los une, un enlace plano. En ese momento, a una distancia estimada de 2.8Å, se produce el ataque nucleofílico por parte de HB-Cys 319 a la nueva unidad en adición. Tras la incorporación de sucesivas unidades de HB obtendremos nuestro polímero de forma helicoidal levógira. Este será acumulado en forma de gránulos de tamaño considerable, 80 veces más pesados que el propio peso en seco del microorganismo.
En el modelo cristalizado y empleando de nuevo la herramienta CAVER se teorizó que la salida de esta cadena naciente tendría lugar por un canal compuesto por residuos hidrofóbicos, que se alejan en dirección a la superficie de la PhaC, hacia una cavidad aledaña al residuo N-terminal de la proteína y un residuo de aspartato, que participaría en la terminación de la cadena de PHA.
Para permitir el paso de este polímero por el canal de salida se cree que hay unos dominios conservados, dispuestos en las cercanías del canal, llevando a cabo una redistribución de esos aminoácidos hidrofóbicos que lo conforman, ensanchándolo hasta lograr el tamaño adecuado para el polímero.
Con objeto de emplear el carbono almacenado, la bacteria usa una enzima capaz de hidrolizar el PHB, la polihidroxibutirato polimerasa. Esta hidrolasa estudiada a través de una estructura cristalina purificada obtenida de P. funiculosum, nos muestra que se compone por un único dominio, con un plegamiento alfa/beta formado por 8 láminas beta: 7 de ellas paralelas y la restante antiparalela, todas ellas rodeadas por alfa hélices. Su dominio catalítico lo forman Ser39, Asp121, y His155, residuos que se encuentran preservados en su dominio catalítico.
Cabe destacar que existen depolimerasas que poseen también la capacidad de degradar plástico extracelular, que no haya sintetizado la bacteria. En este caso la enzima presenta tres dominios esenciales: catalítico, enlazante y de unión al sustrato. Esta se encarga de romper en pequeñas unidades el bioplástico, gracias a la serina de su centro activo, de forma similar a como actúa la tripsina. En el caso de Cupriavidus necator, los monómeros resultantes de la hidrólisis son moléculas de HB, que la bacteria oxida a acetoacetato por medio de una deshidrogenasa, y posteriormente convierte en acetil-CoA (del que obtendrá energía incorporándolo a distintas vías metabólicas).
Fig.12: polihidroxibutirato depolimerasa presente en Cupriavidus necator. Imagen creada con ChimeraX a partir de PDB 2d81.
POLIHIDROXIALCANOATOS Y SU PRODUCCIÓN INDUSTRIAL
Dado que tenemos un amplio abanico de bacterias capaces de producir PHB, las técnicas y sistemas de cultivo son igualmente variados. Todas ellas pueden dividirse en dos grupos, dependiendo de las condiciones que necesitan para producir los PHAs. El primer grupo, donde encontramos a Cupriavidus necator, lo forman microorganismos que necesitan que precisan escasez de nutrientes y alta disponibilidad de carbono. Con respecto al segundo grupo, este no necesita de una situación de escasez, ya que son capaces de acumularlo en grandes cantidades durante su etapa de crecimiento.
GRUPO 1: Limitación de nutrientes
Para realizar un cultivo con las bacterias del primer grupo necesitamos un proceso dividido en 2 etapas: la primera de ellas tiene como objetivo obtener labiomasa necesaria para la producción, por lo que no existe limitación de nutrientes; la segunda etapa comienza una vez alcanzada la biomasa requerida, alguno de los nutrientes esenciales está limitado, promoviendo que las bacterias sinteticen el polímero (lo acumulan en gran cantidad formando esos gránulos tan llamativos).
GRUPO 2: Sin limitación de nutrientes
En el segundo grupo la estrategia que se sigue para obtener el bioplástico consiste en una única etapa, sin limitación de nutrientes. Los microorganismos disponen de ellos en grandes cantidades en el medio de cultivo, lo que les permite mejorar su rendimiento y aumentar de tamaño.
Obtención, proceso industrial
Una de las primeras empresas en tratar de producir el bioplástico en masa fue ICI, una empresa inglesa. Para el proceso de producción finalmente se decidieron por Cupriavidus necator, que lo sintetiza en dos etapas. El cultivo tiene a su disposición glucosa y fosfato limitado, suficiente para alcanzar una biomasa considerable y pasar a la segunda etapa, en la que las bacterias comienzan a almacenar el bioplástico. A la hora de recuperar el polímero empleamos metanol caliente para retirar los restos celulares (lípidos, fosfolípidos, etc.), y se extrae el PHB con cloroformo o cloruro de metileno. Por último, se filtra la mezcla, se enfría y precipita, luego es secada al vacío.
Tras diversas pruebas, finalmente se determinó que para obtener 1g de PHB con la cepa de la bacteria empleada, eran necesarios 3g de glucosa para permitir su desarrollo y proporcionarle la fuente de carbono para formar el plástico.
Una propiedad muy importante, a parte de su relativamente sencillo proceso de obtención, es el hecho de que es un plástico completamente biodegradable. A pesar de la gran cantidad de monómeros posibles capaces de conformar los polihidroxialcanoatos, existen diversas bacterias, e incluso hongos, capaces de degradarlos bajo cualquier condición y sin generar ningún tipo de residuo tras el proceso. Mediante las reacciones de hidrólisis citadas anteriormente en las que participan las PHB depolimerasas de esos microorganismos, se produce la ruptura del bioplástico (que se emplea como fuente de energía dentro de la bacteria descomponedora). Este proceso tiene lugar a una velocidad considerable, que depende de diversos factores (pH, temperatura, dimensión del polímero, etc.).
¿NOS AGUARDA UN FUTURO CON ELLOS?
A día de hoy, tener a nuestra disposición este polímero totalmente biodegradable y que no genera residuos con su producción, nos permitiría llevar un estilo de vida mucho menos dañino para el planeta, puesto que todos aquellos problemas que encontramos con la producción y degradación de los plásticos tradicionales desaparecerían.
Presentan muchas aplicaciones y gracias a sus diversas propiedades pueden ser utilizados en campos muy diversos, como en: farmacia, industria, agricultura, medicina o incluso como materias primas. Es por ello, que tratamos de producirlo en masa y de la forma más económica posible, intentando buscar microorganismos capaces de producirlos, empleando para ello residuos de las propias industrias en algunos casos, como sustrato en el proceso de síntesis. “Biocycle”, una empresa brasileña, es una de las que empleó los residuos de su propia producción como sustrato, en el año 2001.
Actualmente los científicos investigan a fondo la estructura molecular de la PhaC, enzima sin la cual este proceso no tendría lugar, con el fin de conseguir modificar molecularmente el bioplástico para otorgarle funciones mucho más específicas y amplias. Y aunque queda un largo camino por recorrer en cuanto a la investigación sobre estos aspectos, los PHAs nos acercarían un paso más hacia una sociedad sostenible, en la que los residuos generados sean mínimos y lo menos contaminantes posibles. Desgraciadamente, por el momento, se sigue sin poder hacer competencia al plástico tradicional, sobre todo en el ámbito económico.
Fig.13: Diferentes posibilidades que nos ofrece el empleo de bacterias para la síntesis de PHA. Imagen obtenida de: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7137286/ DOI: 10.1186/s12934-020-01342-z
BIBLIOGRAFÍA
1- Chek, M.F., Hiroe, A., Hakoshima, T. et al (2019). PHA synthase (PhaC): interpreting the functions of bioplastic-producing enzyme from a structural perspective. Appl Microbiol Biotechnol 103, 1131–1141. DOI: https://doi.org/10.1007/s00253-018-9538-8.
5- Mezzolla, V.; D’Urso, O.F.; Poltronieri, P (2018).Role of PhaC Type I and Type II Enzymes during PHA Biosynthesis. Polymers, 10, 910. DOI: https://doi.org/10.3390/polym10080910.
6- Tamao Hisano, Ken-ichi Kasuya, Yoko Tezuka, Nariaki Ishii, Teruyuki Kobayashi, Mari Shiraki, Emin Oroudjev, Helen Hansma, Tadahisa Iwata, Yoshiharu Doi, Terumi Saito, Kunio Miki, (2006). The Crystal Structure of Polyhydroxybutyrate Depolymerase from Penicillium funiculosum Provides Insights into the Recognition and Degradation of Biopolyesters, Journal of Molecular Biology, Volume 356, Issue 4, Pages 993-1004, ISSN 0022-2836, DOI: https://doi.org/10.1016/j.jmb.2005.12.028. (https://www.sciencedirect.com/science/article/pii/S0022283605015901)
7- The Protein Data Bank H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne (2000) Nucleic Acids Research, 28: 235-242. DOI: 10.1093/nar/28.1.235
8- Wittenborn, E.C; Jost, M; Wei, Y; Stubbe, J; Drennan, C.L. (2016). Structure of yhe catalityc Domain of the Class I Polyhydroxybutyrate Synthase from Cupriavidus necator*, J Biol Chem, 291: 25264-25277 . DOI: https://doi.org/10.1074/jbc.M116.756833.
9- Yu Jung Sohn, Jina Son, Seo Young Jo, Se Young Park, Jee In Yoo, Kei-Anne Baritugo, Jeong Geol Na, Jong-il Choi, Hee Taek Kim, Jeong Chan Joo, Si Jae Park, (2021). Chemoautotroph Cupriavidus necator as a potential game-changer for global warming and plastic waste problem: A review, Bioresource Technology, Volume 340, 125693, ISSN 0960-8524. DOI: https://doi.org/10.1016/j.biortech.2021.125693. (https://www.sciencedirect.com/science/article/pii/S0960852421010348)
HEXOKINASA: ESTRUCTURA, EVOLUCIÓN Y PAPEL EN EL CÁNCER
escrito por cmfc5_1A | 17 diciembre, 2024
Redactado por María Arrondo Sánchez y Carolina Amil Zamorano
INTRODUCCIÓN
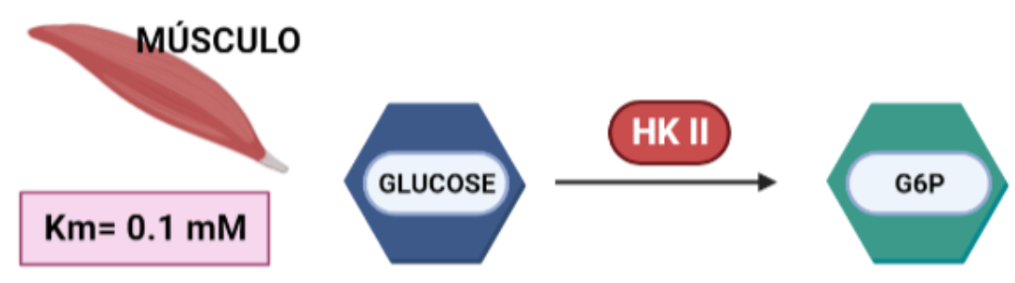
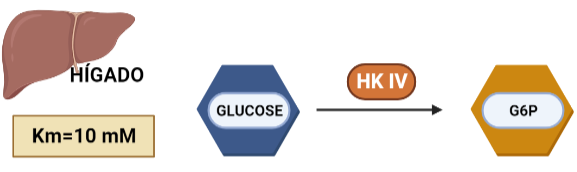
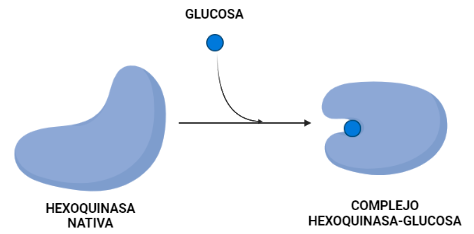
La hexoquinasa es una enzima transferasa (D-hexose-6-phosphotransferase), del grupo de las quinasas, encargada de fosforilar hexosas. Sin embargo, presenta mayor afinidad por la glucosa, puesto que la Km de esta es menor que la de otras hexosas como la fructosa. Esta proteína presenta cuatro isoformas, que han ido surgiendo de forma gradual. En dicho proceso de evolución ocurren determinados cambios que son claves en la estructura y que han permitido que la hexokinasa en dos de sus isoformas se oligomerice. La estructura de la proteína va a contar con dos dominios de unión a los sustratos (glucosa y ATP) y su actividad va a estar regulada alostéricamente mediante un mecanismo de ajuste inducido provocado por la propia glucosa.
Así mismo, esta enzima va a presentar un papel clave en el cáncer. En este artículo se abordará el normal funcionamiento de la hexokinasa así como su papel tumoral por diferentes vías, profundizando, además, en posibles estudios futuros y nuevos campos que se abren en la investigación contra el cáncer que emiten un rayo de esperanza en el estudio biomédico.
PAPEL BIOLÓGICO
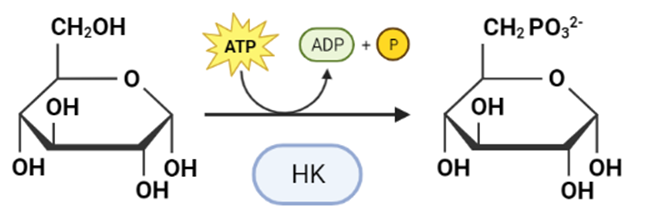
Figura I: reacción esquemática de la Hexokinasa, que fosforila la glucosa, produciendo Glucosa-6-Fosfato.
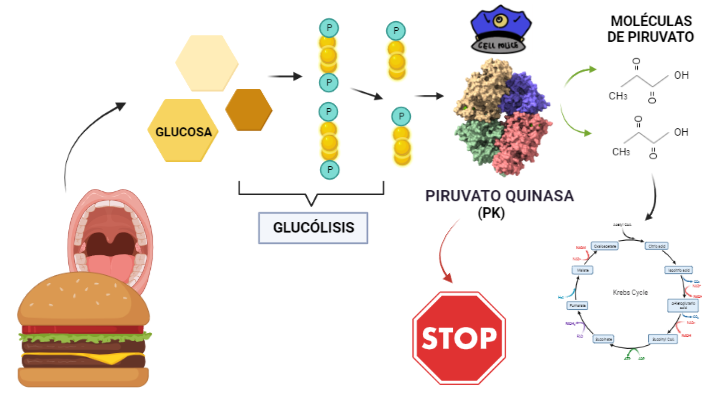
La Hexokinasa participa en la primera reacción irreversible de la glucólisis, que es la primera etapa del metabolismo de la glucosa. Tras hidrolizar el ATP, transfiere el grupo fosfato a la glucosa, para dar glucosa-6-fosfato (G6P), y así manteniendo el gradiente de glucosa que permite que haya un flujo de la misma mediado por los transportadores GLUT. La G6P es un inhibidor competitivo del ATP, por tanto se trata de un fenómeno de feedback negativo, en el que el mismo producto regula alostérica y negativamente su reacción de síntesis. Además, el grupo fosfato (Pi) liberado de la hidrólisis del ATP puede antagonizar la inhibición de G6P o sumarse al efecto inhibidor, según la isoenzima que haya llevado a cabo la reacción.
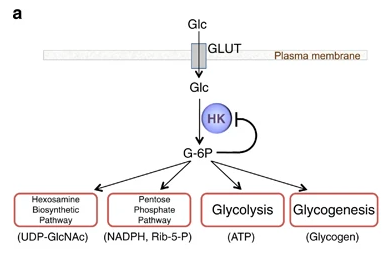
El producto (G6P) puede seguir varias rutas o vías celulares y funcionales:
Metabolismo catabólico: se introduce la glucosa en la glucólisis, para llevar a cabo un metabolismo oxidativo y obtener energía.
Metabolismo anabólico: la G6P es destinada a la vía de las pentosas fosfato, para sintetizar NAPDH y Ribulosa-5-Fosfato; o puede ser convertido a sus formas poliméricas (glucógeno), mediante la gluconeogénesis.
Figura II: vías del producto Glucosa-6-Fosfato, que se introduce en vías como la glucólisis, la ruta de las pentosas fosfato, o la glucogénesis.
