¿Por qué aumenta la infectividad del coronavirus?: Un sencillo experimento virtual con la ‘variante británica’ del SARS-CoV-2
por C. Menor-Salván
En los medios y redes sociales ha causado una gran alarma la aparición, reportada en diciembre de 2020 en Gran Bretaña, de una nueva variante del virus SARS-CoV-2. Esta variante, denominada B.1.1.7, contiene varias mutaciones puntuales respecto de la variante original que se extendió en febrero-marzo de 2020. Según los datos clínicos disponibles, la variante B.1.1.7 tiene mayor infectividad y velocidades de transmisión. Desde diciembre, según el COVID-19 UK Genomics Consortium, más del 50% de los nuevos contagios son de ésta variante, que presenta una transmisibilidad incrementada entre un 50 y un 70% respecto de la variante original que llamaremos WT (por ‘wild type’). Como es lógico, la aparición de variantes con mayor transmisibilidad crean alarma, pero son perfectamente esperables, dado que estamos forzando el proceso de evolución viral: mediante confinamientos y medidas anticontagio, creamos una selección artificial de las variantes más infectivas, que prevalecen sobre las variantes originales.
La infectividad viral es una consecuencia de varios aspectos moleculares del virus; uno de ellos es la afinidad por el receptor celular, que una proteína del virus reconoce, y al que se une, iniciando el proceso de infección. La proteína viral que reconoce el receptor en la célula que va a infectar es la proteína de la espícula. Como si de un puzzle molecular se tratara, la espícula viral ‘encaja’ en el receptor. Os remito a la Noticia Nº 59 para una introducción general al SARS-CoV-2

Como es lógico, si modificamos la proteína de la espícula de modo que mejore la estabilidad de su interacción con el receptor, aumentará la infectividad. ¿como?. Para entenderlo, hay que tener en cuenta que un virus es un agregado supramolecular. Digamos que es una gran molécula heterogénea. No es un ser vivo. Si el virus tiene mayor afinidad por su receptor, quiere decir que la infección se producirá a una concentración menor. Dicho de otra manera, si aumentamos la afinidad del virus modificado por el receptor, se producirá una infección con una cantidad de virus menor que los necesarios con el virus sin modificar. Y, posiblemente, este proceso está detras del efecto de la variante UK B.1.1.7: una de sus mutaciones se encuentra precisamente en la región de la espícula que interacciona con el receptor de la célula a infectar.
En ésta entrada voy a mostraros como nosotros mismos podemos ver éste efecto: voy a tomar los datos de la estructura PDB 60MJ y, a partir de ellos, voy a:
- Calcular las interacciones por puentes de hidrógeno entre la espícula y el receptor. Estas interacciones determinan la estabilidad el complejo espícula-receptor, y, como es lógico, cuando más puentes y más fuertes, mayor estabilidad = mayor afinidad =mayor infectividad. También observaremos otras interacciones evidentes, como el stacking pi: un tipo de enlace que se produce entre aminoácidos aromáticos.
- Voy a introducir la mutación N501Y de la ‘variante británica’, que implica el cambio de la asparagina 501 de la espícula por una tirosina, justamente en la región de interacción entre la espícula y el receptor.
- Después, usando el software, el ordenador calculará la estructura más estable resultante de ése cambio y veremos si la mutación introduce la aparición de nuevas interacciones. Si la mutación implica la aparición de nuevos enlaces, esto explica fácilmente el aumento de infectividad de la ‘variante británica’.
Este procedimiento lo voy a llevar a cabo usando los softwares UCSF Chimera, Pymol y Chem3D.
La mutación N501Y de la ‘variante británica’ del SARS-CoV-2 introduce nuevas interacciones y podría aumentar la afinidad por el receptor y la infectividad
Como hemos comentado, la unión entre la espícula viral y el receptor desencadena el proceso infectivo. Esta unión se lleva cabo gracias a que se establecen una serie de interacciones no covalentes entre las dos proteínas: puentes de hidrógeno e interacciones tipo apilamiento entre anillos aromáticos. Cuanto más fuerte sea la unión, mayor estabilidad tendrá el complejo y, por tanto, mayor afinidad tendrá el virus por el receptor. Al aumentar la afinidad, aumenta la infectividad del virus, dado que se requiere una inoculación de menor cantidad de virus para producir una infección efectiva. Si, por ejemplo, el virus WT necesita (número arbitrario) introducir 1000 unidades de virus en el hospedador para producir una infección, al aumentar la afinidad por el receptor ese número se puede reducir a 500-300.
