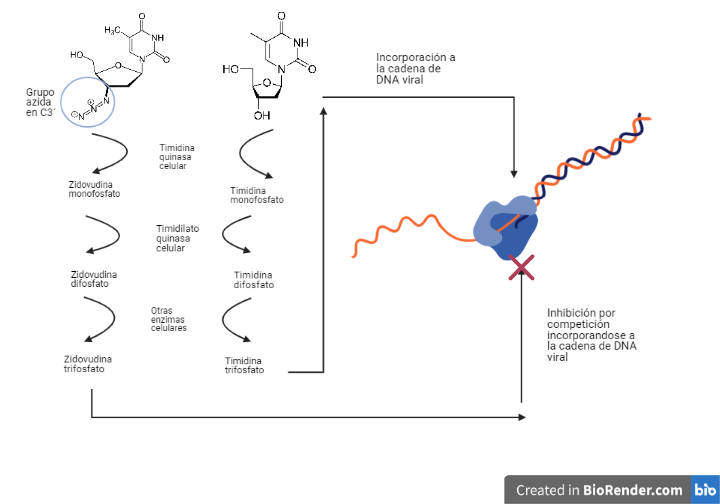
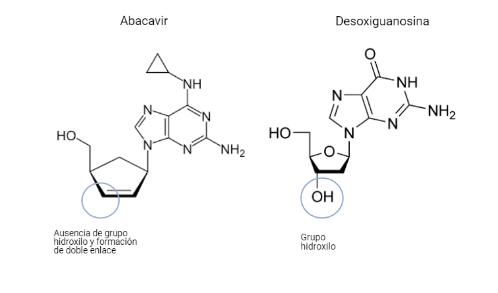
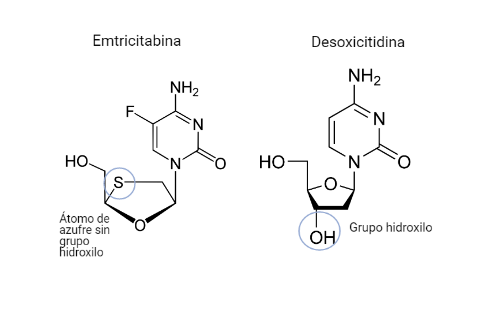
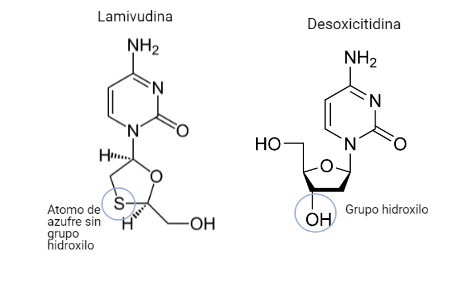
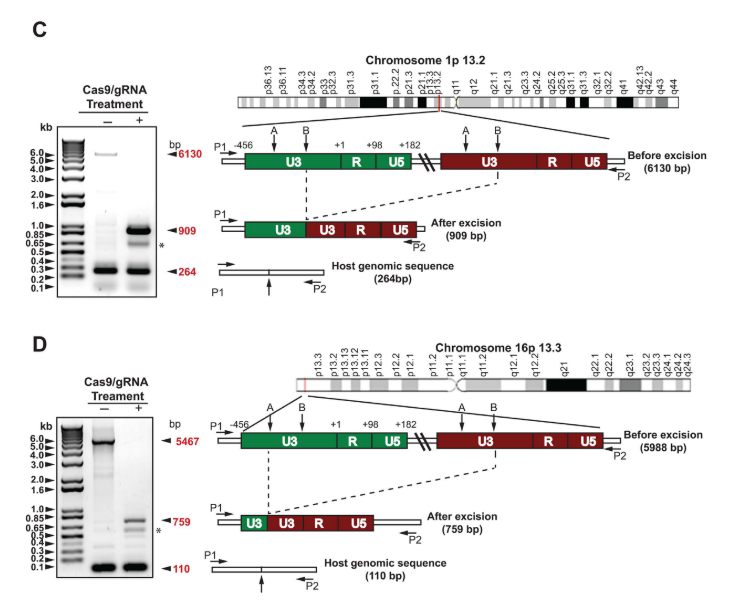
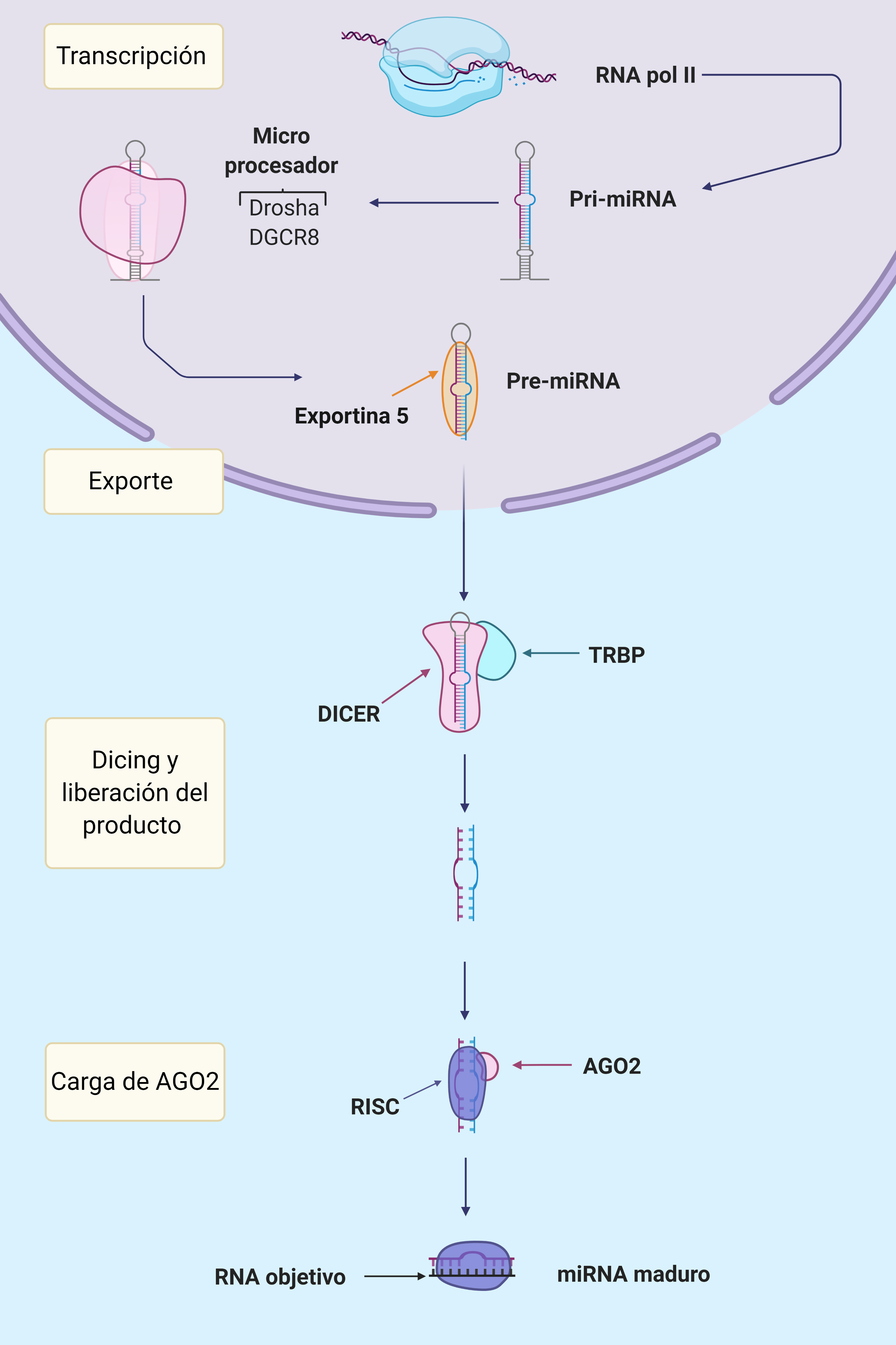
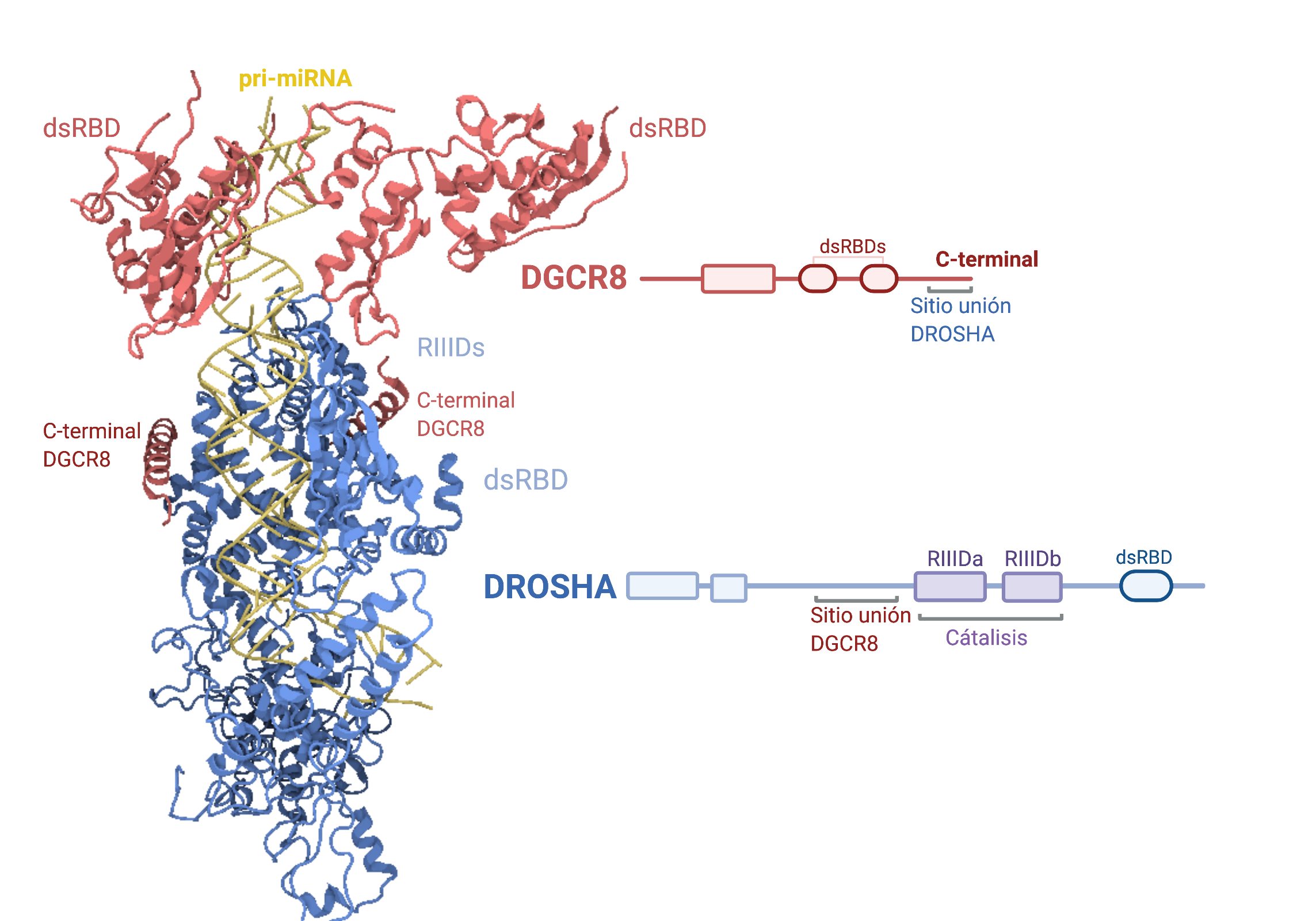
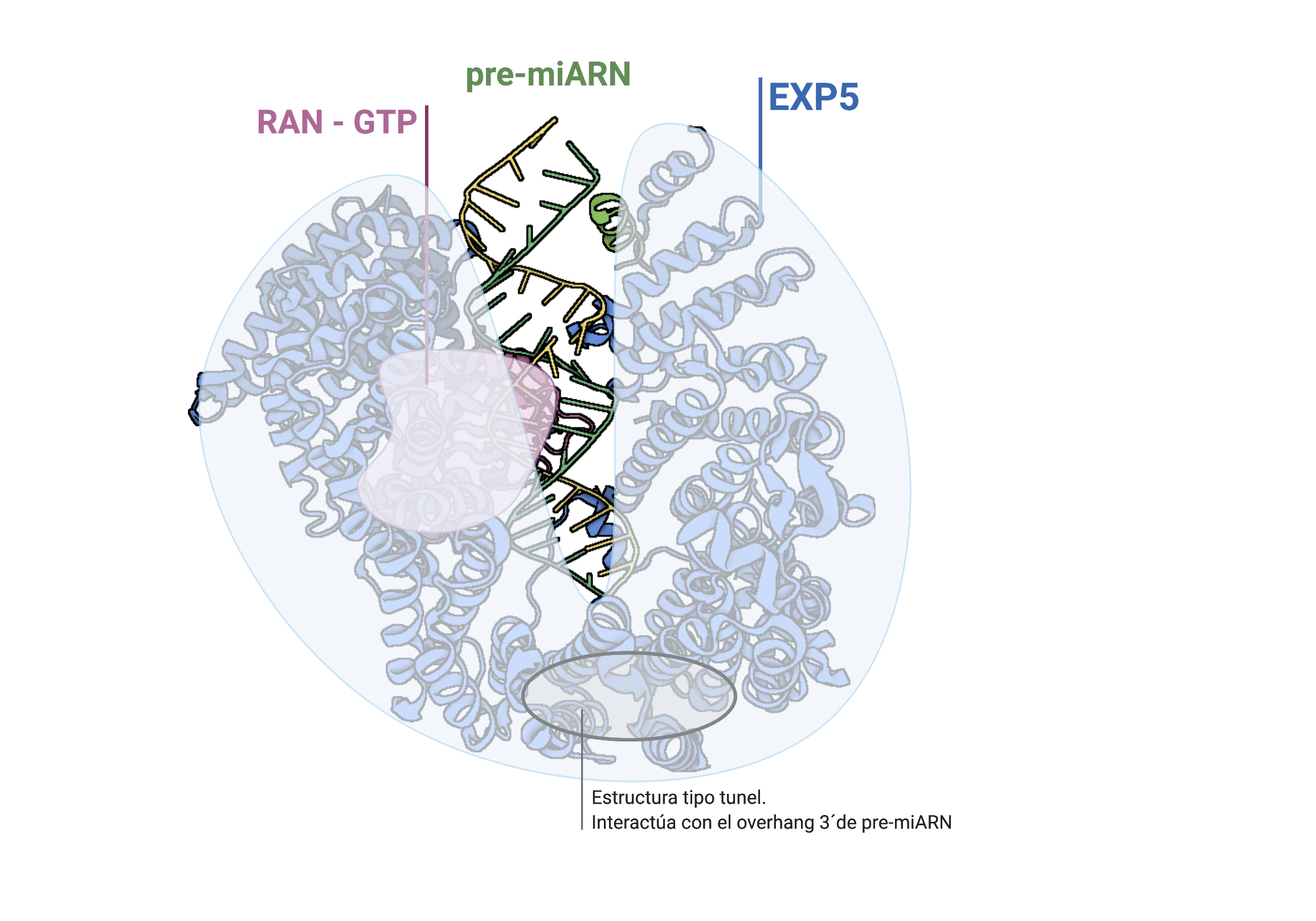