ESTRUCTURA
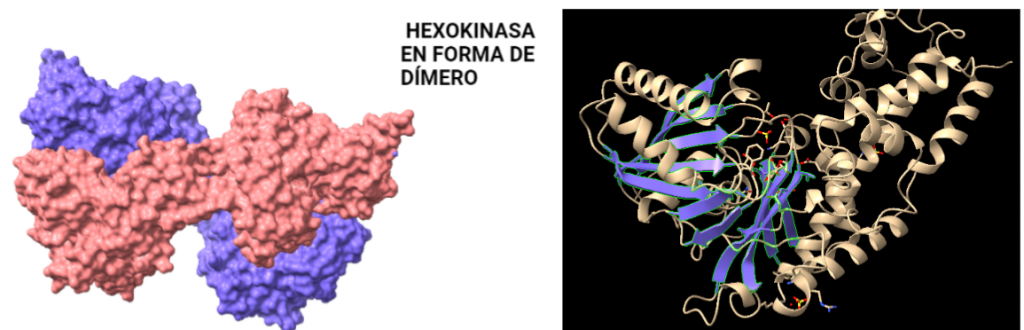
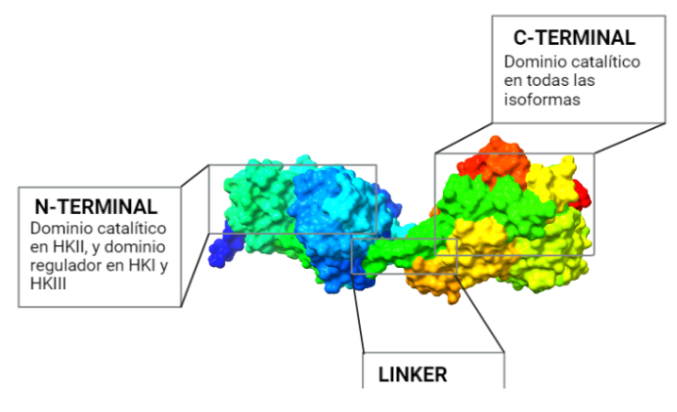
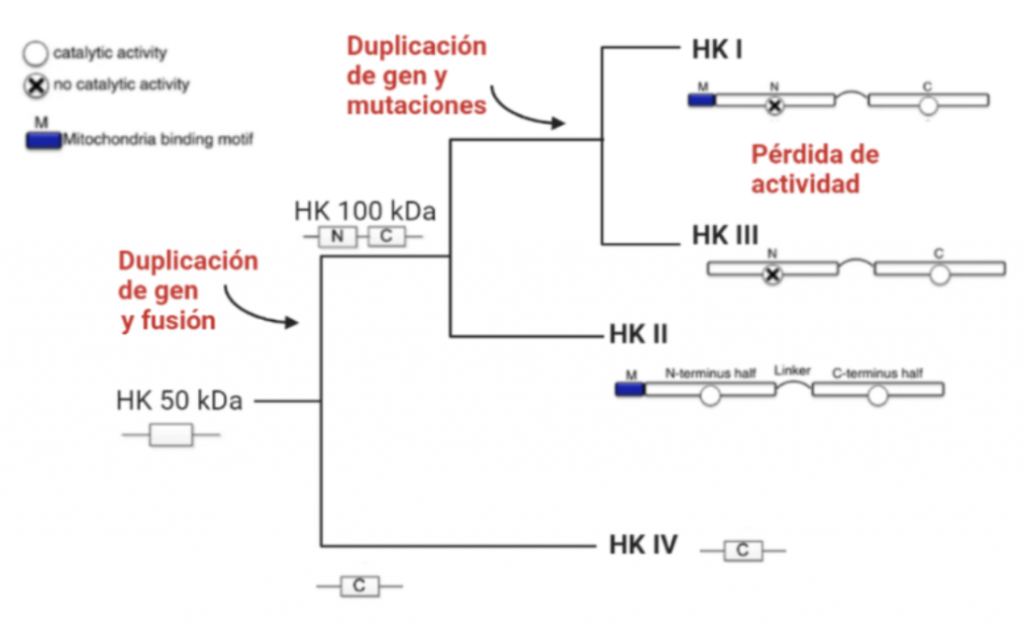
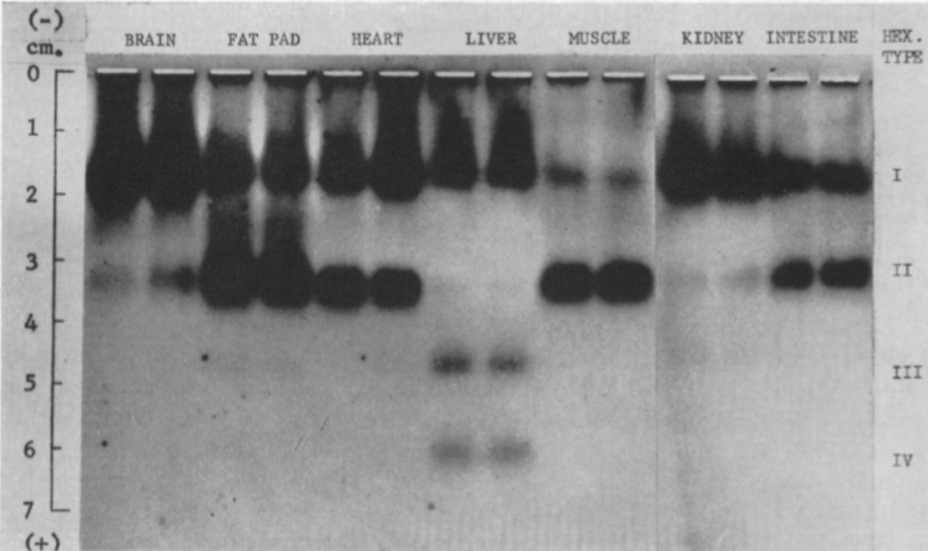
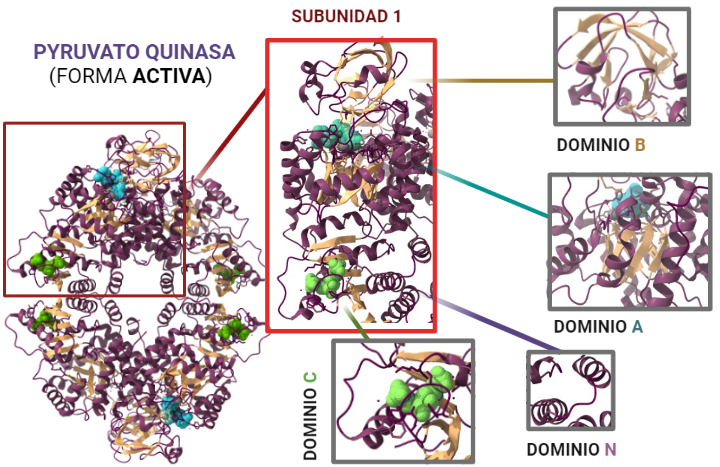
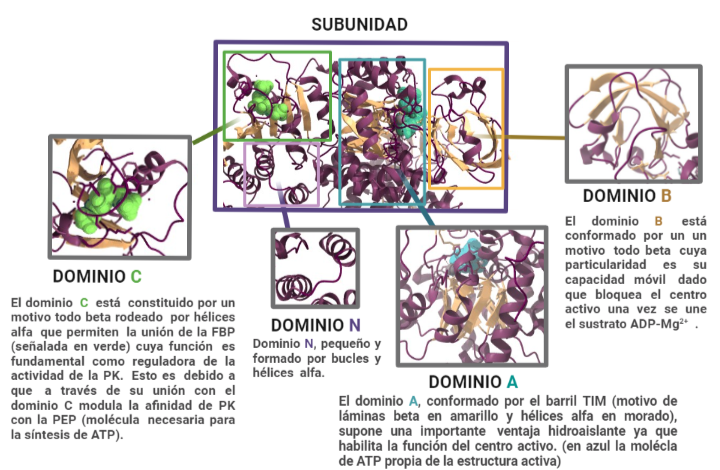
La estructura de las Hexokinasas más comunes (las isoformas I, II y III) cuenta con dos lóbulos muy similares de unos 50KDa cada uno. Algunas de ellas, como la HK I, son monoméricas, pero cuando se une a la membrana externa de la mitocondria se oligomerizan. De esta manera, la Hexokinasa cuenta con dos dominios principales, uno regulador y otro catalítico. La estructura dimérica y por tanto cuaternaria está presente en todas las isoformas salvo en la IV, que es la más ancestral.
Figura III: HexokinasaI dimérica, con cada monómero de un color. Hecho con BioRender y Chimera, a partir de PDB 1BG3. Figura IV: Dominio de unión del ATP, se observan cuatro láminas paralelas y una antiparalela. Hecho con Chimera
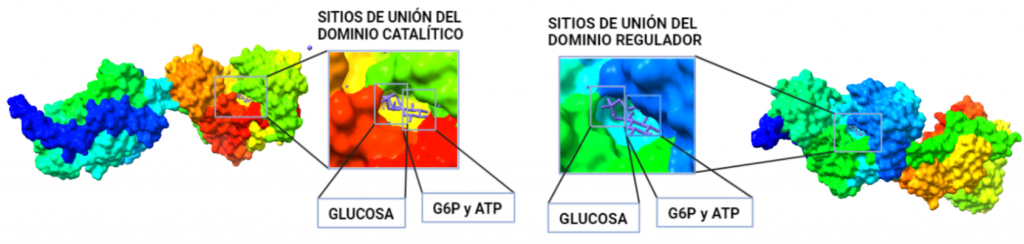
El dominio N-terminal se considera el dominio regulador en las isoenzimas I y II, y contiene el motivo de unión a la mitocondria. Además, está unido al dominio C-terminal (que es el dominio catalítico) a través de una hélice alfa. Ambos dominios presentan sitios de unión con la glucosa, G6P y ATP, y la inhibición de G6P en el dominio regulador se contagia al dominio catalítico por medio del contacto por la hélice alfa entre los dominios. La estructura terciaria de la hexoquinasa se basa en un plegamiento alfa/beta abierto. El dominio de unión al ATP está compuesto por cinco láminas beta y tres hélices alfa en el cual cuatro de las láminas beta son paralelas y una es antiparalela. Por otro lado, la hexoquinasa requiere de iones de magnesio para poder llevar a cabo la actividad catalítica. El magnesio (Mg2+) va a ser el cofactor de la enzima y se encuentra formando un complejo con el ATP (MgATP2-), que estabiliza la catálisis y reduce la energía de activación de la reacción.
Figura V: esquema general de la Hexokinasa en forma de monómero, que presenta un dominio catalítico y otro regulador, unidos por una hélice alfa. Hecho con BioRender, a partir de PDB 1BG3.Figura VI: Sitios de unión de la Hexokinasa con la glucosa y el inhibidor G6P, en ambos dominios. Hecho con BioRender, a partir de PDB 1BG3.
EVOLUCIÓN
Todas las isoenzimas de la Hexokinasa provienen de una Hexokinasa de 50 kDa, susceptible a la inhibición por el producto G6P, por tanto, todas las isoenzimas presentan esta característica. A partir de la duplicación y fusión del gen que codificaba esta forma ancestral, surgieron las isoenzimas I, II y III, que ya son moléculas de 100kDa.
Figura VII: Esquema de la evolución de las isoformas de la Hexokinasa, hecho con BioRender e imágenes de D J Roberts y S. Miyamoto
La isoforma más próxima evolutivamente a la Hexokinasa original es la Tipo IV, que no sufrió la duplicación y fusión génica. Una vez que esto ocurrió, la segunda isoenzima que apareció fue la Hexokinasa II, que mantiene la actividad catalítica en ambos extremos terminales de la proteína, al igual que la Hexokinasa ancestral.
Una consecuente duplicación tuvo como resultado la aparición de la isoforma III. Posteriormente, las mutaciones de genes que codificaban la Hexokinasa 100 kDa, produjeron que el extremo N-terminal se diferenciara funcionalmente, perdiendo la actividad catalítica, y adquiriendo una función reguladora (con un sitio de unión para el inhibidor G6P). Esta diferenciación dio lugar a las en las Hexokinasas I y III.
Además, en las HK I y II, el extremo N-terminal presenta un dominio hidrofóbico que permite a estas integrarse en la membrana de la mitocondria. Concretamente, se unen a las porinas (VDAC) de la membrana mitocondrial externa, las cuales interaccionan con los ANT (Translocadores de Nucleótidos de Adenina). Esto es esencial para el mecanismo enzimático de la HK, puesto que es el sitio de salida del ATP producto de la fosforilación oxidativa (que usará la HK), y el sitio de entrada del ADP resultante de la reacción enzimática de la hexokinasa. Por tanto, existe una coordinación entre la introducción de la glucosa al metabolismo glucolítico y las últimas etapas de este en la mitocondria (la fosforilación oxidativa), para que se den a un ritmo adecuado a las necesidades celulares.
Hay cuatro isoenzimas de la Hexokinasa (HK) en los tejidos de mamíferos, con una estructura similar, pero expresión en diferentes tejidos:
Figura VIII: Electroforesis de las isoformas de la Hexokinasa en diferentes tejidos, que muestra que la HK I se encuentra presente de manera general, mientras que la HK II aparece en el músculo y tejido adiposo, y la HK III y IV, en el hígado. Imagen de H M Katzen and R T Schimk.
HEXOKINASA I (HKI)
Fundamentalmente en el cerebro, donde la tasa metabólica es muy exigente, pero expresada de manera general.
HEXOKINASA II (HKII)
Más limitada en su expresión, aparece en tejidos sensibles a insulina, como es el caso del tejido adiposo y el músculo esquelético. En el músculo, es necesaria una alta tasa de glucólisis, y por tanto, lo que ocurre es que esta isoforma presenta una gran afinidad por la glucosa (menor Km), para que con concentraciones muy bajas de glucosa, se alcancen altas velocidades de la enzima.
Figura IX: Afinidad por la glucosa de la HKII en músculo. Creado con BioRender
La insulina aumenta la actividad de esta isoforma, induciendo la transcripción del gen que la codifica, y de esta forma, favoreciendo la metabolización y eliminación de glucosa. Por esta razón, en los individuos con diabetes de tipo II, la expresión de la HKII se ve reducida, acentuando la hiperglucemia.
Al igual que la isoenzima Tipo I, incluye un dominio hidrofóbico en el extremo N-terminal que permite que se inserte en la membrana externa mitocondrial, y también usa ATP intramitocondrial.
Cabe destacar su predominancia en células tumorales, puesto que estas se caracterizan por presentar un aumento anormal del metabolismo, en el que la reacción que lleva a cabo la Hexokinasa es esencial para la obtención de energía.
HEXOKINASA III (HKIII)
A diferencia de las dos isoenzimas anteriores, la Hexokinasa III no está unida a la mitocondria, puesto que carece del dominio hidrofóbico en el extremo N-terminal. Se piensa que se expresa en el citoplasma, o que incluso tiene una localización perinuclear, en células del hígado.
HEXOKINASA IV (HK IV)
En hepatocitos y células beta pancreáticas, y es conocida como glucoquinasa. La G6P que produce está destinada a la síntesis deglucosa. Esto es un proceso que se lleva a cabo cuando la cantidad de sustrato (glucosa) es alta, por tanto, tiene sentido que esta isoenzima presente una mayor Km, porque necesitará altas concentraciones de glucosa para realizar la reacción a una velocidad alta.
Figura X: Afinidad por la glucosa de la HK IV en hígado. Creado con BioRender.
MECANISMO
La hexoquinasa sufre un cambio conformacional que es regulado por la propia glucosa que va a ser esencial para la catálisis. En este proceso se observa que la superficie en contacto con el solvente del complejo hexoquinasa-glucosa es más pequeña que la hexoquinasa nativa.
Utilizando dicho cambio en el área de superficie que se encuentra expuesta se ha podido estimar la contribución hidrofóbica a los cambios de energía libre tras la unión de la glucosa. De esta manera se descubre que el efecto hidrofóbico por sí solo favorece la conformación activa de la hexoquinasa en presencia y ausencia de azúcar. La estabilidad observada de la conformación inactiva de la enzima en ausencia de sustratos puede resultar de una deficiencia de interacciones complementarias dentro de la cavidad que se forma cuando los dos lóbulos se unen.
El cambio conformacional que sufre la hexoquinasa mantiene la estructura terciaria prácticamente igual excepto por un gran cambio en la orientación de los dos lóbulos. Para demostrar este cambio lo que se hizo fue superponer los carbonos alfa de cada lóbulo usando un procedimiento de mínimos cuadrados y tratando a los carbonos como cuerpos rígidos. Esta superposición mostró que cada lóbulo se comporta como un cuerpo rígido durante el cambio conformacional entre la forma nativa de la proteína y el complejo con la glucosa.
Figura XI: Cambio conformacional de la HK inducido por la glucosa y cambio en la superficie de contacto con el solvente.
Tal y como venimos viendo, la superficie accesible de la hexoquinasa se reduce cuando se une la glucosa para formar el complejo enzima-sustrato (ES) y se reduce aún más por el cambio a E’(inactiva)-S. Por lo tanto, se puede esperar que las fuerzas hidrofóbicas favorezcan la conformación activa en presencia de azúcar, asumiendo que todos los donantes y aceptores de enlaces de hidrógeno están satisfechos en E’-S. Sin embargo, el área superficial también se reduce cuando la conformación activa se forma en ausencia de azúcar (E’ – E). Sin embargo, debido a que hay menos de un factor de dos diferencias entre los cambios en la superficie accesible para las transiciones E-S → E’-S y E’-E, el efecto hidrofóbico no puede explicar la gran diferencia en las constantes de equilibrio conformacional K2 y K3 en presencia y ausencia de azúcar.
Figura XII: Cambios conformacionales de la Hexoquinasa en presencia y ausencia de azúcar. Esquema creado con Biorender.
Otra cuestión que surge es por qué la enzima no permanece en el estado activo, E, en ausencia de ligandos sabiendo que el efecto hidrófobo, de manera individual, predice que la estructura E debería ser más estable. La respuesta a esta pregunta reside en que cuando la enzima carece de la presencia de la glucosa contiene una cavidad en la que entran las moléculas de agua y donde quedan encerradas. Además, tanto los puentes de hidrógeno como las fuerzas de Wan der Waals contribuyen muy poco a la estabilidad de la proteína y del complejo proteína-ligando. El hecho de no obtener estas interacciones complementarias dentro de la cavidad daría como resultado entalpías desfavorables causadas por la pérdida de los puentes de hidrógeno o fuerzas de Van der Waals en relación con los que se producen en la estructura abierta. También puede haber alguna pérdida de entropía traslacional al atrapar una pequeña cantidad de moléculas de agua en la cavidad.
Presuntamente, el agua misma desestabiliza la forma activa mediante la creación de interacciones favorables con la estructura abierta inactiva. Solamente el ligando correcto puede proporcionar las fuerzas de Van der Waals y los puentes de hidrógeno necesarios para que se active la estructura.