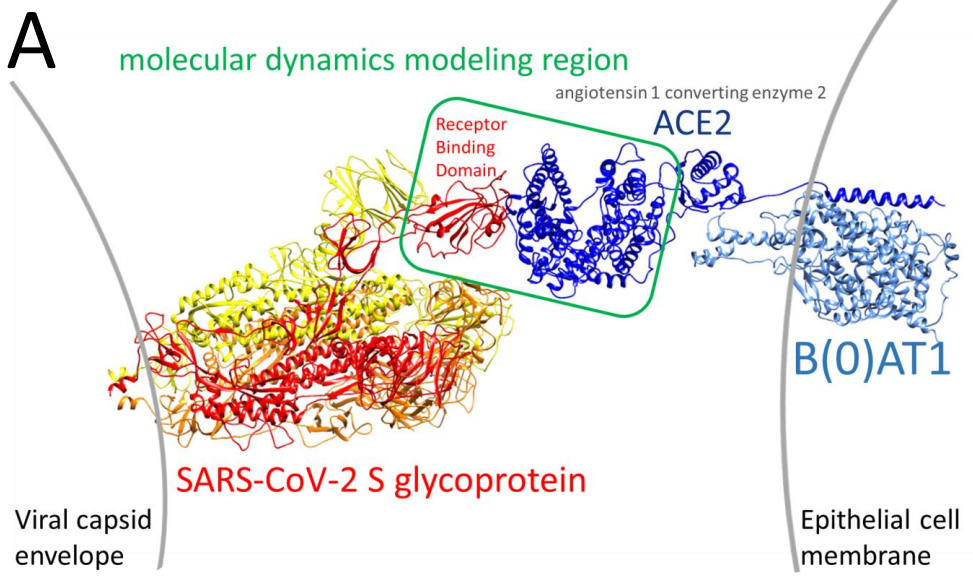
Centrándonos en las estructuras del dominio RBD de la espícula y de la proteína ACE2 (recuadrados en la figura anterior), vamos a ver el resultado que obtenemos usando los datos de la variante original del virus:

Vemos la estructura molecular del dominio RBD, en azul. Este dominio es la parte de la espícula que se une al receptor celular, la proteína ACE2. Las líneas azules representan los puentes de hidrógeno entre aminoácidos. Aquí tienen especial protagonismo el Gln 493 y la lisina 417, que forman puentes con el receptor. La fenilalanina 486 da lugar a un apilamiento pi con una tirosina del receptor. Esta interacción no existía en el SARS de 2003, y es una de las que explica la mayor afinidad (e infectividad) del SARS-CoV-2. En amarillo veis la Asn (asparagina) 501, que ya en su momento se señalaba como un aminoácido potencialmente interesante en la estructura. Vamos a ver qué ocurre cuando introducimos la mutación:

La introducción de la mutación, con la nueva tirosina en la posición 501, tiene consecuencias que son sutiles pero de gran importancia. Primero, aparecen nuevas interacciones entre el nuevo aminoácido y el receptor: nuevos puentes de hidrógeno y un enlace por stacking pi entre la tirosina 501 y una tirosina del receptor. Por otro lado, interacciones que ya existían previamente disminuyen su distancia. Los puentes de hidrógeno entre las lisina 417 y la glutamina 493 se hacen más cortos (y por tanto mas fuertes, ya que es una interacción electrostática). Como resultado la proteína mutante se une con mayor estabilidad al receptor, ya que aumentan el número de puntos de atracción electrostática y disminuye la distancia de las que ya existían. Esto da lugar a una unión más estable, y, por tanto, a un aumento de la afinidad. Esto se traduce en la práctica en un aumento de la infectividad de la variante.
Las representaciones de proteínas son algo abstractas. Para que el lector pueda ver más intuitivamente cómo se produce la unión, vamos a ver las superficies de las moléculas de proteína. Estas interaccionan ‘encajando’ y uniéndose por las interacciones electrostáticas, de modo similar a como si uniéramos unas piezas con imanes.
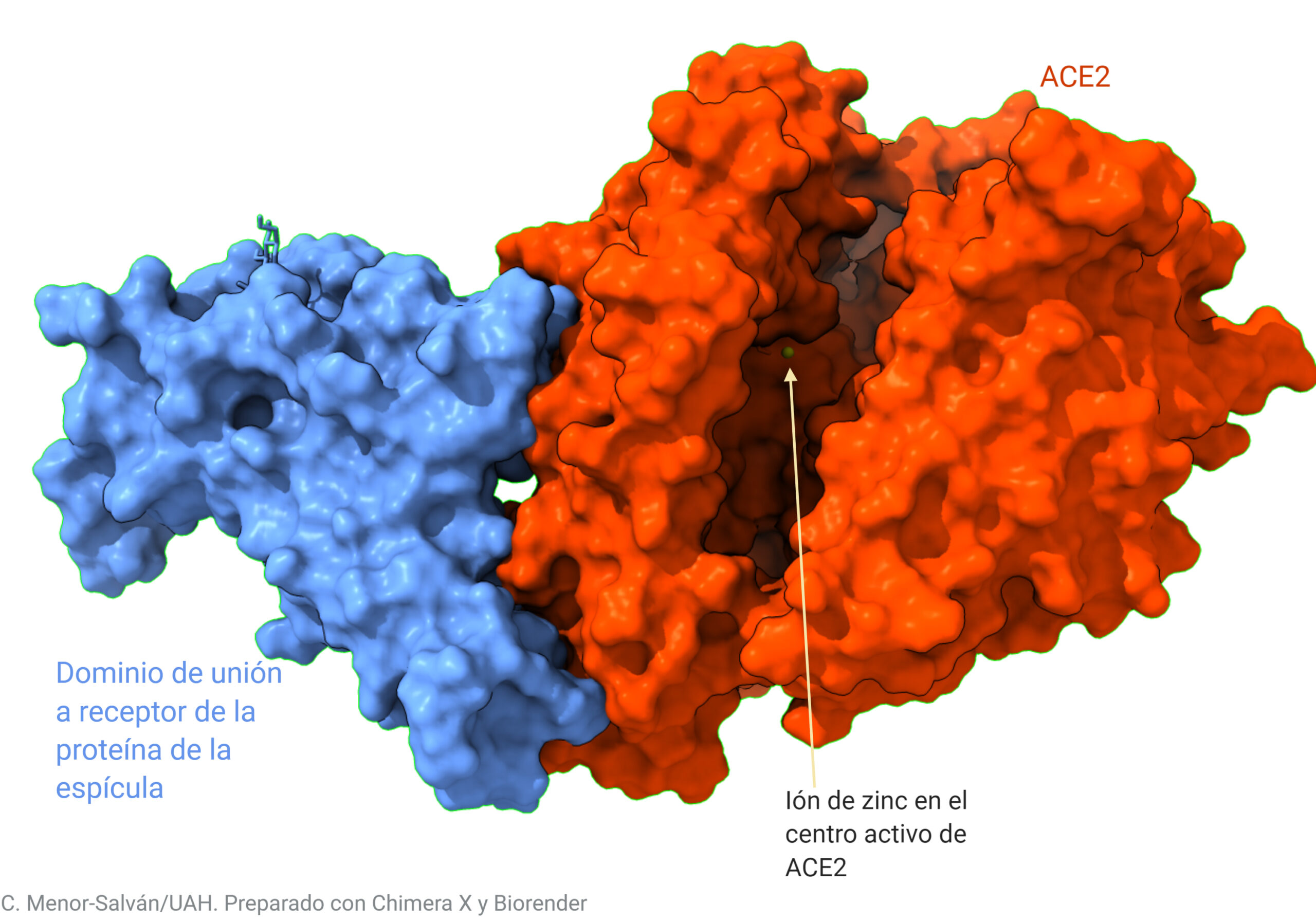
En cierto modo, el virus y su receptor interaccionan de modo similar a las piezas de un conector magnético, pero con una interacción electrostática en lugar de magnética.