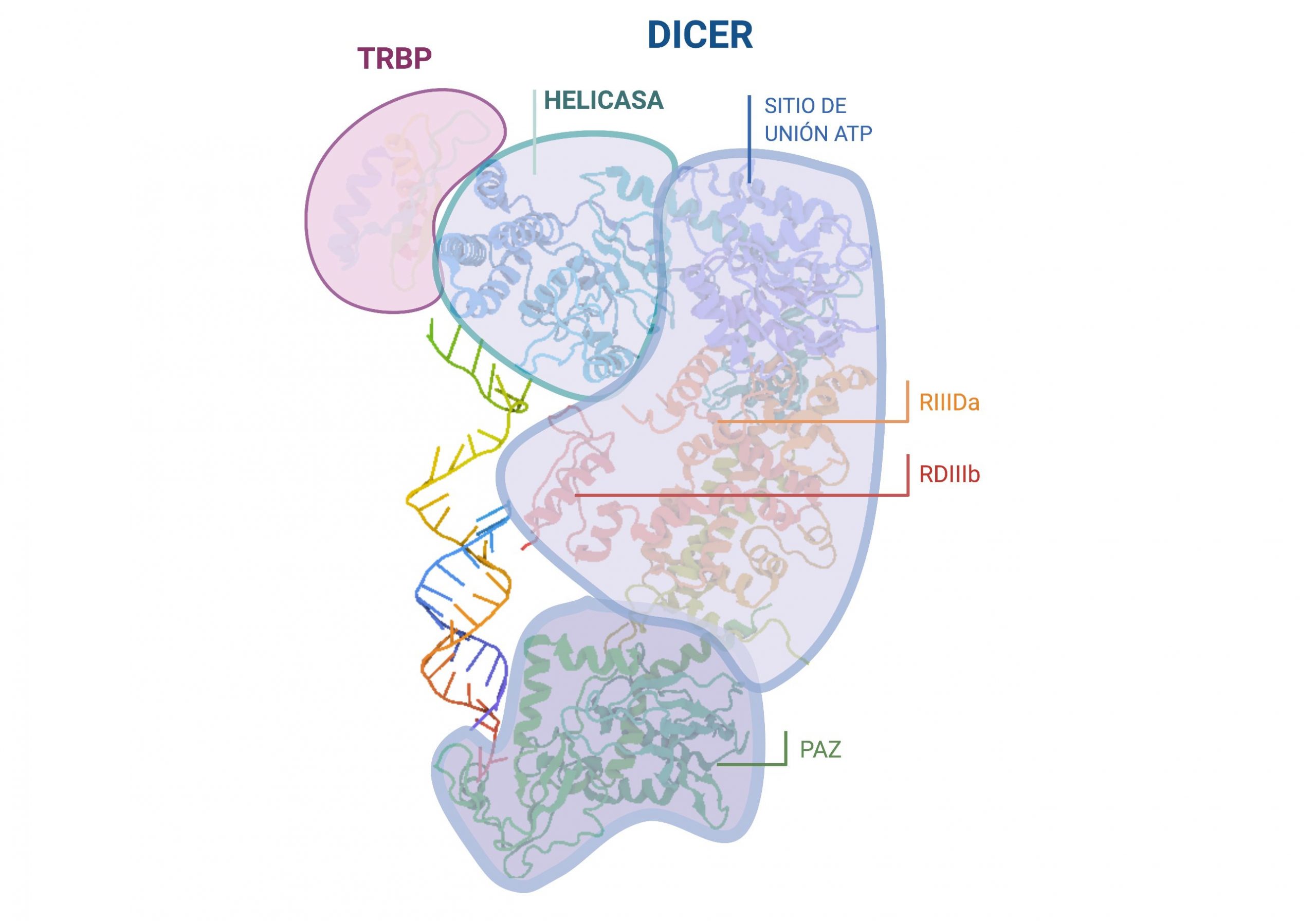
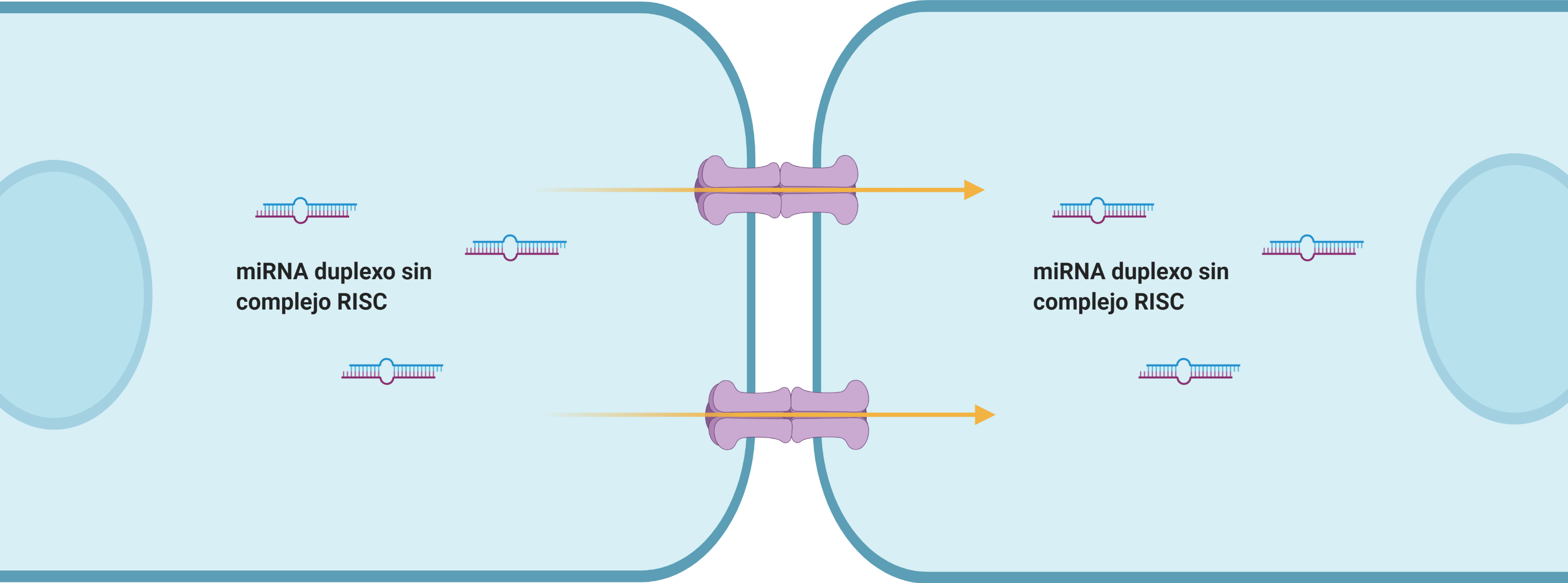
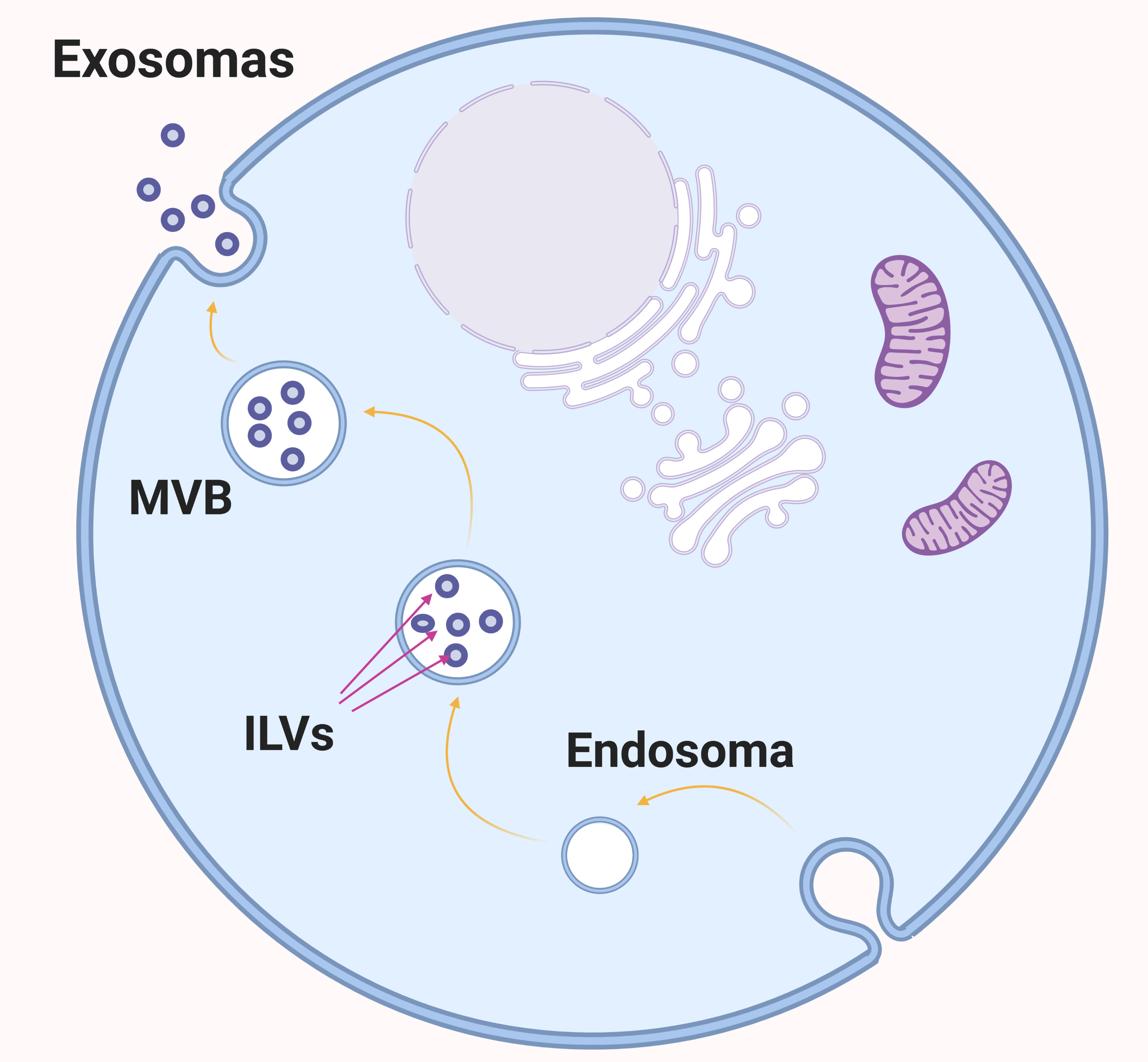
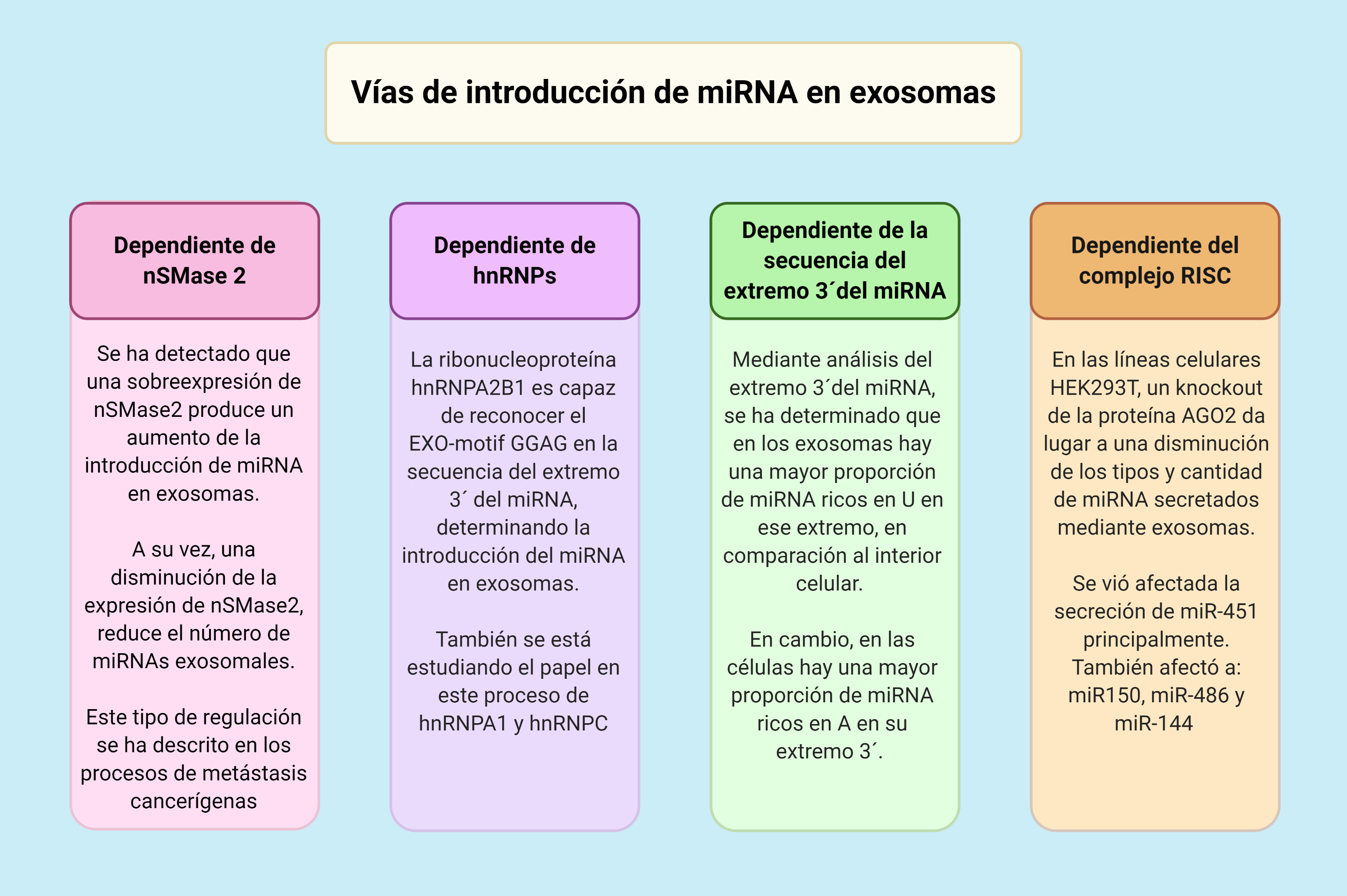
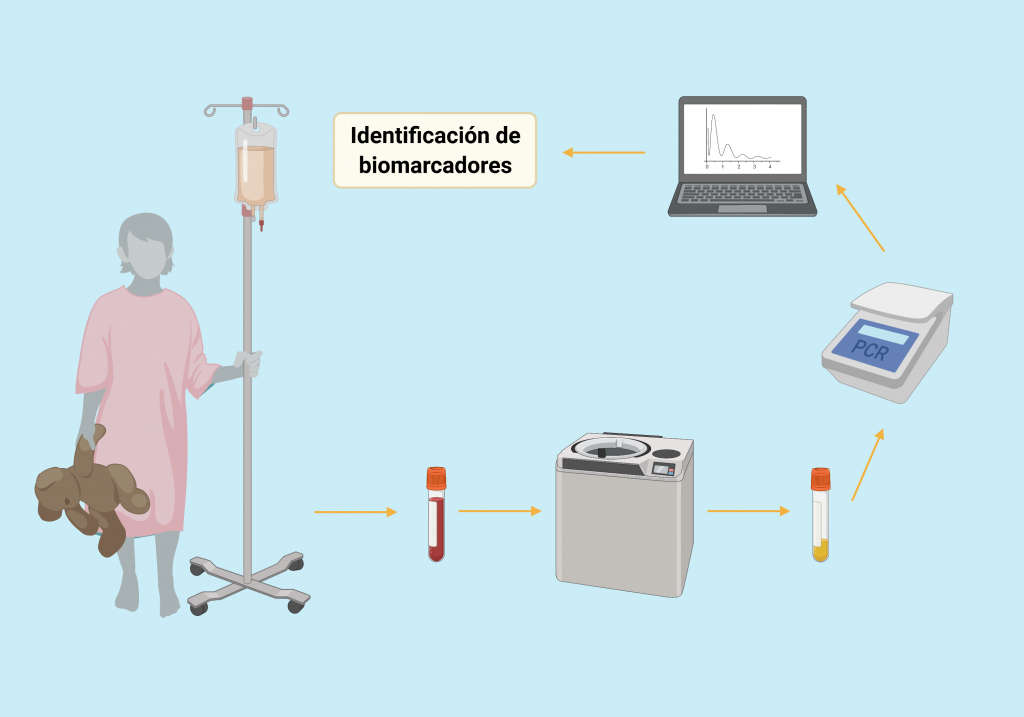
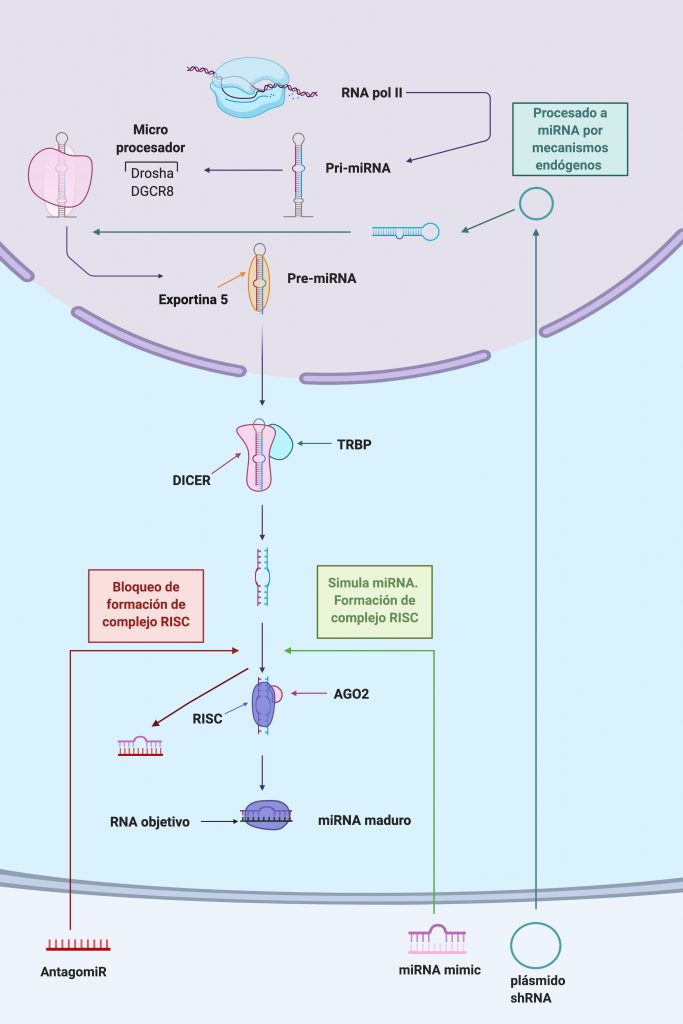
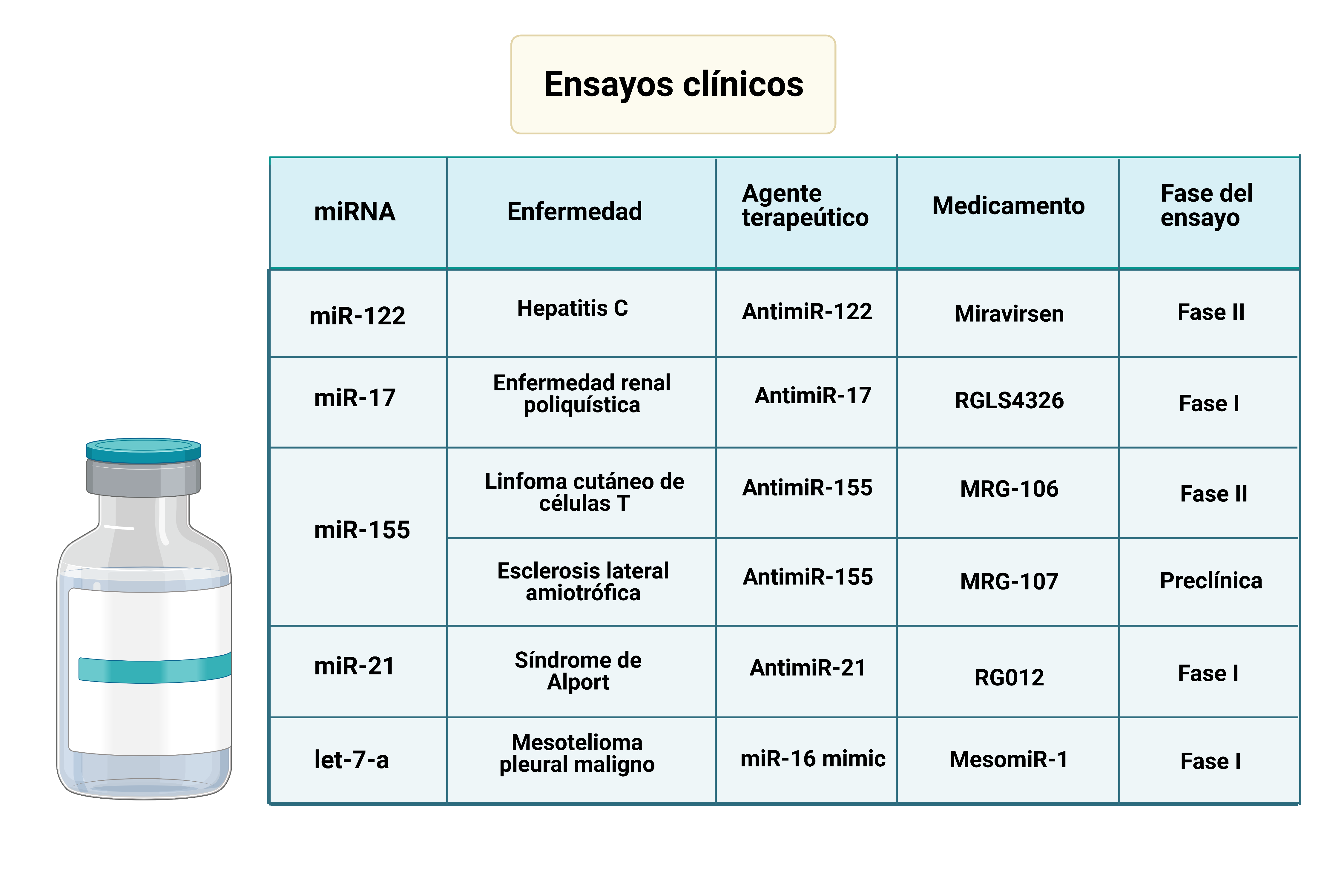
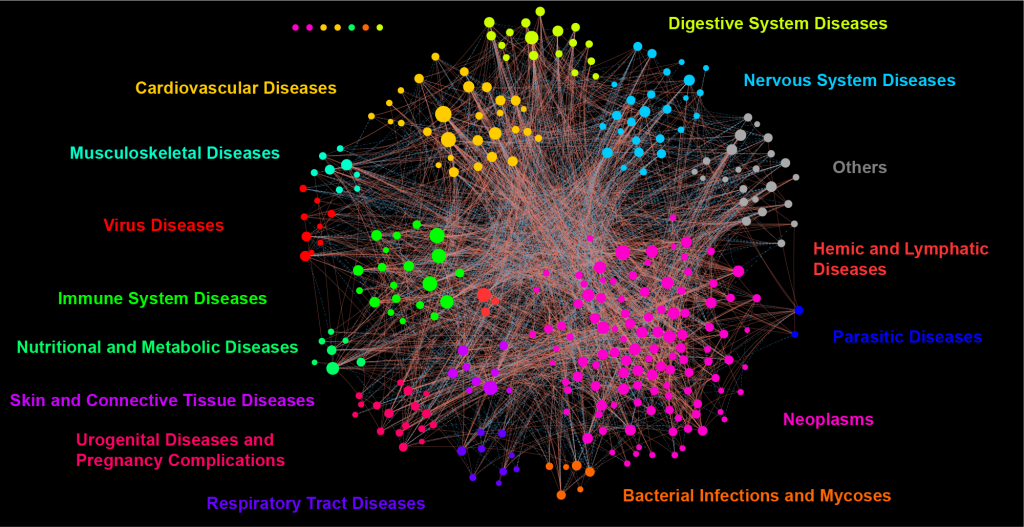
Por: Aitor Gil Iglesias, Milagros González Flores y Álvaro Mellado Cuesta
1.Introducción
Seguramente todos nosotros hemos leído que el actor Chris Hemsworth, más conocido como Thor en su papel dentro de la saga de Marvel, ha decidido retirarse del cine temporalmente al haber descubierto que tiene muchas posibilidades de desarrollar Alzheimer. Este hallazgo fue conocido por el actor cuando se encontraba rodando el documental “Limitless” para National Geographic al realizarse una prueba genética por la cual trataba de estudiar cómo su físico se vería afectado con el paso de los años. Sin embargo, estos estudios, que en un primer momento trataron de analizar el estado físico del actor, acabaron revelando la posibilidad de desarrollar Alzheimer al presentar dos copias del gen ApoE4 que hace que su riesgo de padecerla sea entre 8 y 10 veces más. Ahora bien, ¿qué es el Alzheimer y por qué es tan importante este gen en su desarrollo?