Con ello, concluimos que hay al menos dos posibles funciones para el cambio conformacional inducido por la glucosa: permitir un «mecanismo de acogida» o proporcionar especificidad.
PAPEL BIOMÉDICO: HEXOKINASA II EN CÁNCER
El metabolismo de las células tumorales se caracteriza por una alta actividad glucolítica: metabolizan anaeróbicamente grandes cantidades de glucosa en ácido láctico, incluso en presencia de oxígeno, aumentando la velocidad de la glucólisis y de la síntesis de ATP. Esto es lo que se conoce como el efecto Warburg. Por tanto, la actividad de cualquier enzima glucolítica como la HK será esencial en un tumor.
La Hexokinasa II aparece sobreexpresada en células tumorales, satisfaciendo estas altas velocidades de la glucólisis. La introducción de la glucosa en el metabolismo glucolítico es crucial para la producción de energía, y la síntesis de precursores de nucleótidos (derivados de glucosa) por la vía de las pentosas fosfato, destinados a la síntesis de ADN para la proliferación del tumor.
Fig. XIII: Amplificación del gen de la HKII en hepatoma y hepatocitos. Hay unas 5-10 copias más de lo normal en el hepatoma, ya que la intensidad de una tira de ADN de hepatocito de 30 μg se asemeja a la de ADN de hepatoma de 6-3 μg Imagen: Annette Rempel
La regulación por AKT de la HKII es el factor determinante para que esta isoforma sea la esencial en el metabolismo tumoral, puesto que controla la unión de esta a la mitocondria, y con ello, fija la función de la HK, que varía en función de si aparece unida a la mitocondria o no.
Con el objetivo de crecimiento del tumor, y cuando hay disponibilidad de nutrientes, AKT une la HKII a la mitocondria (fosforilando su residuo Thr-473), conectándola con VDAC. Esto le permite a la enzima tener un acceso privilegiado al ATP que sintetiza la mitocondria, y la célula sigue un metabolismo de proliferación y de producción de energía, mediante la glucólisis.
Figura XIV: Unión de la HKII con el VDAC de la membrana externa de la mitocondria, y cómo esta le posibilita acceder al ATP sintetizado por la ATP sintasa. Esta imagen señala la relación entre el primer paso de la glucólisis y el último paso del metabolismo oxidativo. Imagen: Pedersen, P.L.
La HK II mitocondrial, además, lleva a cabo una función protectora en tumores con acceso a nutrientes, promoviendo la supervivencia celular: inhibe a los miembros pro-apoptóticos de la familia Bcl2, e introduce la G6P en la ruta de las pentosas fosfato, cuyos productos son antioxidantes que reducen las ROS (especies reactivas de oxígeno).
Sin embargo, en una situación de isquemia, en la que no llegan suficientes nutrientes y oxígeno a la célula, disminuye la actividad de AKT y HKII mitocondrial, aumentando la HKII citoplásmica. Entonces, esta isoforma se une a mTORC (mediante el motivo TOS), inhibiéndola, e induciendo la vía autofágica, y la conservación de energía y homeostasis en ausencia de glucosa, pensando en el “bien mayor” del tumor.
Figura XV: Esquema explicativo del papel que juega la HK II en células tumorales, promoviendo la supervivencia celular si tiene acceso a nutrientes, pero induciendo indirectamente la autofagia en el caso contrario, con el fin de conservar la energía y homeostasis tisular.Creado con BioRender.
Esta regulación por AKT posibilita esta compleja acción de la HKII, que puede ser proapoptótica (HKII citoplásmica) o antiapoptótica (HKII mitocondrial), según la disponibilidad de recursos. Sin embargo, AKT no regula la HKI, ya que esta no presenta una secuencia de consenso para esta enzima, y por ello, esta isoenzima no se encuentra prevalentemente en tumores. Además, la HKI no puede satisfacer la alta demanda energética, al perder la actividad catalítica en el extremo N-terminal.
Según todas las vías beneficiosas para el tumor mencionadas anteriormente en las que participa la HKII, la eliminación de esta isoforma perjudicaría a la progresión del tumor, por lo tanto, es un frente esperanzador en terapias oncogénicas. Lo ideal sería encontrar un modo de inhibir únicamente esta isoforma, pero es difícil puesto que todas ellas son bastante similares.
La inhibición de la HKII en células cancerosas puede darse por p53, o por un sustrato análogo a la glucosa, la 2-desoxiglucosa, que favorece la apoptosis. Una combinación de estos dos factores podría ser favorecedora. Otro posible campo a investigar sería la inhibición de la HKII por fosfato inorgánico, que sensibiliza la inhibición por G6P en esta, pero la antagoniza en HKI. Incluso, una alternativa más podría ser el uso de determinados péptidos que desplazaran la HKII de la mitocondria. Este desplazamiento parece producir un aumento de la concentración de Ca2+ citosólica, lo que abriría poros en la membrana mitocondrial e induciría a la célula a apoptosis.
Figura XVI: Posible terapia oncogénica sobre HK II, mediante péptidos que la separen de la mitocondria. Creado con BioRender.
Bennett, W. S., & Steitz, T. A. (1978). Glucose-induced conformational change in yeast hexokinase. Proceedings of the National Academy of Sciences, 75(10), 4848-4852. https://www.pnas.org/doi/abs/10.1073/pnas.75.10.4848
Gahr, M. (1980). Isoelectric Focusing of Hexokinase and Glucose-6-Phosphate Dehydrogenase Isoenzymes in Erythrocytes of Newborn Infants and Adults. British Journal of Haematology, 46(4), 529-535. https://doi.org/10.1111/j.1365-2141.1980.tb06009.x
Haruhiko Osawa, Calum Sutherland, R. Brooks Robey, Richard L. Printz, Daryl K. Granner (1996). Analysis of the Signaling Pathway Involved in the Regulation of Hexokinase II Gene Transcription by Insulin. https://www.sciencedirect.com/science/article/pii/S002192581831932X
Mulichak, A., Wilson, J., Padmanabhan, K. et a (1998)l. The structure of mammalian hexokinase-1. Nat Struct Mol Biol 5, 555–560. https://pubmed.ncbi.nlm.nih.gov/9665168/
R.L. Printz, S. Koch, L.R. Potter, R.M. O’Doherty, J.J. Tiesinga, S. Moritz, D.K. Granner (1993). Hexokinase II mRNA and gene structure, regulation by insulin, and evolution. Journal of Biological Chemistry. Volume 268, Issue 7 https://www.sciencedirect.com/science/article/pii/S0021925818535213
Reference for PDB-101: PDB-101: Educational resources supporting molecular explorations through biology and medicine. Christine Zardecki, Shuchismita Dutta, David S. Goodsell, Robert Lowe, Maria Voigt, Stephen K. Burley. (2022) Protein Science 31: 129-140 doi:10.1002/pro.4200
Reference for RCSB PDB: The Protein Data Bank H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne (2000) Nucleic Acids Research, 28: 235-242. doi:10.1093/nar/28.1.235
Rempel, A., Mathupala, S. P., Griffin, C. A., Hawkins, A. L., & Pedersen, P. L. (1996). Glucose Catabolism in Cancer Cells: Amplification of the Gene Encoding Type II Hexokinase1. American Association for Cancer Research. https://pubmed.ncbi.nlm.nih.gov/11557773/
Roberts, D., Miyamoto, S. (2015) Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ 22, 248–257. https://www.nature.com/articles/cdd2014173
UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE. Protein Sci. 2018 Jan;27(1):14-25.
UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. Protein Sci. 2021 Jan;30(1):70-82.
La Piruvato quinasa (PK) es una enzima que se encuentra al final de la ruta metabólica conocida como glucólisis y su función es permitir la salida del piruvato a la siguiente vía metabólica con el fin de obtener energía. Para entender la relevancia de esta molécula, quizás es conveniente explicar de forma concisa el recientemente popular término, conocido como “metabolismo”.
El metabolismo, al que hacen referencia desde dietistas y entrenadores hasta profesores de bioquímica, es quizás un proceso que para el ciudadano de a pie resulta abstracto en su relación con la ingesta de comida. Brevemente, se trata de un conjunto de reacciones que consisten en tomar una molécula grande (glucosa) con alto potencial energético y dividirla en paquetes más pequeños (piruvatos, en el caso de la glucólisis) con el fin de distribuir la energía por los distintos sistemas celulares y permitir que los organismos vivos se sigan considerando como tales. Esa glucosa, llega a las células por medio de la alimentación y se convierte en energía química útil mediante las distintas rutas metabólicas. De modo, que el correcto funcionamiento de la PK, es fundamental para que estos pequeños paquetes se introduzcan en la siguiente ruta metabólica (el ciclo de Krebs) que se encargará de distribuir la energía. De hecho, modificaciones de esta molécula podrían degenerar en condiciones tan serias como el Cáncer o el Alzheimer, dado que impediría que la energía alcanzase los destinos pertinentes.
PAPEL BIOLÓGICO
El rol que asume la PK a nivel biológico se traduce a la función que tiene por ejemplo el personal de seguridad a la entrada de un museo. Éste, en caso de que el turista no presente ninguna irregularidad (posesión de armas, comida, etc), procede a concederle la entrada al establecimiento; del mismo modo la PK permite el acceso al piruvato a la siguiente ruta, siempre que no suponga una amenaza contra el correcto funcionamiento de la célula. Al tratarse la PK de una quinasa, esto la convierte en una molécula con la capacidad de fosforilar (añadir grupos fosfato a otras moléculas). De modo que para concederle el acceso a la siguiente ruta, la PK retira los fosfatos de las triosas procedentes de la glucosa y los añade a moléculas de ADP, dando lugar a ATP (que es una molécula que transporta energía). Como resultado de este proceso, se sintetizan dos piruvatos, a los que finalmente les será concedido el acceso al ciclo de Krebs.
La PK en humanos, aparte de la función biológica general descrita anteriormente, también tiene una serie de isoformas cuya función e importancia biomédica es más específica. Las cuatro isoformas de la PK (PKM, PKK, PKR, PKL) fueron descubiertas en 1965 y reciben su nombre a partir del nombre tejido donde se encuentra cada una. La PKM (actualmente denotada como PKM1) se encuentra en los tejidos musculares (muscle, M) del corazón y en el cerebro, la PKK (actualmente denotada como PKM2) se encuentra en los tejidos de los riñones (kidneys, K), el intestino y las células cancerosas; la PKR en el tejido sanguíneo (red blood cells, R) y la PKL en el tejido del hígado (liver, L) y en los riñones también. La diferenciación (que ocurre tras la fase fetal, ya que todas provienen de la isoforma PKM2) favorece la aparición de patologías mucho más especializadas debido a ligeros cambios en la conformación proteica.
Ilustración que refleja el papel de la Piruvato quinasa en el metabolismo de la glucosa. La molécula de la que partimos es la glucosa, que tiene 6 carbonos, ésta, mediante una concatenación de reacciones da lugar a 1,3-bifosfoglicerato (las bolas amarillas representan los carbonos y las P los grupos fosfato) que tras otra serie de reacciones, dan lugar a fosfoenolpiruvato (PEP) que es el precursor del piruvato. La PK es la encargada de catalizar la reacción de desfosforilación que da lugar al paso de PEP a piruvato cuyo fosfato restante se utiliza para fosforilar ATP. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
ESTRUCTURA Y CÓMO FUNCIONA
Desglose estructural
Para entender el papel que desempeña la PK es fundamental comprender su estructura, dado que de ésta dependerá el funcionamiento de la misma. ¿Qué constituye a la PK? La piruvato quinasa es una proteína conformada por 531 aminoácidos que dan lugar a un tetrámero, cuyas cuatro subunidades son iguales. Éstos están organizados en motivos de hélices alfa, láminas beta y bucles.
Estas subunidades se organizan a su vez en tres grandes dominios rotulados como A, B y C junto con un dominio N-terminal. El dominio A está conformado por un barril TIM α8/β8 cuyo centro activo se ubica entre el dominio A y el B, éste es además el dominio más grande de la subunidad. El dominio B sin embargo es móvil y bloquea el centro activo una vez que se le une el sustrato ADP-Mg2+. Finalmente, el dominio C contiene la fructosa-1,6-bifosfato (FBP) que es un potente activador alostérico.
Descripción gráfica del desglose estructural que presenta la Piruvato quinasa en estado activo. Los motivos de láminas beta quedan indicados en amarillo, las hélices alfa en púrpura, los bucles en violeta y los ligandos: ATP en azul y FBP en verde. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
Esto significa, que a través de la unión de la FBP al dominio C, se facilitará la unión del fosfoenolpiruvato (PEP) que es fundamental para la regulación de la actividad de la PK, ya que ésta depende de la afinidad con el PEP. En ausencia de activadores alostéricos como la FBP, la PK tiene poca afinidad con el PEP. O sea, si la PK fuera un niño pequeño y su voluntad para realizar los deberes fuera análoga a la actividad de la quinasa, éste necesitaría una motivación para realizarlos. Si se impone la condición de recibir un caramelo a cambio de la tarea, éste cumplirá. De igual forma si la PK presenta la FBP unida al dominio C, ésta aumentará su afinidad a la PEP alterando su actividad. Además, la unión de FBP estabiliza la molécula en estado activo y promueve la tetramerización. Cabe destacar, que todas las isoformas de la PK se unen con la FBP exceptuando la PKM1 que debido a una discrepancia estructural es suficientemente estable por si sola (siendo además insensible a los moduladores alostéricos) y no presenta ni la región de unión a la FBP ni el interfaz dímero-dímero debido a éstas se expresan en los exones específicos de las isoformas.
Desglose de la funcionalidad de las distintas estructuras que conforman cada subunidad de la Piruvato quinasa en estado activo. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
Otro detalle que cabe mencionar de la estructura de la PK es su capacidad de unión a cofactores (K+ y Mg 2+) cuya intervención en la activación de la molécula es esencial. En el caso del Mg 2+, se ha mencionado que está involucrado en bloquear el acceso al centro de activo (gracias al cambio conformacional que desplaza al dominio B) cuando éste forma un complejo de sustrato al unirse al ADP (formando el sustrato ADP-Mg2+). En el caso del K+ sin embargo, se ha observado que ante su presencia, el mecanismo cinético de la PK se mantiene desordenado (forma natural), esto supone que favorece la forma activa de la PK y permite que se unan el PEP o el complejo ADP-Mg2+ de forma independiente (mecanismo aleatorio). En ausencia de K+, por el contrario, el ADP no se pude unir al centro activo hasta que el PEP no haya terminado de formar un centro activo completamente funcional. De forma, que se deduce que el K+ es el encargado de inducir el cierre del centro activo y de que los residuos encargados de la unión al nucleótido adopten la conformación correspondiente.
Ilustración que señala la presencia de cofactores ( K+, Mg2+) en el Dominio A, fundamentales para la adopción de la conformación activa de la PK. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
Función ¿Qué hace?
Entonces, ¿Qué función tiene? Una vez conocida la estructura, es posible dilucidar qué función lleva a cabo, y cómo. La función catalítica de la PK consiste en fosforilar moléculas, debido a su condición de quinasa; en concreto, moléculas de ADP a partir de moléculas de PEP de la etapa anterior de la glucólisis. Todo ello para dar lugar a dos productos. Por un lado, piruvatos estables a partir de sus precursores PEP y por otro, obtener energía en forma de moléculas de ATP a través de la fosforilación del ADP. De modo, que el nombre surge del producto (piruvato) + tipo de enzima (quinasa). Para ello, al centro activo del dominio A se unen el PEP y el complejo ADP-Mg2+ dado que los cationes de Mg2+ median y facilitan la transferencia del grupo fosfato del PEP al ADP dando lugar al ATP y a los piruvatos. Todo ello es posible debido a la alta energía que libera PEP al ser hidrolizada. Al perder el fosfato, el PEP pasa a su forma de enolpiruvato que es menos estable, de modo que se llevará a cabo un proceso de tautomerización que consiste en que el enolpiruvato acepte un protón procedente de una molécula de agua convirtiéndose así en un piruvato estable y favoreciendo la fosforilación del ADP.
Representación gráfica de la reacción que tiene lugar cuando la PK funciona de forma natural.El complejo ADP-Mg2+ se une al centro activo junto con el PEP. Éste le aporta el fosfato que le hace inestable con el fin de utilizarlo para fosforilar al ADP. Como productos se obtienen ATP y piruvato. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
En cuanto a la capacidad alostérica de la PK, aparte de la FBP, que favorece la unión del sustrato PEP, hay otras moléculas que alteran la actividad de la enzima. Por ejemplo, un inhibidor alostérico de esta enzima (PKM1, PKM2) sería la fenilalanina (Phe) cuya unión supone la disminución de afinidad con la PEP mediante la estabilización de la estructura inactiva de la PK. El lugar de unión de Phe también puede albergar a la alanina que actúa como inhibidor, pero solo ante la isoforma PKM2, y esto lo lleva a cabo favoreciendo la conformación dimérica, contraria a la tetramérica a la que se une FBP. A pesar de que en presencia de concentraciones normales de FBP la inhibición de la alanina queda mitigada. La serina sin embargo, también puede ocupar este centro de unión, pero con función activadora no inhibidora, en la PKM2. Dejando a un lado los aminoácidos, hormonas ,como la hormona tiroidea triyodo-L-tironina (T3), actúan también como inhibidor alostérico favoreciendo la conformación monomérica inactiva de la PK. Mientras que el oxalato puede actuar como activador de la PK mediante su interacción con el centro activo por ser análogo al enolpiruvato, en caso de que la concentración de PEP sea baja.