La fuerza y geometría de las interacciones electrostáticas determina la estabilidad de la unión entre el virus y su receptor, y, por tanto, su afinidad e infectividad. La acción de los anticuerpos se basa en un principio similar. Una pregunta habitual es si ésta nueva variante supondrá una pérdida de eficacia de las vacunas o mejorará la evasión inmune del virus. Todavía queda mucho trabajo para los científicos, pero los primeros datos indican que ésta variante no alterará la eficacia de la vacuna ni la respuesta inmune, al ser mutaciones puntuales que no afectan al reconocimiento de los anticuerpos.
Por otro lado, las modificaciones en el dominio de reconocimiento de receptor en la espícula viral, tampoco implican un aumento de la agresividad del virus, por lo que el aumento de infectividad no va asociado a un aumento de gravedad de las infecciones. Como digo, aún queda mucho que investigar y que aprender y todo puede ir cambiando, pues la pandemia evoluciona más rápido que la capacidad de los científicos para obtener resultados y avances.
Estos sencillos resultados que he mostrado, obtenidos mediante un no menos sencillo análisis computacional de datos de estructuras de proteínas, no son mas que una aproximación muy sencilla. Un estudio mas complejo y riguroso requeriría muchos mas medios y tiempo de los que dispongo, pero espero que sirva para entender la mecánica molecular que hay tras la infección del coronavirus.
Referencias
Kupferschmidt, K. Fast-spreading UK virus variant raises alarms. Science (New York, NY), 371(6524), 9-10.
Lauring, A. S., & Hodcroft, E. B. Genetic Variants of SARS-CoV-2—What Do They Mean?. JAMA.
Rynkiewicz, P., Babbitt, G. A., Cui, F., Hudson, A. O., & Lynch, M. L. A comparative survey of Betacoronavirus binding dynamics relevant to the functional evolution of the highly transmissible SARS-CoV-2 variant N501Y. bioRxiv, 2021: https://doi.org/10.1101/2020.09.11.293258
Tang, J. W., Tambyah, P. A., & Hui, D. S. (2020). Emergence of a new SARS-CoV-2 variant in the UK. Journal of Infection.
UCSF Chimera–a visualization system for exploratory research and analysis. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004 Oct;25(13):1605-12
Volz, E., Mishra, S., Chand, M., Barrett, J. C., Johnson, R., Geidelberg, L., … & Ferguson, N. M. (2021). Transmission of SARS-CoV-2 Lineage B. 1.1. 7 in England: Insights from linking epidemiological and genetic data. medRxiv, 2020-12.
Zhou, Q., Yan, R., Zhang, Y., Li, Y., & Xia, L. (2020). Structure of dimeric full-length human ACE2 in complex with B0AT1. BioRxiv: https://www.biorxiv.org/content/10.1101/2020.02.17.951848v1.abstract