Figura I: fotos del actor Chris Hemsworth, en su papel como Thor en la saga Marvel y en la cartelera del nuevo documental que él protagoniza Limitless.
2.-¿Qué es el Alzheimer?
El Alzheimer (EA) es una enfermedad neurodegenerativa lentamente progresiva que se caracteriza por la pérdida de memoria de manera gradual. En ella también se describen otros síntomas como demencia irreversible y afectación global del resto de las funciones cognitivas (pensar, razonar…) que conllevan a un deterioro progresivo y que afectan al funcionamiento laboral y social.
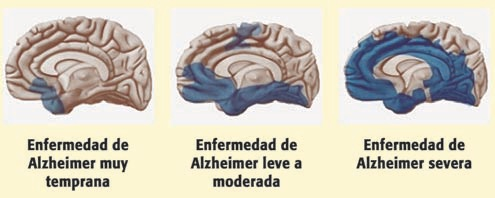
Cabe destacar que la progresión de la enfermedad a nivel cerebral es la misma en todos los pacientes. Comienza en el neocórtex basal temporal, avanza hacia el hipocampo y se extiende por todo el neocórtex.
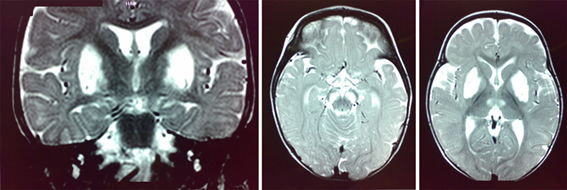
Figura II: modelo de progresión cerebral de la Enfermedad de Alzheimer. Áreas cerebrales afectadas (en azul) en las distintas fases de la enfermedad (temprana, leve-moderada y severa). Colegio Oficial de Enfermería de Cantabria (2013)
La mayoría de los casos aparecen a partir de los 70 años (“late onset”) , aunque puede haber una aparición temprana en menores de 60 (“early onset”). Afecta a alrededor de 50 millones de personas en el mundo y el principal factor de riesgo es la edad.
La early onset es autosómica dominante y hereditaria y, aunque la mayoría suceden como consecuencia de mutaciones esporádicas sobre el gen de la apolipoproteína (APOE, cromosoma 19), otros casos se deben a mutaciones en los genes que codifican para la proteína precursora amiloide (APP, cromosoma 21, por ello, las personas con Síndrome de Down, trisomía del 21, son más propensos a desarrollar la enfermedad de Alzheimer), para la presenilina 1 (PSEN1, cromosoma 14) y para la presenilina 2 (PSEN2, cromosoma 1).
No obstante, algunos otros factores de riesgo que podrían estar implicados son las enfermedades vasculares, infecciones, factores ambientales y lesiones encefálicas.
Figura III: Relación entre el riesgo de padecer la enfermedad del Alzheimer y la frecuencia alélica de algunos genes de interés. Las frecuencias alélicas más comunes se relacionan con un menor riesgo de sufrir la enfermedad y las menos habituales con un riesgo mayor. El caso de Chris Hemsworth se encuentra entre ambas, el gen de ApoE4 tiene un frecuencia alélica baja y el riesgo de desarrollar la enfermedad es medio-alto, pero como posee dos copias del gen ApoE4, se sitúa en un riesgo mayor y la frecuencia alélica ApoE4/ApoE4 es menor. Lane, et al (2018)
3.Etiopatología
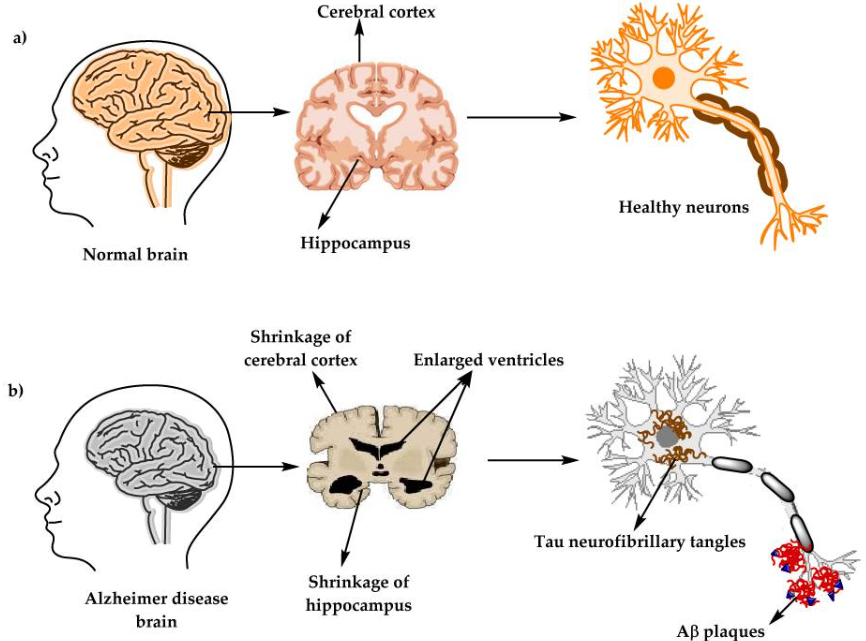
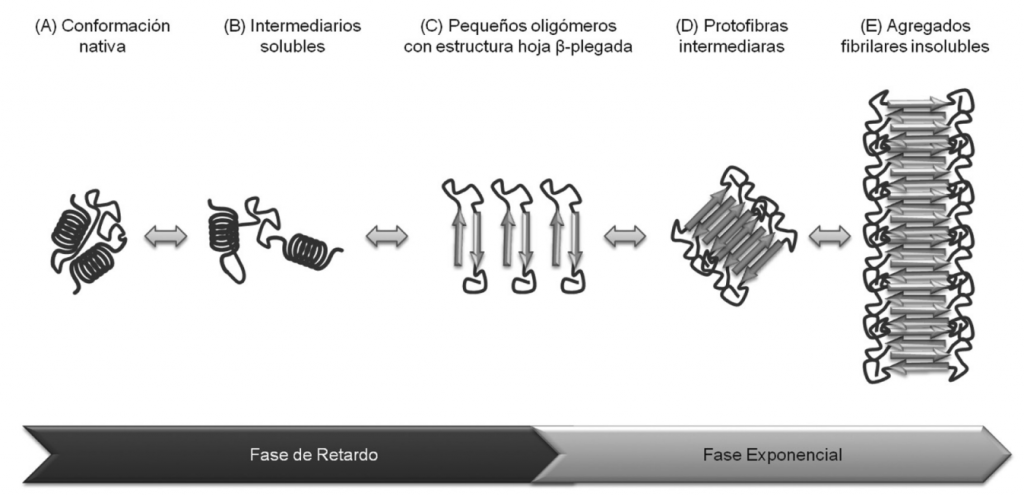
Bioquímicamente, esta enfermedad se produce como consecuencia de dos elementos neuropatológicos: las placas seniles o placas Aβ y los ovillos neurofibrilares (NFT), esenciales para entender cómo se desarrolla el Alzheimer.