¿PORQUÉ PODRÍA ACABAR CONTIGO?
EL PELIGRO RESIDE EN LA ISOFORMA
Las isoformas de una proteína son proteínas que provienen del mismo gen que la proteína original, dicho gen se duplica y comienza a acumular mutaciones para dar lugar a las distintas isoformas. En el caso de la PK, tras este proceso de duplicación y modificación por mutaciones se han obtenido 4 isoformas distintas: PKM1, PKM2, PKR y PKL. La importancia de las isoformas recae en que a pesar de realizar la misma función que la proteína inicial, cada una presenta ligeramente distintas: propiedades cinéticas, estructurales, de regulación o de localización en la célula. Estas ligeras diferencias atienden a las necesidades metabólicas del tejido al que pertenecen. O sea, las modificaciones que sufra PKL (L hace referencia a liver, hígado en inglés) afectarán en principio al hígado dado que la estructura de la PKL ha resultado ser la más eficaz a la hora de catalizar las reacciones que precisa este órgano. A pesar de esto, existe una isoforma que destaca en su implicación en numerosas patologías inflamatorias (como la Sepsis, IBD o Arterosclerosis) o enfermedades como el Cáncer o el Alzheimer. Ésta es la PKM2.
PKM2 Y UN COMPENDIO DE LO QUE PUEDE SALIR MAL
PKM2 en el Cáncer
Ante el desarrollo de células neoplásicas, la PKM2 tiene un comportamiento que podría clasificarse como moonlighting. Esto se debe a que en condiciones normales la PK transforma PEP en piruvato y este sigue la ruta metabólica normal hacia el ciclo de Krebs, mientras que, ante una situación de estrés, como puede ser el desarrollo de células cancerígenas, ésta altera su forma tetramérica natural y pasa a su forma dimérica. Al dimerizarse mediante la fosforilación de su tirosina 105 la proteína deja de realizar su función natural y divierte el proceso de la glucólisis hacia la síntesis de metabolitos necesarios para la síntesis de serina. Esto se debe a que dicho aminácido regula a mTORC1 ( mammalian target of rapamacyn complex 1), que es fundamental para favorecer la proliferación celular, característica de las células cancerígenas. Otra de las modificaciones que sufre, es la acetilación de su lisina 305, junto con la oxidación de su cisteína 358 que provoca una alteración en la ruta de la glucólisis haca la PPP (pentose phosphate pathway, vía de la pentosa fosfato) que favorece la síntesis de nucleótidos para sufragar los efectos de la interrupción de la ruta glucolítica.
Descripción gráfica de la dimerización y modificación de la piruvato quinasa en presencia de cáncer. Este proceso consiste en dimerizar la proteína mediante la fosforilación de su tirosina 105 y alterar sus propiedades mediante la oxidación de su cisteína 358 y la acetilación de su lisina 305. A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
Estas modificaciones de la PK se conocen como el efecto Warburg o Glucólisis Aeróbica. El efecto Warburg supone que las células cancerígenas conviertan el piruvato en lactato, disminuyendo su producción de ATP. La disminución de síntesis de ATP se soluciona mediante los procesos mencionados anteriormente, mientras que el aumento de lactato supone la aparición de un ambiente tumorigénico dado que es excretado, reduciendo así el pH extracelular (esto favorece las condiciones para la proliferación celular) y provocando inflamación. El aumento de lactato también se utiliza para acceder a un recurso de energía alternativo, como es la glutamina. Esto es posible, dado que se disminuye la entrada de piruvato en el ciclo de Krebs, por lo que comienzan a introducirse metabolitos de glutamina en su lugar y aumenta por lo tanto la síntesis de lípidos y aminoácidos. Finalmente, la PKM2 es translocada al núcleo donde se une a HIF1-α (hypoxia-inducible factor-1 alfa) promoviendo la transactivación de HIF1 que favorece la aparición de un ambiente tumorigénico y la desviación de la glucólisis hacia la vía de la pentosa fosfato.
Diagrama que dilucida la modificación de la vía metabólica regulada por la piruvato quinasa en presencia de cáncer (efecto Warburg). Las flechas rosas indican los procesos que surgen bajo condiciones de dicho efecto, mientras que las verdes indican el curso habitual de la vía. A partir de PDB 1a3w y 6wp3(en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
PKM2 en el Alzheimer
Debido a que el origen y funcionamiento del Alzheimer todavía no se ha esclarecido, se están explorando nuevas vías de investigación. Entre ellas, destacan factores como el estrés oxidativo, fallos en el transporte y metabolismo de la glucosa, y la inflamación. Dado que la piruvato quinasa juega un papel central en el metabolismo de la glucosa, su función se ve afectada por la enfermedad. Como consecuencia de la alteración de la ruta metabólica de la glucosa (debida a la acumulación de β-amiloides), se da la inactivación HIF1-α lo que supone que la PKM2 en dímero no puede unirse a ella, quedando inhibida. Otra forma en la que la PKM2 queda inhibida (en presencia de esta enfermedad) es mediante la inactivación o la regulación insuficiente de PPIasa (peptidil-prolil cis-trans-isomerasa) o la elevada concentración de ROS (especies reactivas de oxígeno), ya que ambas impiden la translocación de la PKM2 al núcleo, y consecuentemente esta no se une a HIF1-α, dando lugar en este caso a un desequilibrio en el metabolismo mediante una cascada de PEP. Respecto al papel de la PKM2 en la respuesta inflamatoria, la PKM2 regula la respuesta de los macrófagos, que son esenciales a la hora de retirar los amiloides para evitar la formación de placas. Esto se debe a que ante la inflamación, los macrófagos adoptan un fenotipo que favorece la síntesis de ATP y se ha observado que algunos incluso activan la Glucólisis Aeróbica (efecto Warburg), que se da cuando hay una sobreactivación de la PKM2. La Glucólisis Aeróbica mediada por PKM2 también promueve la activación de inflamasomas mediante la fosforilación de EIF2AK2 (factor 2 de iniciación de traducción de la α quinasa 2). Por otro lado, mediante la interacción con HIF1-α se promueve la transcripción de genes que activan la glucólisis en macrófagos. Finalmente, la PKM2 también es responsable de la glucólisis de la α-sinucleína que es una proteína presináptica asociada a la fisiopatología del Alzheimer.
Esquema que ilustra las consecuencias de la enfermedad del Alzheimer sobre la actividad de la piruvato quinasa. A la izquierda se indican las principales formas mediante las que se inhibe a la PK en condiciones patológicas (inactivación de las moléculas HIF1-α, PPIasa y la elevada concentración de radicales libres). A la derecha sin embrago, se exponen los procesos que se activan como consecuencia de la patología. A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
PKM2 en diversas enfermedades inflamatorias
La respuesta inflamatoria, como se ha expuesto en los dos casos anteriores, requiere de un alto consumo de energía. Células del sistema inmune, como los macrófagos, activan mecanismos similares al denominado efecto Warburg en el cáncer para aumentar la síntesis de energía y poder responder de manera eficaz. Esto está íntimamente relacionado con la glucólisis por lo que la implicación de la PK es crucial. En el caso de la Sepsis la implicación de la PKM2 en la glucólisis anaeróbica que adoptan las células bajo la patología supone un perjuicio para la supervivencia del individuo. Esto se ha observado tras inhibir su actividad comprobando que de este modo también quedaban inhibidas moléculas como el inflamasoma NLRP3 y HMGB1 (proteína de alta movilidad del grupo 1) cuya represión favorece la supervivencia del individuo. En el caso de la Arterosclerosis se ha observado que la inhibición de PKM2 en linfocitos B y T favorece la atenuación de la inflamación. Esto está unido a que parte de los productos que se sintetizan durante la glucólisis aeróbica (adoptada por las células del sistema inmune bajo condiciones de estrés) producen inflamación como se expone en la segunda imagen de la PKM2 en el cáncer. Incluso en la IBD (enfermedad inflamatoria de Bowel) la PKM2 se está valorando como posible bioindicador de la enfermedad según estudios recientes. Una vez más, la supresión de la PKM2 favoreció el control sobre la inflamación ya que en su versión nuclear dimérica contribuye a la adaptación de las necesidades metabólicas de los linfocitos. Esto significa que la presencia (en muestras) de esta proteína en estado dimérico podría ayudar a identificar la enfermedad. También cabe mencionar, que se ha observado que la PKM2 regula a Bcl-xL impidiendo la apoptosis de las células del epitelio intestinal.
Ilustración que refleja concisamente la influencia que tiene la PK en enfermedades inflamatorias como son: la Sepsis, la Arterosclerosis y la IBD. A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
Israelsen, W. J., & Vander Heiden, M. G. (2015). Pyruvate kinase: Function, regulation and role in cancer. Seminars in cell & developmental biology, 43, 43–51. https://doi.org/10.1016/j.semcdb.2015.08.004
June 2022, Faiza Ahmed, Jonathan Ash, Thirth Patel, Auriel Sanders, David Goodsell, Shuchismita Dutta
Oria-Hernández, J., Cabrera, N., Pérez-Montfort, R., & Ramírez-Silva, L. (2005). Pyruvate kinase revisited: the activating effect of K+. The Journal of biological chemistry, 280(45), 37924–37929. https://doi.org/10.1074/jbc.M508490200
Patel, S., Das, A., Meshram, P., Sharma, A., Chowdhury, A., Jariyal, H., … & Shard, A. (2021). Pyruvate kinase M2 in chronic inflammations: a potpourri of crucial protein–protein interactions. Cell Biology and Toxicology, 37(5), 653-678.
The RCSB PDB «Molecule of the Month»: Inspiring a Molecular View of Biology D.S. Goodsell, S. Dutta, C. Zardecki, M. Voigt, H.M. Berman, S.K. Burley (2015) PLoS Biol13(5): e1002140. doi: 10.1371/journal.pbio.1002140
Yang, L., Venneti, S., & Nagrath, D. (2017). Glutaminolysis: a hallmark of cancer metabolism. Annual review of biomedical engineering, 19, 163-194.
ApoE4: La kriptonita de Thor
escrito por trifosfato_3C | 17 diciembre, 2024
Por: Aitor Gil Iglesias, Milagros González Flores y Álvaro Mellado Cuesta
1.Introducción
Seguramente todos nosotros hemos leído que el actor Chris Hemsworth, más conocido como Thor en su papel dentro de la saga de Marvel, ha decidido retirarse del cine temporalmente al haber descubierto que tiene muchas posibilidades de desarrollar Alzheimer. Este hallazgo fue conocido por el actor cuando se encontraba rodando el documental “Limitless” para National Geographic al realizarse una prueba genética por la cual trataba de estudiar cómo su físico se vería afectado con el paso de los años. Sin embargo, estos estudios, que en un primer momento trataron de analizar el estado físico del actor, acabaron revelando la posibilidad de desarrollar Alzheimer al presentar dos copias del gen ApoE4 que hace que su riesgo de padecerla sea entre 8 y 10 veces más. Ahora bien, ¿qué es el Alzheimer y por qué es tan importante este gen en su desarrollo?
Figura I: fotos del actor Chris Hemsworth, en su papel como Thor en la saga Marvel y en la cartelera del nuevo documental que él protagoniza Limitless.
2.-¿Qué es el Alzheimer?
El Alzheimer (EA) es una enfermedad neurodegenerativa lentamente progresiva que se caracteriza por la pérdida de memoria de manera gradual. En ella también se describen otros síntomas como demencia irreversible y afectación global del resto de las funciones cognitivas (pensar, razonar…) que conllevan a un deterioro progresivo y que afectan al funcionamiento laboral y social.
Cabe destacar que la progresión de la enfermedad a nivel cerebral es la misma en todos los pacientes. Comienza en el neocórtex basal temporal, avanza hacia el hipocampo y se extiende por todo el neocórtex.
Figura II: modelo de progresión cerebral de la Enfermedad de Alzheimer. Áreas cerebrales afectadas (en azul) en las distintas fases de la enfermedad (temprana, leve-moderada y severa). Colegio Oficial de Enfermería de Cantabria (2013)
La mayoría de los casos aparecen a partir de los 70 años (“late onset”) , aunque puede haber una aparición temprana en menores de 60 (“early onset”). Afecta a alrededor de 50 millones de personas en el mundo y el principal factor de riesgo es la edad.
La early onset es autosómica dominante y hereditaria y, aunque la mayoría suceden como consecuencia de mutaciones esporádicas sobre el gen de la apolipoproteína (APOE, cromosoma 19), otros casos se deben a mutaciones en los genes que codifican para la proteína precursora amiloide (APP, cromosoma 21, por ello, las personas con Síndrome de Down, trisomía del 21, son más propensos a desarrollar la enfermedad de Alzheimer), para la presenilina 1 (PSEN1, cromosoma 14) y para la presenilina 2 (PSEN2, cromosoma 1).
No obstante, algunos otros factores de riesgo que podrían estar implicados son las enfermedades vasculares, infecciones, factores ambientales y lesiones encefálicas.
Figura III: Relación entre el riesgo de padecer la enfermedad del Alzheimer y la frecuencia alélica de algunos genes de interés. Las frecuencias alélicas más comunes se relacionan con un menor riesgo de sufrir la enfermedad y las menos habituales con un riesgo mayor. El caso de Chris Hemsworth se encuentra entre ambas, el gen de ApoE4 tiene un frecuencia alélica baja y el riesgo de desarrollar la enfermedad es medio-alto, pero como posee dos copias del gen ApoE4, se sitúa en un riesgo mayor y la frecuencia alélica ApoE4/ApoE4 es menor. Lane, et al (2018)
3.Etiopatología
Bioquímicamente, esta enfermedad se produce como consecuencia de dos elementos neuropatológicos: las placas seniles o placas Aβ y los ovillos neurofibrilares (NFT), esenciales para entender cómo se desarrolla el Alzheimer.
Figura IV: Comparación entre el estado del cerebro y las neuronas de una persona sana (imagen A) y una persona con Alzheimer (imagen B). Se puede observar que el principal área afectado sería el hipocampo, el cual se encuentra contraído en personas con EA. Además, estos pacientes presentan una corteza cerebral más encogida y unos ventrículos más grandes. Por su parte, en la EA las neuronas presentan acumulaciones intracelulares de la proteína Tau (marrón) y acumulaciones extracelulares del péptido Aβ (rojo). Breijyeh et al (2020)
3.1.-¿Qué son las placas seniles?
Estas consisten en acumulaciones extracelulares de la proteína beta-amiloide (Aβ) anómala que en principio no es tóxica pero que se agrega con otras proteínas beta-amiloide anómalas hasta formar oligómeros tóxicos que precipitan y se depositan como placas amiloides insolubles y que son realmente tóxicas.
Su síntesis tiene lugar por medio de la proteínaprecursora amiloidea (APP) cuyo gen se encuentra localizado en el cromosoma 21. Esta, es una proteína transmembrana que contiene una región amino-terminal extracelular larga y una región carboxilo terminal intracelular más corta que se sintetiza en el retículo endoplasmático, se glicosila en el aparato de Golgi y por medio de vesículas se inserta en la membrana celular.
Figura V: Estructura de APP no mutada y completa (con todos sus dominios). El dominio E1 actúa como receptor de señales. El péptido beta-amiloide enlaza los dominios extracelulares de la proteína con el dominio intracelular AICD. Una familia de endopeptidasas, las secretasas, cortan la APP, liberando el dominio AICD, que alcanza el núcleo, promoviendo la expresión de diversos genes. En este corte puede liberarse el péptido beta-amiloide y, si se forma en exceso (debido a mutaciones, por ejemplo) dar lugar a la formación de placas amiloides.
Una vez en la membrana celular, esta proteína puede ser procesada fisiológicamente por medio de dos vías:
Vía no amiloidogénica (90% de los casos): por medio de esta vía, la proteína APP es escindida por la enzima α-secretasa en un fragmento extracelular soluble neuroprotector (SAPPα, Soluble APPα) compuesto por 665 aminoácidos. Posteriormente la región no escindida que ha quedado insertada en la membrana, es procesada por la enzima γ-secretasa, quien se encarga de liberar la región C-terminal para posteriormente poder ser degradada. De esta manera, se libera al medio extracelular un péptido de 36-40 aminoácidos denominado P3, que no es neuroprotector, pero tampoco patógeno. El fragmento restante C-terminal se desprende hacia el citosol y se denomina AICD (Amyloid Intracellular Domain), que se degrada enseguida. Este proceso es constitutivo (APP tiene una vida media de 1h) y fundamental para el funcionamiento neuronal.
Vía amiloidogénica (10% de los casos): por medio de esta vía, la enzima β-secretasa o BACE libera un fragmento N-terminal de la proteína APP (SAPPβ) que no es neuroprotector. Posteriormente, el péptido que queda en la membrana será procesado por la enzima γ-secretasa dando lugar a diferentes péptidos que presentan diferentes solubilidades: el péptido Aβ (42 aá) y el AICD, que tiene un tiempo de vida mayor, va al núcleo y favorece la transcripción de genes como la propia APP, GSK3 (Ser/Thr kinasa), BACE, NEP (nefrilisina) y LRP1 (lipoprotein receptor-related protein 1).
Figura VI: Esquema sobre el procesado fisiológico de la proteína APP, enzimas que actúan y productos que se obtienen en la vía no amiloidogénica (sobre fondo azul) y en la vía amiloidogénica en condiciones normales y durante el Alzheimer (sobre fondo rojo). En la vía amiloidogénica, el dominio transmembrana de APP sufre una escisión diferente con respecto la vía no amiloidogénica tanto en condiciones normales como durante el Alzheimer. Como consecuencia se genera un producto de mayor tamaño, el péptido Aβ. En el caso del Alzheimer, mutaciones en Aβ, en APP, en ApoE y en los dominios PSEN1/PSEN2 de la secretasa favorecen el incremento de la via amiloidogénica. Esto hace que se dirija hacia el espacio extracelular y se agregue hasta formar placas amiloides y que pase al interior celular por endocitosis y favorezca la fosforilación de Tau.
Los péptidos más comúnmente formados en la vía amiloidogénica son los péptidos Aβ40, los cuales presentan una estructura en forma de hélice alfa que puede ser fácilmente degradada. Sin embargo, en situaciones patológicas predomina la síntesis del péptido Aβ42 y Aβ43, los cuales son mucho más insolubles, tienden a formar láminas β y a interaccionar entre ellos formando fibrillas que pueden acumularse y formar placas extracelulares amiloides insolubles que se diseminarán por otras estructuras como el hipocampo, la amígdala y la corteza cerebral resultando realmente dañinas.
Figura VII : péptido Aβ-42 agregado, formado placas amiloides. Los residuos poco polares de los aminoácidos tienden a apilarse en paralelo favoreciendo el plegamiento incorrecto en láminas β. Esto genera una estructura insoluble que precipita. Imagen obtenida de Chimera. Código PDB: 5KK3 PDB DOI:10.2210/pdb5KK3/pdb
Ahora bien, ¿por qué es tan importante Aβ?
De manera normal, esta proteína se encarga de modular la transmisión del impulso eléctrico entre las neuronas, por lo que, un mal funcionamiento o no funcionamiento supondría la pérdida de la comunicación neuronal, es decir, una pérdida de sinapsis. Esto se debe a que puede alterar la actividad de quinasas, producir señales de estrés oxidativo, daños mitocondriales e incluso defectos en el transporte axonal que son los que finalmente conducen a una degeneración neuronal. Todo ello es lo que acabaría produciendo en estos pacientes una gran deficiencia cognitiva.
Además, Aβ en bajas concentraciones es quelante de metales, mejora la memoria y el aprendizaje, es antioxidante, interviene en el mantenimiento de la barrera hematoencefálica etc.
Esta acumulación de Aβ anómala en condiciones patológicas, en las que no solo se incluye el Alzheimer, es como consecuencia de las mutaciones en algunos genes como APP, PSEN1 (proteína que se encarga de activar el complejo de la γ-secretasa) y PSEN2 (poco frecuentes), los cuales, afectan al catabolismo y anabolismo de Aβ y como consecuencia producen una rápida progresión de neurodegeneración.
Concretamente, dichas mutaciones afectan al procesamiento alternativo de APP, y como consecuencia se generan fundamentalmente proteínas Aβ truncadas, Aβ42 y Aβ43. Estas presentan residuos poco polares que tienden a apilarse en paralelo induciendo a un plegamiento incorrecto de las láminas beta las cuales, son muy insolubles y al no ser reconocidas por los sistemas de degradación acaban precipitando para formar fibrillas amiloides que generan una gran neurotoxicidad.
Figura VIII : Formación de los péptidos Aβ insolubles. Su agregación da lugar a oligómeros y finalmente acaba originando las placas seniles. Duran-Aniotz, et al (2013)
3.2-¿Qué son los ovillos neurofibrilares (NFT)?
Son lesiones de tipo intracelular formadas por filamentos helicoidales emparejados (PHF) que se producen como consecuencia de la hiperfosforilación de las proteínas Tau y que puede deberse a la aparición de proteínas Aβ, al estrés oxidativo o a mediadores inflamatorios. La formación de estos ovillos producen muerte neuronal por apoptosis.
Estas proteínas se encuentran de manera mayoritaria en neuronas y células de la Glía y son esenciales para promover el ensamblaje de los microtúbulos neuronales así como para mantener la integridad estructural neuronal y por tanto para permitir el crecimiento de los axones y el transporte axonal. Concretamente, la proteína Tau se encarga de facilitar la polimerización de la tubulina para permitir que se formen los microtúbulos.
Figura IX: Estructura de la proteína Tau no mutada. Imagen obtenida con Chimera. Código PDB: 2MZ7. PDB DOI: 10.2210/pdb2MZ7/pdb
El gen que codifica para la proteína Tau, localizado en el gen 17, contiene 16 exones y en el cerebro adulto sufre diferentes tipos de empalme alternativo en los exones 2, 3 y 10 originando finalmente 6 isoformas de Tau. Estas isoformas contienen entre 3-4 repeticiones peptídicas de 31-32 residuos en la región carboxilo terminal que se corresponde con el dominio de unión a los microtúbulos. Así mismo, el estado de fosforilación del gen varía durante el desarrollo y es lo que conduce a un cambio en el empalme del ARN.
Figura X: Regiones donde se puede fosforilar a Tau (señaladas con flechas negras). Las regiones señaladas en rojo son aquellas que se encuentran anormalmente fosforiladas en el Alzheimer, las regiones verdes aquellas en las cuales Tau está fosforilada de manera normal y en azul las fosforilaciones encontradas de Tau en el Alzheimer y en condiciones normales. Por tanto, las regiones señaladas en rojo serían aquellas que no deberían encontrarse fosforiladas y las que conllevan a una agregación de Tau en el Alzheimer. Luna-Muñoz, et al (2012)
Tau en el embrión aparece fosforilada, pero en adultos está muy poco fosforilada. Se puede fosforilar por más de 80 sitios Ser/Thr y 5 de Tyr, gracias a GSK-3, PKA, la cuales son Ser/Thrk o Fyn (TyrK). En la enfermedad de Alzheimer, está hiperfosforilada.
Además, cuando Tau está fosforilada cambia de localización y, en lugar de localizarse preferentemente en los axones, se dirige hacia las dendritas y se lleva a Fyn secuestrada consigo. Fyn fosforila a receptores de glutamato (Glu) (receptores NMDA sobre todo) lo que producirá una muerte neuronal por sobreexcitación.
De esta manera, y dado la importancia de la fosforilación en este gen, se ha podido observar que en los procesos de neurodegeneración Tau sufre una fosforilación anormal irreversible que impide su función normal y su agregación. Como consecuencia, no puede unirse correctamente a los microtúbulos y todo ello junto con la formación de las fibrillas por su agregación, impiden que la neurona pueda transmitir señales eléctricas. Además todo ello favorece un mal transporte axonal de nutrientes, lo cual contribuye a la degeneración sináptica y a la muerte neuronal.
Figura XI: Tinción inmunohistoquimica de ovillos neurofibrilares (marcado en marrón). Castellani, et al (2010)
Además, parece ser que, una vez Tau está fosforilada y ha perdido su estructura, se empieza a transmitir de unas neuronas a otras, induciendo el cambio conformacional patológico en proteínas Tau normales por contacto (como los priones). Gracias a ello, la enfermedad puede propagarse y afectar a diferentes estructuras.
4.-¿Qué es la ApoE?
La ApoE es una proteína de 299 aminoácidos, que está dentro del grupo de las apolipoproteínas, las cuales están implicadas en el metabolismo de los lípidos (forman las lipoproteínas), siendo la ApoE la única que encontramos en el cerebro.
En la ApoE hay varios dominios de unión a destacar, como por ejemplo el de unión a lípidos, al receptor, a heparina o la región bisagra. Todos ellos con gran importancia en las funciones de la ApoE como veremos a continuación.
Sin embargo, el interés reciente hacia la ApoE no es por su papel en el metabolismo de los lípidos, sino por su relación con el péptido Aβ y la proteína Tau, ambos implicados en el inicio y avance de la enfermedad de Alzheimer.
4.1- Relación ApoE – péptido Aβ
La ApoE es sintetizada por astrocitos y la microglía, debido a la alta importancia de los lípidos para el desarrollo y mantenimiento de las neuronas, pero, ¿cuál es su función dentro del metabolismo de lípidos?¿y que nexo tiene esto con el péptido Aβ y la enfermedad de Alzheimer?
FUNCIÓN DE LA ApoE
La ApoE se encuentra circulando por la matriz extracelular nerviosa, y es ahí dónde se une al colesterol, que proviene del interior de las células nerviosas (sale a través de transportadores de la familia ABC), y ambos forman lipoproteína discoidal. Esta se une a recetores celulares para lipoproteínas (LRP1 principalmente) y entra en las células por endocitosis mediada por receptor. Ya en el interior, esta lipoproteína entra en el metabolismo de los lípidos.
Sin embargo, lo más interesante de esto es que el péptido Aβ es capaz de unirse a la ApoE circulante (a su dominio de unión a heparina concretamente), formando una molécula que es capaz de entrar en las células al igual que la lipoproteína que hemos mencionado antes (endocitosis mediada por LRP1).
Una vez dentro, el endosoma donde está la molécula ApoE-péptido Aβ se fusiona con lisosomas, y esto hace que por aumento del pH y acción de proteasas lisosomales ApoE y el péptido Aβ se separen. El destino de cada uno es bien distinto:
ApoE queda en una vesícula que se fusionará con la membrana plasmática, permitiendo que salga al exterior y se recicle.
El péptido Aβ es sustrato para proteasas lisosomales, por lo que acaba siendo degradado.
FIGURA XII: Modelo explicativo sencillo de la degradación del péptido Aβ a través de su unión a ApoE. Imagen creada en Paint
Pero yendo más allá, lo que más ha despertado el interés científico de todo esto y lo que está relacionado con el caso concreto de Chris Hemsworth son las distintas isoformas de ApoE que aparecen en las poblaciones, cada una de ellas con un papel distinto en la predisposición a sufrir Alzheimer.
4.2- Isoformas de ApoE y su importancia en el Alzheimer
Hay 3 isoformas de la ApoE, las cuales sólo se diferencian en los residuos 112 y 158:
ApoE2: posee cisteínas (en 112 y 158). Se la considera «protectora» frente a la enfermedad de Alzherimer. Se cree que su eliminación del péptido Aβ es más eficaz.
ApoE3: posee cisteína (en 112) y arginina (en 158). Es la isoforma mayoritaria en la población y se considera que su papel es neutro frente a la enfermedad de Alzheimer.
ApoE4: posee argininas (en 112 y 158). Su presencia aumenta la probabilidad de sufrir la enfermedad de Alzheimer hasta 4 veces si aparece en heterocigosis y hasta 10 veces si aparece en homocigosis.
FIGURA XIII: Distintas isoformas de ApoE con sus diferencias estructurales. Fernandez, et al (2011)FIGURA XIV: ApoE2: (2 Cys en verde, en posiciones 112 y 158). Imagen obtenida con Chimera. Código PDB: 1NFO. PDB DOI: 10.2210/pdb1NFO/pdbFIGURA XV: ApoE3: (Cys en verde, en posición 112 y Arg en rosa en posición 158). Imagen obtenida con Chimera. Código PDB: 1NFN. PDB DOI: 10.2210/pdb1NFN/pdbFIGURA XVI: ApoE4: (2 Arg en rosa, en posiciones 112 y 158). Imagen obtenida con Chimera. Código PBD: 1B68. PDB DOI: 10.2210/pdb1B68/pdb
Pero, ¿por qué ApoE4 aumenta tanto la probabilidad de desarrollar Alzheimer?, ¿en qué afecta el cambio de tan solo 2 aminoácidos a una proteína que posee 299? Ahora mismo te lo explicamos
4.3- ApoE4
Su única diferencia estructural frente a la isoforma protectora es el cambio de dos cisteínas (aminoácidos polares sin carga), por dos argininas (aminoácidos polares con carga positiva). En principio parece un cambio insignificante, pero este pequeño cambio hace que la ApoE4 tenga una conformación algo más «cerrada» que el resto de ApoE, afectando más de lo que creemos en su función.
La conformación que adopta ApoE4 hace que se una más deficientemente a sus receptores, por lo que ApoE4 se encuentra durante más tiempo circulante por la matriz extracelular. Al disminuir la endocitosis mediada por receptor, disminuye la proteólisis intracelular y para compensarlo, aumenta la proteólisis extracelular.
Esta proteólisis favorece la acumulación del péptido Aβ, su unión con otros péptidos Aβ (formando oligómeros Aβ) y el inicio y desarrollo de la formación de placas amiloides y de la enfermedad de Alzheimer. De hecho se han descubierto restos de esta ApoE en las placas amiloides.
Pero esto no es todo, ya que recientemente se ha visto que ApoE también interactúa con la proteína Tau, una proteína muy importante en el desarrollo de la enfermedad de Alzheimer y cuya hiperfosforilación explica muchos de los signos clínicos de la enfermedad.
4.4- Relación ApoE – proteína Tau
Se ha visto que ApoE está relacionada con la acumulación de Tau y Tau fosforilada, especialmente ApoE4 es quien favorece en mayor medida está acumulación, la cual deriva en la formación de los ovillos neurofibrilares. Para explicar está relación de manera más detallada vamos a presentaros un experimento realizado con ratones en 2017.
En este experimento se quiso conocer más acerca del papel de ApoE en la neurodegeneración a través de Tau. Para ello se utilizaron ratones que sobreexpresaban Tau humana (ratones P301S) y las distintas isoformas de ApoE, creando distintos grupos de ratones:
Tau-ApoE2 (TE2): expresaban Tau y ApoE2.
Tau-ApoE3 (TE3): expresaban Tau y ApoE3.
Tau ApoE4 (TE4): expresaban Tau y ApoE4. Es el grupo de ratones en el que mayor neurodegeración se observó a los 9 meses.
Sin Tau y con ApoE (KI): no se observó neurodegeneración a los 9 meses. Esto confirmaba que ApoE por sí sola no ocasiona neurodegeneración.
Con Tau y sin ApoE (TEK0): se observó neurodegeneración a los 9 meses, pero en menor nivel que en cualquiera de los grupos con Tau y las distintas formas de ApoE.
Con esto se confirmó que ApoE (especialmente ApoE4) juega un papel importante en la neurodegeneración a través de Tau. Además en este experimento se observó que ApoE4 no influye en la síntesis de Tau, sino que la acumulación de Tau se debe a que ApoE4 afecta a su degradación por autofagia.
Por último se vio que cuando había patología de Tau (hiperfosforilación y acumulación), ApoE4 provoca neuroinflamación, promoviendo la activación de la microglía y la expresión de genes A1 en astrocitos.
FIGURA XVII: Resumen de los efectos de ApoE4 a nivel cerebral, metabólico, vascular y hormonal. ApoE4 favorecería la fosforilación de Tau, los procesos inflamatorios y la agregación de Aβ. Por el contrario, disminuye la eliminación de Aβ, la función neuronal sináptica y vascular, la señalización de la insulina y VEGF (factor de crecimiento endotelial vascular), el metabolismo mitocondrial y el transporte y aclaramiento de lípidos y colesterol. Safieh et al (2019)
4.5- Genes implicados
En la prueba genética que se realizó el actor Chris Hemsworth, se analizo el gen que da lugar a la síntesis de la ya mencionada ApoE. El gen de la ApoE está formado por 4 exones y se encuentra en el cromosoma 19.
Las mutaciones de mayor interés en este gen son las que afectan a los codones que dan lugar a los aminoácidos 112 y 158, y por tanto a las distintas isoformas ya vistas (ApoE2, ApoE3 o ApoE4) y su influencia en la probabilidad de desarrollar la enfermedad de Alzheimer.
FIGURA XVIII: Representación del gen de la ApoE y su lugar en el cromosoma. Dibujo de la proteína ApoE, incluyendo sus principales dominios y tabla donde se compara el genotipo y aminoácidos que distinguen a las tres isoformas. Abondio, et al (2019)
5.Referencias
Breijyeh, Z., & Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules (Basel, Switzerland), 25(24), 5789. https://doi.org/10.3390/molecules25245789
Lane, C. A., Hardy, J., & Schott, J. M. (2018). Alzheimer’s disease. European journal of neurology, 25(1), 59–70. (2020) https://doi.org/10.1111/ene.13439
Lin, Y. T., Seo, J., Gao, F., Feldman, H. M., Wen, H. L., Penney, J., Cam, H. P., Gjoneska, E., Raja, W. K., Cheng, J., Rueda, R., Kritskiy, O., Abdurrob, F., Peng, Z., Milo, B., Yu, C. J., Elmsaouri, S., Dey, D., Ko, T., Yankner, B. A., … Tsai, L. H. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron, 98(6), 1141–1154.e7. (2018) https://doi.org/10.1016/j.neuron.2018.05.008
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., … & Holtzman, D. M. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature, 549(7673), 523-527. (2017)
Spinney, L. The forgetting gene: for decades, most researchers ignored the leading genetic risk factor for Alzheimer’s disease. That is set to change. Nature, 510(7503), 26-29. (2014) https://doi.org/10.1038/510026a
Figura III: Lane, C. A., Hardy, J., & Schott, J. M. Alzheimer’s disease. European journal of neurology, 25(1), 59–70. (2018) https://doi.org/10.1111/ene.13439
Figura IV: Breijyeh, Z., & Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules (Basel, Switzerland), 25(24), 5789. (2020)
Figura VIII: Duran-Aniotz, C., Moreno-Gonzalez, I., & Morales, R. Agregados amiloides: rol en desórdenes de conformación proteica. Revista médica de Chile, 141(4), 495-505. (2013)
Figura X: Luna-Muñoz, J., Zamudio, S., De La Cruz, F., Minjarez-Vega, B., & Mena, R. Acción protectora de la proteína tau en la enfermedad de Alzheimer. Revista Mexicana de Neurociencia, 13(3), 160-167. (2012)
Figura XI: Castellani, R. J., Rolston, R. K., & Smith, M. A. Alzheimer disease. Disease-a-month : DM, 56(9), 484–546 (2010)
Figura XIII: Fernandez, Celia & Hamby, Mary & McReynolds, Morgan & Ray, William. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Frontiers in Aging Neuroscience. (2011) 11. 10.3389/fnagi.2019.00014
Figura XVII: Safieh, M., Korczyn, A. D., & Michaelson, D. M. ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC medicine, 17(1), 1-17. (2019)
Figura XVIII: bondio, P., Sazzini, M., Garagnani, P., Boattini, A., Monti, D., Franceschi, C., Luiselli, D., et al. The Genetic Variability of APOE in Different Human Populations and Its Implications for Longevity. Genes, 10(3), 222. (2019)
Veneno: nuestro aliado
escrito por lorea_3B | 17 diciembre, 2024
Por Lorea Ayape, 3º Biología Sanitaria. Universidad de Alcalá.