Figura IV: Comparación entre el estado del cerebro y las neuronas de una persona sana (imagen A) y una persona con Alzheimer (imagen B). Se puede observar que el principal área afectado sería el hipocampo, el cual se encuentra contraído en personas con EA. Además, estos pacientes presentan una corteza cerebral más encogida y unos ventrículos más grandes. Por su parte, en la EA las neuronas presentan acumulaciones intracelulares de la proteína Tau (marrón) y acumulaciones extracelulares del péptido Aβ (rojo). Breijyeh et al (2020)
3.1.-¿Qué son las placas seniles?
Estas consisten en acumulaciones extracelulares de la proteína beta-amiloide (Aβ) anómala que en principio no es tóxica pero que se agrega con otras proteínas beta-amiloide anómalas hasta formar oligómeros tóxicos que precipitan y se depositan como placas amiloides insolubles y que son realmente tóxicas.
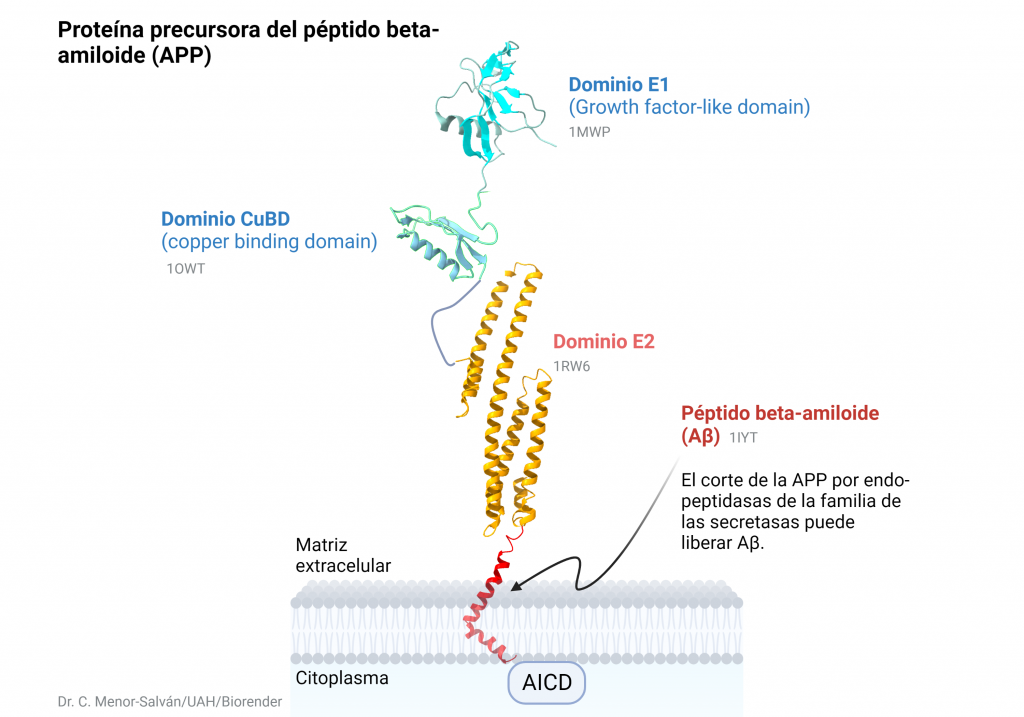
Su síntesis tiene lugar por medio de la proteínaprecursora amiloidea (APP) cuyo gen se encuentra localizado en el cromosoma 21. Esta, es una proteína transmembrana que contiene una región amino-terminal extracelular larga y una región carboxilo terminal intracelular más corta que se sintetiza en el retículo endoplasmático, se glicosila en el aparato de Golgi y por medio de vesículas se inserta en la membrana celular.
Figura V: Estructura de APP no mutada y completa (con todos sus dominios). El dominio E1 actúa como receptor de señales. El péptido beta-amiloide enlaza los dominios extracelulares de la proteína con el dominio intracelular AICD. Una familia de endopeptidasas, las secretasas, cortan la APP, liberando el dominio AICD, que alcanza el núcleo, promoviendo la expresión de diversos genes. En este corte puede liberarse el péptido beta-amiloide y, si se forma en exceso (debido a mutaciones, por ejemplo) dar lugar a la formación de placas amiloides.
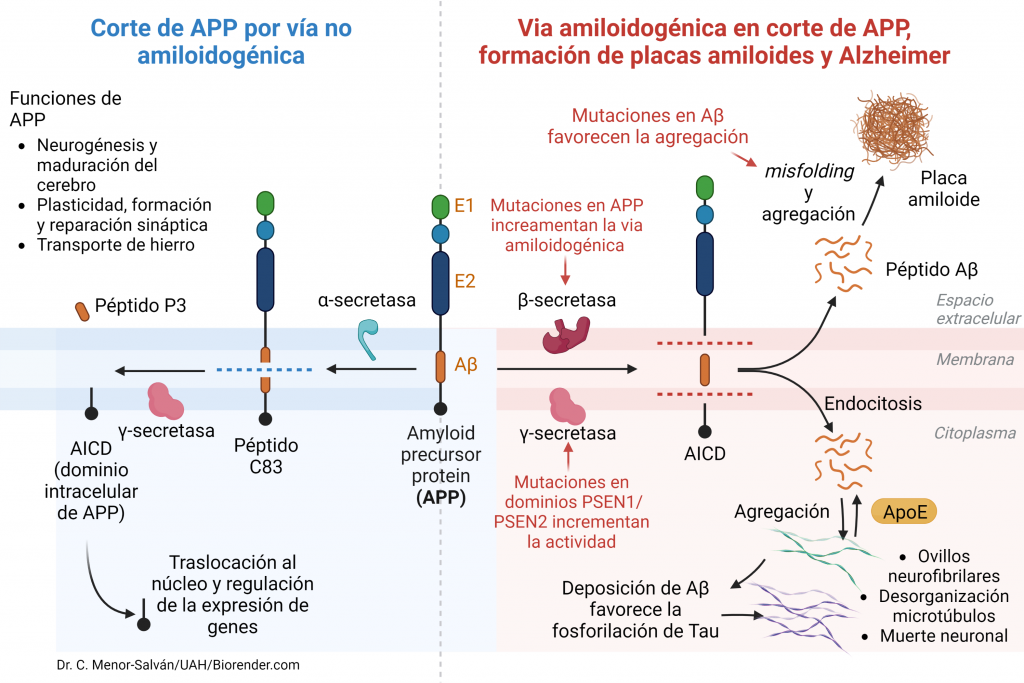
Una vez en la membrana celular, esta proteína puede ser procesada fisiológicamente por medio de dos vías:
Vía no amiloidogénica (90% de los casos): por medio de esta vía, la proteína APP es escindida por la enzima α-secretasa en un fragmento extracelular soluble neuroprotector (SAPPα, Soluble APPα) compuesto por 665 aminoácidos. Posteriormente la región no escindida que ha quedado insertada en la membrana, es procesada por la enzima γ-secretasa, quien se encarga de liberar la región C-terminal para posteriormente poder ser degradada. De esta manera, se libera al medio extracelular un péptido de 36-40 aminoácidos denominado P3, que no es neuroprotector, pero tampoco patógeno. El fragmento restante C-terminal se desprende hacia el citosol y se denomina AICD (Amyloid Intracellular Domain), que se degrada enseguida. Este proceso es constitutivo (APP tiene una vida media de 1h) y fundamental para el funcionamiento neuronal.