Introducción
Desde la introducción de la insulina hace casi un siglo, se han comercializado más de 80 fármacos peptídicos para el tratamiento de una amplia gama de enfermedades, como la diabetes, el cáncer, la osteoporosis, la esclerosis múltiple, la infección por VIH y el dolor crónico (Muttenthaler et al., 2021). Los grandes avances en biología molecular y química peptídica siguen haciendo progresar este campo, así como la venómica integrada, una estrategia emergente que crea nuevas vías para el descubrimiento de fármacos peptídicos (Muttenthaler et al., 2021).
A continuación, explicaremos un ejemplo de cómo los venenos, esas sustancias dañinas que presentan ciertos animales de los que muchos huimos despavoridos, pueden convertirse en nuestros aliados con ayuda de la ciencia, pues contienen determinados péptidos que podemos usar en nuestro beneficio. Para ello, le propongo al lector que visualice a nuestra protagonista: la tarántula, ese animal grande y peludo de 8 patas que tan rápido se mueve con, aparentemente, enormes ganas de morder a su víctima humana cuando nos encontramos frente a él. No obstante, las tarántulas tienen poca importancia clínica debido a que no son muy agresivas y evitan el contacto con el entorno humano (Murray, Rosenthal and Pfaller, 2021). Sin más dilación, comencemos a tratar esta curiosidad científica y, quizás, incluso les terminemos cogiendo un poquito de cariño a las arañas.
Tarántula. Foto: Pixabay
Necesidad de un nuevo analgésico
Recientemente, se han descubierto determinados componentes del veneno de tarántulas capaces de bloquear la ruta por la que se envían las señales de dolor al cerebro.
El estudio de Klint et al. ha sido realizado debido a la gran necesidad clínica que tenemos de crear analgésicos eficaces para tratar el dolor crónico, puesto que la mayoría de los fármacos disponibles actualmente tienen una eficacia limitada y efectos secundarios que limitan la dosis (Klint et al., 2015). Por su parte, el dolor crónico es un importante problema de salud en todo el mundo que afecta a una media del 15% de las personas adultas (Gaskin and Richard, 2012). Además, tan solo en Estados Unidos, la carga económica anual que supone el dolor crónico es de unos 600.000 millones de dólares, lo que supera el coste económico en conjunto del cáncer, la diabetes y los accidentes cerebrovasculares (Gaskin and Richard, 2012).
En definitiva, la prevención en dolor crónico es muy importante al ser un problema muy extendido, producir una inestimable cantidad de sufrimiento y constituir un coste económico enorme (Rodríguez-Marín, Bernabeu and Hofstadt, 2021).
Representación de múltiples puntos donde puede afectar el dolor crónico y su definición. Imagen creada por Lorea Ayape con BioRender.
Fisiología de los canales de sodio dependientes de voltaje
La comunicación celular en el sistema nervioso se basa en fenómenos de señalización eléctrica y química mediados por canales iónicos (Boron, 2017). En las células con dicha propiedad es posible desencadenar un impulso eléctrico denominado potencial de acción, que actúa como una señal capaz de propagarse a grandes distancias a lo largo de las fibras nerviosas o musculares. La conducción de los potenciales de acción permite que se pueda transmitir información a través de nervios desde los órganos hacia el cerebro (Boron, 2017). En el ejemplo que tratamos, las señales que se envían al cerebro viajan mediante la vía del dolor.
Por otro lado, debemos conocer los canales de sodio dependientes de voltaje (NaV), unas proteínas situadas en la membrana de estas células especiales que permiten el paso de los iones sodio a su través. Si no hay canales de sodio, no se producen potenciales de acción y, por tanto, tampoco se transmiten las señales.
Representación de transmisión del impulso nervioso: Se alcanza el umbral, por lo que se abren los canales de sodio sensibles al voltaje, de manera que entra sodio (Na+) en la neurona. A continuación, se cierran los canales de sodio y se abren los de potasio (K+), saliendo éste desde el interior de la neurona. Después se inactivan los canales de potasio y se alcanza el potencial de reposo. Finalmente, se activa la bomba sodio-potasio con gasto de ATP (energía) para restablecer los niveles iniciales de los iones. Diagrama elaborado a partir de un modelo de BioRender.
En concreto, el hNaV1.7 es un canal de sodio presente en las neuronas nociceptivas. Estudios anteriores ya han demostrado que las mutaciones de pérdida de función en el gen que da lugar al hNaV1.7 generan una insensibilidad a todas las formas del dolor (Cox et al., 2006).
Estos hallazgos sugieren que el hNaV1.7 podría ser una buena diana en la investigación de nuevos fármacos para el tratamiento del dolor (Boron, 2017). Es por todo ello que Klint et al. se han centrado en los canales de sodio, específicamente en el hNaV1.7.
Mutaciones del canal de Na+ en enfermedades genéticas humanas. Los símbolos muestran lugares en una subunidad α del canal de Na+ genérico con los 4 dominios (Dominio I-IV), cada uno con 6 segmentos transmembrana (Drenth and Waxman, 2007). C indica el extremo C-terminal de la cadena peptídica, mientras que N indica el extremo N-terminal de la cadena peptídica. Se representan algunos ejemplos de las localizaciones de mutaciones pertenecientes a tres genes de canales de Na+ (SCN4A, SCN5A y SCN9A) (Boron, 2017). Todas estas mutaciones patológicas dan lugar a cambios o deleciones de aminoácidos, salvo en el caso de la insensibilidad al dolor, que se debe a mutaciones que ocasionan la falta de expresión funcional del canal NaV1.7 (Boron, 2017). Imagen obtenida de Boron, W. F. (2017) ‘Excitabilidad eléctrica y potenciales de acción’, in Fisiología médica. third, pp. 172–189.
Relación entre las arañas y los analgésicos
Las arañas son el grupo que más animales venenosos alberga. A su vez, existe un total de 9 millones de péptidos de veneno de araña, de los cuales solo se ha explorado un 0,01% (Klint et al., 2015); por lo tanto, estos venenos suponen una gran fuente para buscar nuevos moduladores del canal hNaV1.7.
En total se analizaron los venenos de 206 especies de arañas, de los cuales un 40% presentaban algún compuesto capaz de bloquear al canal hNaV1.7 (Klint et al., 2015). Finalmente, la estructura del Hd1a (un péptido de la araña Haplopelma doriae) demostró que se trataba del mejor candidato a posible analgésico, puesto que presenta una gran estabilidad química, térmica y biológica (Klint et al., 2015). Además, inhibió el hNaV1.7 con un alto nivel de selectividad sobre todos los demás subtipos, excepto el hNaV1.1 (Klint et al., 2015).
Gráfico de los venenos de araña estudiados por Klint et al. Elaborado por Lorea Ayape mediante Word y BioRender.
Otros canales de sodio como diana de analgésicos
A partir de una serie de venenos que activan neuronas sensoriales de ratón, Osteen et al. aislaron dos péptidos (Hm1a y Hm1b) del veneno de la tarántula Heteroscodra maculata que activaban tan sólo a unas pocas neuronas sensoriales (Osteen et al., 2016). Después comprobaron que el efecto de dichos péptidos era bloqueado mediante la tetrodotoxina (TTX), la cual actúa inhibiendo a los canales de sodio activados por voltaje (NaV) (Osteen et al., 2016). Esto llevó al descubrimiento de que los péptidos aislados son agonistas de los canales NaV1.1 y, a su vez, de que éstos están implicados en la señalización del dolor, lo cual se desconocía hasta entonces (Osteen et al., 2016).
También se demostró que el NaV1.1 regula la excitabilidad de unas fibras nerviosas que transmiten señales de dolor a la médula espinal, con lo que este canal iónico también se convirtió en una posible diana analgésica (Osteen et al., 2016). De hecho, se ha comprobado que los inhibidores del NaV1.1 reducen la hipersensibilidad mecánica en varios modelos de dolor visceral crónico (Salvatierra et al., 2018).
Es decir, en este caso los péptidos de araña mencionadas no se usarían como analgésico contra NaV1.1 (como veíamos en el caso anterior), puesto que realmente contribuyen a que dichos canales produzcan su efecto. No obstante, Hm1a y Hm1b permitieron descubrir que el NaV1.1 está relacionado con la señalización del dolor, posicionándolo así como un objetivo más contra el que aplicar analgésicos.
Conclusiones
El proceso de elaboración de un nuevo fármaco requiere muchísimos años, no solo por las fases que debe superar dicho medicamento hasta aparecer en las farmacias, sino también por el trabajo previo que implica detectar la diana correcta y el compuesto capaz de dirigirse hasta ella afectándola específicamente, como hemos podido comprobar.
Por su parte, todavía quedan millones de péptidos de veneno sin ser explorados, debido a la lentitud del método tradicional de descubrimiento, en el que las fracciones crudas de veneno se analizan frente a dianas conocidas, seguidas de purificación y secuenciación para dilucidar la secuencia de una diana (Muttenthaler et al., 2021).
En esta entrada del blog nos hemos centrado en algunos estudios con péptidos de arañas, pero los escorpiones y los caracoles cono también proporcionan una de las más ricas diversidades químicas (Muttenthaler et al., 2021). Afortunadamente, los nuevos avances tecnológicos están sentando las bases de un potente enfoque denominado «venómica integrada» (Muttenthaler et al., 2021).
Los compuestos de veneno pueden utilizarse como herramientas farmacológicas, terapéuticas, insecticidas o para combatir vectores de enfermedades humanas u organismos patógenos (Herzig et al., 2020). Imagen creada por Lorea Ayape con Canva.
Referencias
Bonica, J. J. (1976) ‘Organization and function of a multidisciplinary pain clinic’, in Pain. Springer, pp. 11–20.
Boron, W. F. (2017) ‘Excitabilidad eléctrica y potenciales de acción’, in Fisiología médica. third, pp. 172–189.
Cox, J. J. et al. (2006) ‘An SCN9A channelopathy causes congenital inability to experience pain’, Nature, 444, pp. 894–898.
Drenth, J. P. H. and Waxman, S. G. (2007) ‘Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders’, Journal of Clinical Investigation, 117(12), pp. 3603–3609. doi: 10.1172/JCI33297.
Gaskin, D. J. and Richard, P. (2012) ‘The economic costs of pain in the United States’, Journal of Pain, 13(8), pp. 715–724. doi: 10.1016/j.jpain.2012.03.009.
Herzig, V. et al. (2020) ‘Animal toxins — Nature’s evolutionary-refined toolkit for basic research and drug discovery’, Biochemical Pharmacology, 181(May), p. 114096. doi: 10.1016/j.bcp.2020.114096.
Klint, J. K. et al. (2015) ‘Seven novel modulators of the analgesic target NaV1.7 uncovered using a high-throughput venom-based discovery approach’, British Journal of Pharmacology, 172(10), pp. 2445–2458. doi: 10.1111/bph.13081.
Li, T. and Chen, J. (2018) ‘Voltage-Gated Sodium Channels in Drug Discovery’, Ion Channels in Health and Sickness. doi: 10.5772/intechopen.78256.
Murray, P. R. ., Rosenthal, K. S. . and Pfaller, M. A. (2021) ‘Artrópodos’, in Elsevier (ed.) Microbiología médica. ninth, pp. 791–807.
Muttenthaler, M. et al. (2021) ‘Trends in peptide drug discovery’, Nature Reviews Drug Discovery, 20(4), pp. 309–325. doi: 10.1038/s41573-020-00135-8.
Osteen, J. D. et al. (2016) ‘Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain’, Nature, 534(7608), pp. 494–499. doi: 10.1038/nature17976.
Rodríguez-Marín, J., Bernabeu, P. and Hofstadt, C. J. van-der (2021) ‘Dolor crónico, enfermedades crónicas y enfermedades terminales’, in Psicología médica. second, pp. 391–412.
Salvatierra, J. et al. (2018) ‘NaV1.1 inhibition can reduce visceral hypersensitivity.’, JCI insight, 3(11). doi: 10.1172/jci.insight.121000.
¿Por qué el número de la suerte es el 7?. Sobre dados, entropía, vida y orden.
escrito por C. Menor-Salvan | 17 diciembre, 2024
C. Menor-Salván. UAH. Actualizado Octubre 2023
El concepto de entropía y su relación con la Bioquímica es uno de los más complicados de entender por parte de los estudiantes. En este artículo, vamos a tratar, sin entrar en formalismos matemáticos, de aclarar el concepto de entropía, el significado del Segundo Principio de la Termodinámica y la relación con la vida y el metabolismo.
Introducción: Entropía NO es desorden
El concepto de entropía, una de las funciones de estado termodinámicas (es decir, una función que describe las características térmicas y energéticas de un sistema), es uno de los que más dificultades presenta para su comprensión. En mi enfoque de la enseñanza de la Bioquímica, la entropía tiene un papel esencial, pues es una de las claves que explican la propia vida y sus estructuras. La entropía es esencial para entender el funcionamiento de las enzimas y para entender por qué el metabolismo no es una colección de reacciones diseñada para fastidiar a los estudiantes, sino que tiene una lógica comprensible.
Una de las razones por las que los estudiantes tienen dificultades con el concepto de entropía es debido a una simplificación confusa que se transmite constantemente en la enseñanza: que la entropía es una medida del desorden. Mayor desorden=mayor entropía. En un texto de 2º de Bachillerato he podido leer:
La entropía S es una magnitud que mide el grado de desorden de un sistema físico o químico
Esta definición es errónea. Además suele acompañarse con imágenes como esta:
Ilustrando la supuesta relación entre entropía y desorden. Este tipo de imágenes DEBEN EVITARSE al introducir la entropía (en esta imagen, podríamos decir que la imagen de la derecha tiene mayor entropía, pero no por la razón que imaginas)
¿cómo puedo explicar a los estudiantes que la entropía es generadora de orden, y que el orden o estructura biológica es resultado del incremento de entropía, si previamente les hemos engañado con una simplificación errónea?.
Tratando de hacerlo lo más simple y conceptual posible y sin entrar en las matemáticas de la Termodinámica formal, vamos a buscar alternativas que nos permitan acercarnos al significado de la entropía y a entender por qué la entropía genera el orden biológico. Ello es necesario para entender, a su vez, por qué el metabolismo es tal como es.
Dados y estados: buscando otras analogías para entender la entropía
¿sabes por qué el 7 es considerado un número de suerte?. ¿si lanzas DOS dados, crees que hay la misma probabilidad de que salga cualquiera de los resultados posibles (la suma de lo que sale en cada dado)?. Hagamos el experimento: si lanzamos dos dados, el resultado puede ser desde 2 hasta 12. Si los lanzamos muchas veces (he estado una tarde de sábado haciéndolo), obtendremos una distribución como esta:
Distribución de frecuencias en el lanzamiento de dos dados de seis caras. Resultado experimental
Según esta distribución, el 7 es el número mas probable. La razón de la preferencia por el 7 en el lanzamiento de dos dados es, en realidad, bastante obvia. Podemos usarla como una analogía del concepto de entropía: la probabilidad de obtener el número 7 es la mayor, por lo que el número 7 es el estado de mayor entropía. 2 y 12 representan los estados de mínima entropía, razón por la cual son las apuestas mas arriesgadas. Antes de explicar por qué es así, vamos a dejarnos enseñar por el físico aleman Arnold Sommerfeld.
¿por qué encendemos una estufa en invierno [igual este invierno lo vamos a tener difícil…]. Una persona común dirá: para que la habitación esté mas caliente; un físico se expresará hablando de suministrar energía. En este caso, tendrá razón el profano y no el físico
Según Sommerfeld, este ejemplo lo proponía el físico suizo Robert Emden. El caso es que, en efecto, encendemos la estufa para que la habitación esté caliente. Este calor se disipa constantemente a través de las ventanas y las paredes; por ello si apagamos la estufa, la temperatura comienza a disminuir. Aquí no os estoy descubriendo nada nuevo.
Como la habitación es mayoritariamente aire, sabemos que la energía contenida va a depender de la temperatura. Esto se expresa a través de esta ecuación:
Esta ecuación nos dice que la energía U contenida en la habitación es proporcional al número de átomos que hay en la habitación, N, y a la temperatura de la habitación, T. Parece lógico: cuanta más temperatura, más energía.
También sabemos que, si calentamos un gas, va a sufrir o un aumento de presión o una expansión. Como en la habitación la presión va a ser constante, mantener la temperatura constante implica que vamos a mantener constante la energía contenida en la habitación. Pero, mientras la estamos calentando, el aire se va a expandir, saliendo por las rendijas e intercambiándose con el exterior.
¿qué ocurriría si la habitación estuviera perfectamente aislada? Lógicamente, al no poder expandirse, aumentaría la presión y la temperatura. Pero, una habitación normal es un sistemaabierto, es decir, puede intercambiar energía con el exterior, y también materia, pues hay intercambio de aire a través de rendijas. Por lo que el aislamiento perfecto no va a existir.
Rudolf Clausius nos explicó que la variación de entropía de un sistema es el cociente entre el calor suministrado dividido entre la temperatura. Es decir, la entropía no es una medida de desorden, sino una medida de la relación entre energía suministrada y temperatura.
Como queremos mantener nuestra habitación a temperatura y presión constante (no queremos morir, y hay rendijas, por lo que el aire puede expandirse), hemos deducido, a partir de la ecuación anterior y de las propiedades de los gases, que a presión constante, la variación de entropía en nuestra habitación es:
Es decir, que la variación de entropía que tenemos en la habitación es proporcional al incremento de la temperatura desde A a B, y al incremento de volumen del aire desde A a B. Si aumentamos la temperatura, entonces, aumentamos la entropía. n es el número de moles de aire, y cv es el calor específico. Idealmente, una vez que hemos alcanzado una temperatura constante, ya no hay aumento de entropía. Y por tanto, no sería necesario suministrar más calor. Observad que no hablo de orden. El incremento de entropía va asociado al incremento de temperatura y/o volumen. Las moléculas del aire ¿están igual de ordenadas o más ordenadas?. No lo se. Pero si sabemos que, si aumentamos el volumen, por ejemplo, al calentar, les estamos dando a nuestras moléculas más libertad para moverse. Vamos a volver a esta idea de ‘libertad’ más adelante.
Pero, en nuestra habitación ocurre un proceso irreversible: el calor se disipa al exterior de dos formas: irradiándose a través de los materiales y las ventanas, y mediante intercambio de aire caliente del interior con aire frío del exterior.
El famoso dibujo de I. Prigogine, que ilustra la el Segundo Principio de la Termodinámica aplicado a sistemas abiertos. El sistema (habitación), produce entropía y ésta siempre va a ser positiva. O, como mucho, va a ser cero. Pero, hay un proceso de disipación a través de las paredes, y asociado a ese proceso hay una variación de entropía también.
El intercambio de materia y energía de la habitación (nuestro sistema) con sus alrededores, implica que tenemos que descomponer la entropía en dos términos: ΔiS es la entropía generada en el interior de la habitación, que es siempre cero o positiva; ΔeS es la entropía del proceso de intercambio con el exterior y puede ser negativa, cero o positiva. Como la entropía es el cociente entre calor y temperatura, y queremos mantener la temperatura y presión constante en la habitación, entonces:
La temperatura del exterior de la habitación Te es menor que la temperatura del interior Ti. Por tanto, si perdemos todo el calor, o sea, Qi=Qe, entonces el término de entropía de intercambio con el exterior es mayor en valor absoluto. Como el intercambio con el exterior está sacando calor de la habitación, entonces, el término es negativo. Por tanto, la variación global ΔS=ΔiS–ΔeS<0. Hemos visto antes que la entropía depende de la temperatura, por lo que si sacamos el mismo calor que metemos, la habitación se enfría hasta que Ti=Te. Esto es lo que ocurre en una habitación cualquiera: en el momento que apagamos la calefacción, la habitación se enfría. Al final, Qi se igualará con Qe. La clave para mantener la habitación caliente es suministrar energía constantemente y llegar a un estado estacionario en el que Qi>Qe durante un lapso de tiempo.
Total, que para mantener la temperatura constante, y, por tanto, ΔS=0, necesitamos que Qi>Qe, es decir, suministramos más energía de la que perdemos durante el tiempo que queremos estar calientes. Lógico, ¿no?. Por ello nos interesa minimizar Qe, utilizando aislamientos, de modo que podamos minimizar Qi, y reducir la factura de la luz. Aquí voy a añadir una nota: estamos simplificando mucho la discusión, pues nos estamos centrando en el aire, un gas, y además lo consideramos un gas ideal. Cuando hablemos de reacciones químicas, cambios de fase, etc. la discusión se complica, y hablaremos de energía libre, entalpía y entropía.
El término ΔeS negativo lo denominó Schrödingerentropía negativa o neguentropía: la entropía asociada al proceso de disipación. Es decir, estamos neutralizando la acumulación de entropía en la habitación al calentarla, sacándola fuera y aumentando la entropía del exterior.
Pérdidas de calor de una casa. Estas pérdidas representan la entropía negativa: aumentan la entropía del exterior, disminuyendo la entropía del interior.
Esto es lo que define el SEGUNDO PRINCIPIO DE LA TERMODINÁMICA: la entropía total del universo (nuestra habitación + el exterior) siempre aumenta. Por ello, para poder mantener la entropía de la habitación constante (que mantenga su temperatura y presión constantes) necesitamos estar constantemente generando calor en ella y aumentar la entropía del universo.
Nuestra habitación (sistema, puede ser el salon de casa o una célula) y el exterior. Dentro de la habitación aumentamos la entropía (enciendiendo el radiador por ejemplo). Las condiciones de contorno controlan el flujo de entropía con el exterior. Si ambos se igualan, la presión y temperatura de la habitación se mantendrán constantes. Eso requiere un gasto de energía.
Con esto hemos explicado físicamente lo que todos sabemos por experiencia: que si apago el radiador, la habitación se enfría. Por este principio, los ingenieros han hecho enormes esfuerzos en reducir los procesos irreversibles en máquinas y edificios, tales como reducir la disipación de calor mediante aislamiento, disminuir el rozamiento y desgaste en los motores, etc. En la vida real, como vamos a tener procesos irreversibles, siempre tenemos que consumir energía para ‘pagar’ esta especie de impuesto entrópico. Esta es la razón por la que no existe el móvil perpetuo y por la que, hagamos lo que hagamos, siempre se va a enfriar la casa al apagar la calefacción.
Aplicación en la bioquímica
La discusión anterior se puede aplicar exactamente igual a un organismo vivo, ya sea una célula o un animal.
En 1992 se publicó un interesante experimento: los investigadores midieron, usando un calorímetro, la producción de entropía de cerditos en una granja y observaron que la producción de entropía iba creciendo durante el periodo infantil, hasta alcanzar un máximo. Cuando el cerdito entraba en edad adulta y cesaba su desarrollo, la producción de entropía disminuía, e iba disminuyendo gradualmente hasta la vejez.
Es decir, mientras estaba en proceso de morfogénesis activa, generando estructura biológica, la disipación de entropía crecía. En la edad adulta entra en estado estacionario.
La ecuación que vimos antes:
la podemos expresar así:
En la que la velocidad o la variación de la producción de entropía es el balance entre la producción de entropía interna del sistema (es decir, debida al metabolismo y procesos vivos del cerdito) y la disipación de energía al entorno en forma de calor (Q) que es una función que varía con el tiempo (estamos en un sistema dinámico), en relación con la temperatura del sistema. Aquí, el calor Q no hay que confundirlo con temperatura. El calor engloba la irradiación de energía y la disipación de energía transportada por moléculas, como el agua, CO2 y otras moléculas emitidas por el organismo, que ‘esparcen’ o ‘dispersan’ la energía. En estado estacionario, ambos términos se igualan.
Por el contrario, si la tasa de producción interna de entropía es menor que la disipación de calor, la variación de entropía es negativa. Tenemos la ‘neguentropía’ y, en el proceso, es cuando se produce la auto-organización de las estructuras de los organismos vivos.
Ilya Prigogine entra en escena: explicando la vida con el teorema de mínima entropía, sistemas no lineales y estructuras disipativas.
Resulta que estamos disminuyendo la entropía de nuestro sistema, mediante la disipación de energía y el aumento de entropía del exterior del sistema. Hay varios tipos de este sistema peculiar que, espontáneamente, se ha alejado del equilibrio.
Uno de estos sistemas es el metabolismo.
El metabolismo es un conjunto conectado y coordinado de reacciones químicas que permite, según el teorema de mínima entropía de Prigogine, minimizar la entropía producida durante la disipación de energía libre, maximizando la negentropía. Ello lleva a la reducción de entropía interna del sistema y la aparición de estructuras emergentes. Este tipo de sistemas se denominan estructuras disipativas (mas adelante volveremos a este concepto).
El metabolismo permite que los organismos vivos se ‘alimenten de entropía negativa‘ (como expresó Schrödinger). Este concepto aún está en discusión y también se ha sugerido que la evolución marca un camino hacia la maximización de la producción de entropía, provocando un aumento de complejidad que aumente la neguentropía para mantener la estabilidad dinámica. En fin, aquí entramos en un área muy compleja. Tanto que lo llamamos la Ciencia de la Complejidad. Pero como sumario, podemos decir que hay una diferencia fundamental entre un sistema vivo y una máquina: en las máquinas, los ingenieros quieren reducir los procesos irreversibles para aumentar su eficiencia disminuyendo la entropía. En la vida, en cambio, DEPENDE de los procesos irreversibles y la producción de entropía es la que hace que funcione. ¿por qué depende de ellos? porque la vida surge a costa de alejarse del equilibrio y de aumentar la disipación de entropía (el segundo término en la última ecuación)
Un ejemplo relativamente simple de estructura disipativa, muy similar al metabolismo, es la reaccion de Belousov-Zhabotinsky:
La aparición espontánea de patrones durante la reacción, como los de la imagen, que son zonas con diferentes parámetros físicos y químicos, es resultado de la disminución de entropía, mediante la transformación de la energía contenida en el ‘combustible’ de la reacción y el aumento de entropía del exterior. Es una estructura disipativa que depende del aumento de entropía del exterior. Por ello, una estructura disipativa, tal como el metabolismo, sólo puede producirse en un sistema abierto. En un sistema cerrado en el que no pueda intercambiar materia y energía con el exterior, el sistema ‘muere’ rápidamente.
¿qué tienen en común la reacción de Belousov con el metabolismo? Para que un sistema químico tenga estas propiedades, deben establecerse una serie de ciclos y reacciones encadenadas, en los que se alternen reacciones químicas reversibles e irreversibles y se produzca un fenómeno de autocatálisis o de catálisis recíproca. Aquí ya entramos en una complejidad excesiva (alguien pillará el chiste), por lo que lo dejamos en éste punto. Pero, la idea es que para que se de la vida tal como la conocemos, el metabolismo tiene que ser tal como es, y debe contener un sistema de reacciones como la glucólisis, que hacen posible que la disipación de entropía permita formar las estructuras biológicas.
Alguien podría preguntar: entonces, termodinámicamente, ¿la reacción de Belousov es vida?. No. La diferencia fundamental es que un organismo vivo es capaz de regular internamente su comportamiento estructural. La estructura en la reacción de Belousov es resultado de los cambios e influencia de su entorno, sin control interno. Este control estructural y regulación interno, que está relacionado con la información biológica, es lo que llamamos biología molecular. Así, podemos establecer un criterio de demarcación entre la biología molecular y la bioquímica, si queremos.
Reacción de Belousov-Zhabotinsky: un modelo del metabolismo.
Vamos a ver de nuevo el ejemplo de la reacción de Belousov:
Belousov descubrió, en 1951, su famosa reacción tratando de encontrar un modelo de laboratorio para el metabolismo central (glucolisis+ciclo de Krebs). Lo que encontró fué fascinante: una reacción que no era ni un equilibrio ni irreversible, sino que oscilaba entre el estado inicial y el final. Si la reacción se lleva a cabo sin agitación y en una capa fina, tiene lugar la formación de bellas estructuras llamadas patrones de Turing.
Belousov estudió exhaustivamente la reacción y propuso un esbozo de su mecanismo (que tardaría décadas en resolverse); envió sus resultados y los detalles experimentales para ser publicados en una importante revista científica, en 1951. Los revisores recomendaron contra su publicación y el editor respondió a Belousov que su “supuesto descubrimiento” era “imposible”. Seguramente, la idea de que una mezcla química se comportase como un péndulo, o que en un líquido homogéneo surgieran estructuras de composición química diferenciada, violando en apariencia el Segundo Principio de la Termodinámica, pesaba más que el repetir la simple receta de la reacción y verlo con sus propios ojos. El editor dijo a Belousov que tenía que aportar más pruebas. Boris entonces volvió al laboratorio y tras otros seis años de trabajo (la Ciencia requiere tiempo…), envió un nuevo artículo, más elaborado y con un estudio profundo del mecanismo. La respuesta que recibió fue de rechazo y un escepticismo aún mayor. Ni los revisores ni los editores se molestaron en repetir por ellos mismos una reacción extremadamente sencilla de reproducir. Simplemente se dejaron llevar por sus prejuicios. Belousov, que ya tenía 64 años, estaba tan enfurecido y decepcionado que decidió no volver a publicar ningún artículo jamás ni relacionarse con otros científicos. Publicó su receta de la reacción en un simposium, quedando solo un oscuro resumen en ruso en el fondo de algunas bibliotecas.
Cuando Zhabotinsky tomó el testigo, la receta de la reacción de Belousov era conocida en diversos centros en la URSS, aunque se desconocía su autor y origen. Zhabotinsky comenzó, como tema de tesis, a estudiar esa curiosa reacción que circulaba por ahí. En los años 60, ya había realizado un extenso estudio y había descubierto al autor de la reacción original: Boris Belousov. Informó a éste de su trabajo y progresos y Boris le remitió, muy agradecido por recuperar su vieja receta, su manuscrito rechazado original y otro material. Cuando la reacción empezó a ser conocida fuera de la URSS, a finales de los 60 y principios de los 70, aún era conocida como la “reacción de Zhabotinsky”, aunque por el esfuerzo de éste, el crédito para Belousov fué finalmente reconocido. Zhabotinsky mantenía informado de sus progresos a Belousov, pero éste siempre rechazó un encuentro personal, así como asistir al simposio sobre reacciones oscilantes que se había organizado en Praga en 1968. Así, Zhabotinsky y Belousov nunca se conocieron personalmente. Belousov murió en 1970, sin llegar a conocer las implicaciones que tendría su trabajo ni disfrutar del reconocimiento que actualmente se le brinda.
Desde el punto de vista de la termodinámica, podemos usar la reacción de Belousov para entender cómo funciona un sistema metabólico:
En primer lugar no es una sola reacción, sino que se genera un sistema multicomponente, abierto y genera una red o ciclos de reacciones. Uno de estos ciclos es un ciclo autocatalítico en el que los productos son, a su vez, reactivos. Estas características las comparten tanto un sistema metabólico como la reacción de Belousov.
Los patrones de Turing son impredecibles, jamás son exactamente iguales y su estructura depende de pequeñas diferencias en las condiciones iniciales. Son el resultado del acoplamiento entre las reacciones químicas y los procesos de difusión. Esta relación es no lineal y da lugar a múltiples estados estacionarios, igualmente probables. La formación de las estructuras, que manifiestan los estados estacionarios, depende de las fluctuaciones microscópicas. Prigogine lo llamaba «orden a través de la fluctuación». En un sistema biológico, las fluctuaciones son cambios ambientales, epigenética o variaciones genéticas, y las estructuras son el fenotipo.
El sistema (ya sea la reacción de Belousov o el metabolismo) alcanza un estado alejado del equilibrio, en el que maximiza la disipación de energía libre de los reactivos, minimiza la producción de entropía y maximiza la disipación de entropía fuera del sistema, de modo que el sistema se sostiene con la neguentropía y, aunque la entropía global aumenta, dentro del sistema se produce una reducción temporal de entropía. En el vídeo podéis ver uno de los modos de disipación de entropía: las burbujas de CO2, el residuo que produce la reacción, y que se eliminan. Este es el principio de mínima producción de entropía de Prigogine. El sistema evoluciona reduciendo su producción interna de entropía y maximizando la disipación. Pero, la reacción de Belousov no es un buen sistema abierto, y termina acumulando demasiada entropía y «muriendo»
Estado final de la reacción de Belousov: se ha disipado la energía libre, la variación de entropía es cero (está en equilibrio) y se destruyen las estructuras. La composición es homogénea y la energía libre final es inferior a la inicial.
Otro modelo, el modelo de máxima disipación de entropía, sugiere el sistema tenderá al estado estacionario en el que la producción de entropía sea máxima. Es decir, la evolución tiende a maximizar el aumento de entropía del entorno, lo cual puede hacerse mediante el balance entre la producción interna de entropía (que siempre aumenta) y el intercambio con el exterior.
La reacción de Belousov o la vida misma no son más que disipadores de energía libre ygeneradores de entropía. Esto permite la evolución compleja de estructuras durante el proceso de disipación de la energía libre (nutrientes) hacia los resíduos (CO2). Prigogine las llamó estructuras disipativas.
Esto nos lleva al concepto de línea del tiempo: el proceso no se puede revertir. Percibimos el tiempo en cuanto partimos de un estado inicial de baja entropía y/o alta energía libre, para terminar en un estado que ha maximizado la entropía y ha disipado la energía libre en forma de calor. Esta es la razón por la que no se puede viajar al pasado, pues ello violaría el segundo principio de la Termodinámica; viajar al pasado implicaría moverse hacia una reducción de entropía del universo.
La vida surge, como una perturbación temporal, en el proceso de disipación de la energía y del aumento de la entropía del universo. Por ello, la vida y estructuras complejas no surgen en los estados de mínima entropía o de máxima entropía, sino de modo dinámico durante el proceso de disipación de energía y aumento de la entropía del universo.
Otro aspecto interesante de estos sistemas es la adaptación y auto-reparación. Las estructuras disipativas son estables a perturbaciones y pueden absorber éstas y ‘auto-sanarse’. La capacidad de autoreparación se puede observar en la reacción de Belousov si se introducen perturbaciones:
Vida y disipación de entropía en el planeta Tierra
Hay una idea interesante al considerar la vida globalmente: la vida surge en la Tierra como una consecuencia del teorema de la maximización de la entropía. La Tierra recibe energía del Sol y debe disiparla. Si no se disipase, el planeta se habría calentado hasta fundirse. En el proceso de disipación, surge la vida como una estructura que contribuye a mejorar esa disipación (es decir, el segundo término de la última ecuación que escribimos).
La Tierra, globalmente, constituye una gran estructura disipativa en la que:
donde T1 es la temperatura de la superficie del Sol (aproximadamente 6000 K) y T2 es la temperatura de equilibrio del planeta, que es a grosso modo la temperatura a la que la Tierra irradia energía, y que es aproximadamente 300 K. Dado que la energía no se acumula en la Tierra (si así fuera, el planeta se calentaría gradualmente), el segundo término de la ecuación es aproximadamente 3×1024/300 cal/grad g
La Tierra recibe mucha energía y poca entropía, es decir, energía de alta ‘calidad’, y se disipa al espacio con un flujo de entropía mucho más elevado. Ello es lo que asegura que se sostenga la vida en la Tierra. Sin embargo, esta cuenta global no es suficiente para entender cómo surgió y cómo funciona la autoorganización molecular que da la vida.
Pero, en términos globales, la biosfera funciona como ciclos de Morowitz (ver más adelante) en los que los organismos fotosintéticos capturan energía solar, la convierten en hidrógeno y genera un gradiente de potencial, que se disipa, impulsando la generación de biomasa. La energía del hidrógeno se almacena y transporta en forma de azúcares, que son utilizados por otros organismos para reconvertirlos en hidrógeno y generar un gradiente, que impulsa a su vez la generación de biomasa o la producción de entropía en estado estacionario. En cada paso, la energía solar se ‘degrada’, o se ‘esparce’, creando un eficiente flujo de entropía hacia el exterior.
La vida en el planeta no es más que otra estructura asociada al principio de máxima producción de entropía. En un planeta como la Tierra, los flujos de fluidos y energía (Sol, calor interno del planeta) han provocado la autoorganización de estructuras (incluyendo la vida) que maximizan el flujo de entropía. Esto no ocurre en todos los planetas. En el Sistema Solar, el principio de máxima producción de entropía se aplica además en Marte, Titán, Europa o Encelado. Por ello, son lugares de interés astrobiológico.
Obsérvese que en ningún momento hemos relacionado la entropía con una medida del orden a pesar de haber estado un rato hablando de entropía. Hemos hablado de la aparición de estructuras (como es la estructura celular), y del macroestado, es decir, el balance global de calor y la temperatura de la habitación, pero no hemos hablado de ‘orden’. Ahora ha llegado el momento de volver a los dados de nuestro juego.
Si lanzamos dos dados, la apuesta más segura es el 7. ¿Por que?. Podemos considerar el resultado de la tirada como el macroestado del sistema, que es el resultado de una serie de microestados, que definimos como el resultado de cada dado en particular (es decir, el estado termodinámico de cada componente del sistema). Al final, el estado global del sistema es el resultado de los estados particulares de cada uno de sus componentes. Por ello, en la primera ecuación hablábamos que la energía del sistema (macroestado) dependía de N, el número de átomos, cada uno de ellos en un ‘microestado’ determinado.
El número 7 es el que resulta del mayor número de combinaciones de los dos dados, por ello es el que más frecuencia presenta en la distribución.
Cuantos más microestados posibles tenga un macroestado, más probabilidad tenemos de tener el macroestado resultante. Esto se llama probabilidad termodinamica. En el caso del número 7, es el macroestado resultante de seis microestados posibles. Y, lo que nos descubrió Boltzmann es que la mayor entropía tiene lugar en el macroestado al cual conducen el mayor número posible de microestados.
Ecuación de Boltzmann, que, en realidad, fue formulada por Planck.
La ecuación de Boltzmann dice que la entropía es igual al producto de una constante (la constante de Boltzmann, determinada por Planck) por el logaritmo de la probabilidad de ese estado. El principio de Boltzmann nos dice entonces que el aumento de entropía en un proceso es la consecuencia de la transición del sistema del estado menos probable al más probable. En nuestro juego de dados, el estado más probable, el de mayor entropía, es el número 7, y por ello el sistema de dos dados tiene la máxima frecuencia en 7 en su distribución de estados.
Veamos otro ejemplo para ilustrar el principio de Boltzmann:
Si pregunto a los estudiantes, a los que se ha engañado con la idea intuitiva de que la entropía mide el grado de desorden, la mayoría responden que la imagen de la derecha se corresponde con el estado de mayor entropía. Pero NO. La probabilidad de que en una distribución de puntos en la cuadrícula (es decir los microestados) no haya ningún punto al lado de otro es MÍNIMA, y, de hecho, esta distribución no se ha generado al azar. Si distrubimos puntos al azar, siempre va a haber alguno pegado a otro.
El estado de la imagen de la izquierda es, por tanto, más probable, no importa cómo estén ordenados los puntos. Y es más probable, porque por azar resultan mas combinaciones de puntos (microestados) con el mismo número de contactos de unos puntos con otros. Por tanto, la imagen de la izquierda sería el estado de mayor entropía.
Entropía: una medida de la ‘utilidad’ de la energía y la burocracia como análogo
En todo proceso, hay un intercambio energético. Esta energía se invierte en hacer ‘algo’, como mover un motor, o hacer una transformación química. Esta energía que invertimos e intercambiamos la llamamos energía libre.
Pero claro, para que se produzca el proceso, hay que pagar una especie de impuesto: No toda la energía se invierte en hacer algo útil (como mover el motor o transformar moléculas), sino que una parte se «disipa» sin que haga nada de utilidad. Es el término entrópico de la energía libre.
Esta idea de ‘utilidad’ de la energía la podemos ilustrar con una analogía con la burocracia.
Todos odiamos la burocracia. En mi trabajo como profesor, la burocracia requiere una cantidad considerable de esfuerzo y dedicación. Podríamos definir la burocracia como el trabajo que no se invierte en nada productivo, pero se disipa para mantener la estructura. Como veis, volvemos a la idea de entropía como generadora de orden.
La burocracia es entropía que se genera y se disipa. En la disipación de esta entropía reside el mantenimiento de toda una estructura organizativa. Es decir, cuanto más grande y compleja es una organización, más estructura requiere mantener y más burocracia, es decir, disipación de entropía requiere. ¿como aplicamos aquí el principio de mínima producción de entropía de Prigogine?. Muy sencillo: intentamos minimizar el tiempo que dedicamos a la burocracia, maximizando la disipación de ésta. Un sistema biológico funciona de modo parecido, manteniendo así su estructura. El problema es que un sistema disipativo, ya sea un organismo, una estructura o un estado, va a depender de este proceso de generación y disipación de entropía. Ello lleva a que la complejidad vaya aumentando hasta que el sistema no es capaz de disipar más entropía de la que produce, teniendo lugar el envejecimiento y degradación de la organización.
En un estado o empresa es igual: si los requerimientos burocráticos aumentan en exceso, el sistema pierde eficiencia en la disipación de entropía que se acumula, dando lugar a la degradación de la estructura y su muerte. Todos conocemos este proceso cuando estamos sometidos a burocracia paralizante, que hace que sea muy dificil llevar a cabo tareas útiles y todas las estructuras se degradan.
La entropía: una medida de la ‘degradación’ y ‘dispersión’ de la energía
Podemos ilustrar esta idea de degradación de la energía usando una analogía: billetes y moneda fraccionaria
Si tomo un billete de 20 euros y lo cambio en 40 monedas de 50 céntimos, voy a tener el mismo valor monetario (es decir, la energía se conserva). Sin embargo, estoy «esparciendo» o «dispersando» ese valor: he aumentado la entropía (ΔiS>0). Si yo ahora doy una moneda a cada persona que pasa por la calle, ese valor (esa energía) la he «esparcido» y la he expulsado del sistema: he aumentado la entropía del entorno, y he reducido la entropía de mi sistema (ΔeS<0), pero a costa de pagar 20 euros. Cada persona que se ha llevado 50 céntimos, realmente poco puede hacer con ellos. Habría hecho más con los 20 euros, ¿verdad?. Es decir, con el aumento de entropía, al repartir y «esparcir» la energía, ésta se ‘degrada’.
Este proceso, en el que, globalmente, me he quedado igual que antes, pero con 20 euros menos, es un proceso cercano al equilibrio. Ahora imaginad que yo hago lo mismo, pero le pido a cada persona a la que doy una moneda que me suba una cosa que he comprado en el súper y la coloque correctamente en la cocina. Estoy estableciendo unos ciclos y la disipación de los 20 euros lleva a que surja una estructura ordenada en mi sistema. Entonces estaríamos hablando de una estructura disipativa: El empleo de energía y la disipación de entropía, generan la estructura. Imaginad que cada moneda es el CO2 que expulso en cada ‘ciclo’ metabólico, y que el billete original era la glucosa que he consumido en el proceso y tendremos una analogía para relacionar la entropía con el metabolismo. Como con las monedas, el CO2 tiene escaso valor (es una molécula muy estable y con un sobrepotencial muy elevado, que la hace poco reactiva, de ahí la dificultad para convertirla en otros materiales), comparado con la glucosa original, de la que se puede extraer bastante energía. Ahora necesitamos un sistema que recopile las monedas y las convierta de nuevo en billetes de 20 euros. Este sistema son los organismos autótrofos, que, utilizando la luz o la energía química de los minerales, fijan el CO2, convirtiéndolo de nuevo en ‘billetes’ de alto valor: los azúcares, que a su vez se transforman en otros muchos productos.
Esta idea de los ciclos la expresó Harold Morowitz en su Teorema de los Ciclos de Morowitz:
Esto nos permite definir la entropía de un modo intuitivo: es una medida de la dispersión de la energía: cuanto más dispersa, es decir, cuantas más formas tengamos de distribuir la energía entre los componentes del sistema, mayor es la entropía. Lógicamente, si en los productos de la reacción tengo mucho CO2, tengo muchas formas de distribuir la energía entre cada una de las moléculas de CO2; por tanto, tengo más microestados que se manifiestan como el macroestado, y eso aumenta la probabilidad y la entropía.
También decimos que al aumentr la entropía, aumentamos los grados de libertad del sistema. Cuantos más grados de libertad, mayor entropía. Veámoslo con un ejemplo clásico de Lehninger:
¿cual de los tres casos, a, b o c, tiene mayor entropía?. ¿puedes definirlo según el concepto de «estado ordenado» que te contaron? En el caso (a) tenemos dos reactivos separados. Podemos distribuir energía entre las dos moléculas y todas las posibles vibraciones entre sus enlaces. Tened en cuenta que, en la molécula, la energía se distribuye haciendo vibrar y rotar los enlaces, y en el caso (a) hay muchas más formas de hacerlo que en los otros (tenemos más microestados). En el caso (b) tenemos una sóla molécula, pero esta molécula tiene tres enlaces capaces de rotar completamente, con lo que tenemos más grados de libertad, es decir, más formas de distribuir la energía, es decir, más microestados, que en caso (c), en el que tenemos una molécula rígida, por lo que las posibilidades de rotaciones y vibraciones disminuyen. Así, el caso (c) es el de menor entropía y el caso (a) el de máxima entropía. Esto hace que la reacción (a) sea mas lenta que la (c). Mucho más lenta. Y aquí entramos en cómo la entropía gobierna la velocidad de las reacciones y la acción de las enzimas, lo cual es contenido para otro tema.
Intuitivamente, podemos expresar que en el caso (a), podemos ‘esparcir’ la energía mucho mejor que en el caso (c). Entonces, una habitación desordenada no tiene por qué ser el estado más probable, y por tanto, el de mayor entropía. Depende de cómo se distribuya la energía entre sus componentes y del número de microestados que den lugar a ese estado desordenado. Jaque mate, amigos que usais la entropía como excusa para no ordenar la habitación.
Veamos otro ejemplo intuitivo, el de una tostada con mantequilla:
Yico…
Mientras la temperatura se mantenga baja y no proporcionemos calor, estaremos en un estado de baja entropía y el trozo de mantequilla se mantendrá sólido sobre la tostada. Al calentar, ¿qué es lo más probable? que la mantequilla se ‘esparza’ por la tostada. Este será un estado de mayor entropía: las moléculas de la mantequilla se separan (tiene mas ‘libertad’) y distribuyen por la tostada y la energía se reparte, ‘esparciéndose’. Aquí no hay sorpresas: tenemos un proceso irreversible (una vez esparcida la mantequilla, no podemos revertir el proceso), que se mueve, tal como nos decía Boltzmann, hacia su estado de mayor probabilidad. La mantequilla no se ‘desordena’, eso es nuestra percepción de orden y desorden, ya que antes de calentar la mantequilla no estaba formando una estructura cristalina, por ejemplo. Solo era más viscosa, dando la apariencia de ser sólida. Simplemente, la energía se ‘reparte’ siguiendo el camino más probable, aumentando los grados de libertad moleculares, o aumentando el número de microestados. Como la mantequilla es un fluido no compresible, entonces la variación de entropía sólo depende de la temperatura. Al calentarla de A a B, la entropía se incrementa según:
En la ecuación, n es el número de moles y Cp el calor específico. En la tostada siempre estamos cerca del equilibrio termodinámico, por lo que aquí no vamos a tener un fenómeno de estructura disipativa, que resulta de la combinación de reacciones reversibles e irreversibles interconectadas.
Estos conceptos intuitivos que relacionan la entropía no con el orden, sino con la probabilidad y con la distribución de la energía entre componentes del sistema e intercambio con el exterior, son útiles para entender la relación entre entropía, orden biólogico o para entender cómo funcionan las enzimas o la ribozima esencial que da vida a todos los organismos vivos de la Tierra: la peptidil transferasa.
Así, finalmente, la vida, como una estructura ordenada, depende de la generación de entropía y del aumento de entropía del entorno. Consumimos moléculas de alta energía para mantener nuestra estructura y, la entropía del entorno es el precio que hay que pagar por la estructura. Como dijo Schrödinger:
Cuanto más suntuoso es un palacio, mayor es el montón de basura que genera
La emisión de CO2, orina y desechos es esencial para ‘dispersar’ o ‘esparcir’ energía, aumentando la entropía del exterior. Por ello, toda organización basada en el consumo de energía siempre, por definición, es un orden temporal, cambiante y destinado a desaparecer, altamente dependiente de cual es el mecanismo por el que disipa la energía de sus fuentes de energía y del balance entre producción y disipación de entropía. Ya sea una célula, un organismo vivo o una sociedad, todas son estructuras disipativas, temporales y dependientes de la producción de entropía. Debemos estar preparados para ello.
Referencias
Cassani, A., Monteverde, A. & Piumetti, M. (2021) ‘Belousov-Zhabotinsky type reactions: the non-linear behavior of chemical systems’, Journal of Mathematical Chemistry, 59(3), pp. 792–826. doi: 10.1007/s10910-021-01223-9.
De Castro, C. & McShea, D. W. (2022) ‘Applying the Prigogine view of dissipative systems to the major transitions in evolution’, Paleobiology, 48(4), pp. 711–728. doi: 10.1017/pab.2022.7.
Harisch, K. (no date) ‘How the mind comes to life : The physics and metaphysics of dissipative structures’, pp. 1–18.
Hooker, C. (2011) ‘Introduction to Philosophy of Complex Systems: A’, in Philosophy of Complex Systems. Elsevier, pp. 3–90. doi: 10.1016/B978-0-444-52076-0.50001-8.
Howell, L., Osborne, E., Franklin, A. & Hébrard, E. (2021) ‘Pattern recognition of chemical waves: Finding the activation energy of the autocatalytic step in the Belousov-Zhabotinsky reaction’, Journal of Physical Chemistry B, 125(6), pp. 1667–1673. doi: 10.1021/acs.jpcb.0c11079.
Kondepudi, D. K., De Bari, B. & Dixon, J. A. (2020) ‘Dissipative structures, organisms and evolution’, Entropy, 22(11), pp. 1–19. doi: 10.3390/e22111305.
Naderi, M. (2020) ‘On the evidence of thermodynamic self-organization during fatigue: A review’, Entropy, 22(3), pp. 1–32. doi: 10.3390/E22030372.
Ota, K., Mitsushima, S., Matsuzawa, K. & Ishihara, A. (2014) ‘Assessing the environmental impact of hydrogen energy production’, in Advances in Hydrogen Production, Storage and Distribution. Elsevier, pp. 32–42. doi: 10.1533/9780857097736.1.32.
Shanks, N. (2001) ‘Modeling Biological Systems: The Belousov–Zhabotinsky Reaction’, Foundations of Chemistry, 3, pp. 33–53.
Tsuda, I., Yamaguti, Y. & Watanabe, H. (2016) ‘Self-organization with constraints-A mathematical model for functional differentiation’, Entropy, 18(3), pp. 1–13. doi: 10.3390/e18030074.