Vía amiloidogénica (10% de los casos): por medio de esta vía, la enzima β-secretasa o BACE libera un fragmento N-terminal de la proteína APP (SAPPβ) que no es neuroprotector. Posteriormente, el péptido que queda en la membrana será procesado por la enzima γ-secretasa dando lugar a diferentes péptidos que presentan diferentes solubilidades: el péptido Aβ (42 aá) y el AICD, que tiene un tiempo de vida mayor, va al núcleo y favorece la transcripción de genes como la propia APP, GSK3 (Ser/Thr kinasa), BACE, NEP (nefrilisina) y LRP1 (lipoprotein receptor-related protein 1).
Figura VI: Esquema sobre el procesado fisiológico de la proteína APP, enzimas que actúan y productos que se obtienen en la vía no amiloidogénica (sobre fondo azul) y en la vía amiloidogénica en condiciones normales y durante el Alzheimer (sobre fondo rojo). En la vía amiloidogénica, el dominio transmembrana de APP sufre una escisión diferente con respecto la vía no amiloidogénica tanto en condiciones normales como durante el Alzheimer. Como consecuencia se genera un producto de mayor tamaño, el péptido Aβ. En el caso del Alzheimer, mutaciones en Aβ, en APP, en ApoE y en los dominios PSEN1/PSEN2 de la secretasa favorecen el incremento de la via amiloidogénica. Esto hace que se dirija hacia el espacio extracelular y se agregue hasta formar placas amiloides y que pase al interior celular por endocitosis y favorezca la fosforilación de Tau.
Los péptidos más comúnmente formados en la vía amiloidogénica son los péptidos Aβ40, los cuales presentan una estructura en forma de hélice alfa que puede ser fácilmente degradada. Sin embargo, en situaciones patológicas predomina la síntesis del péptido Aβ42 y Aβ43, los cuales son mucho más insolubles, tienden a formar láminas β y a interaccionar entre ellos formando fibrillas que pueden acumularse y formar placas extracelulares amiloides insolubles que se diseminarán por otras estructuras como el hipocampo, la amígdala y la corteza cerebral resultando realmente dañinas.
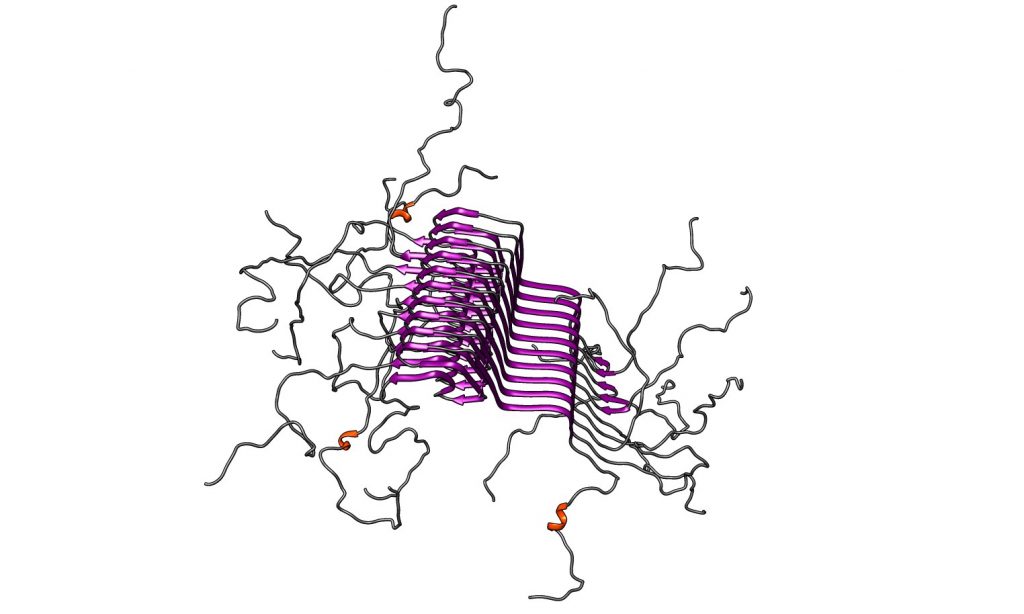
Figura VII : péptido Aβ-42 agregado, formado placas amiloides. Los residuos poco polares de los aminoácidos tienden a apilarse en paralelo favoreciendo el plegamiento incorrecto en láminas β. Esto genera una estructura insoluble que precipita. Imagen obtenida de Chimera. Código PDB: 5KK3 PDB DOI:10.2210/pdb5KK3/pdb
Ahora bien, ¿por qué es tan importante Aβ?
De manera normal, esta proteína se encarga de modular la transmisión del impulso eléctrico entre las neuronas, por lo que, un mal funcionamiento o no funcionamiento supondría la pérdida de la comunicación neuronal, es decir, una pérdida de sinapsis. Esto se debe a que puede alterar la actividad de quinasas, producir señales de estrés oxidativo, daños mitocondriales e incluso defectos en el transporte axonal que son los que finalmente conducen a una degeneración neuronal. Todo ello es lo que acabaría produciendo en estos pacientes una gran deficiencia cognitiva.
Además, Aβ en bajas concentraciones es quelante de metales, mejora la memoria y el aprendizaje, es antioxidante, interviene en el mantenimiento de la barrera hematoencefálica etc.
Esta acumulación de Aβ anómala en condiciones patológicas, en las que no solo se incluye el Alzheimer, es como consecuencia de las mutaciones en algunos genes como APP, PSEN1 (proteína que se encarga de activar el complejo de la γ-secretasa) y PSEN2 (poco frecuentes), los cuales, afectan al catabolismo y anabolismo de Aβ y como consecuencia producen una rápida progresión de neurodegeneración.
Concretamente, dichas mutaciones afectan al procesamiento alternativo de APP, y como consecuencia se generan fundamentalmente proteínas Aβ truncadas, Aβ42 y Aβ43. Estas presentan residuos poco polares que tienden a apilarse en paralelo induciendo a un plegamiento incorrecto de las láminas beta las cuales, son muy insolubles y al no ser reconocidas por los sistemas de degradación acaban precipitando para formar fibrillas amiloides que generan una gran neurotoxicidad.
Figura VIII : Formación de los péptidos Aβ insolubles. Su agregación da lugar a oligómeros y finalmente acaba originando las placas seniles. Duran-Aniotz, et al (2013)
3.2-¿Qué son los ovillos neurofibrilares (NFT)?
Son lesiones de tipo intracelular formadas por filamentos helicoidales emparejados (PHF) que se producen como consecuencia de la hiperfosforilación de las proteínas Tau y que puede deberse a la aparición de proteínas Aβ, al estrés oxidativo o a mediadores inflamatorios. La formación de estos ovillos producen muerte neuronal por apoptosis.
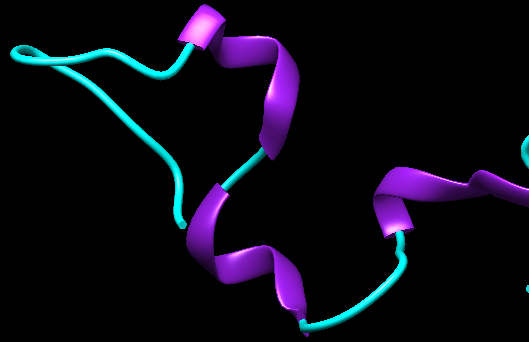
Estas proteínas se encuentran de manera mayoritaria en neuronas y células de la Glía y son esenciales para promover el ensamblaje de los microtúbulos neuronales así como para mantener la integridad estructural neuronal y por tanto para permitir el crecimiento de los axones y el transporte axonal. Concretamente, la proteína Tau se encarga de facilitar la polimerización de la tubulina para permitir que se formen los microtúbulos.
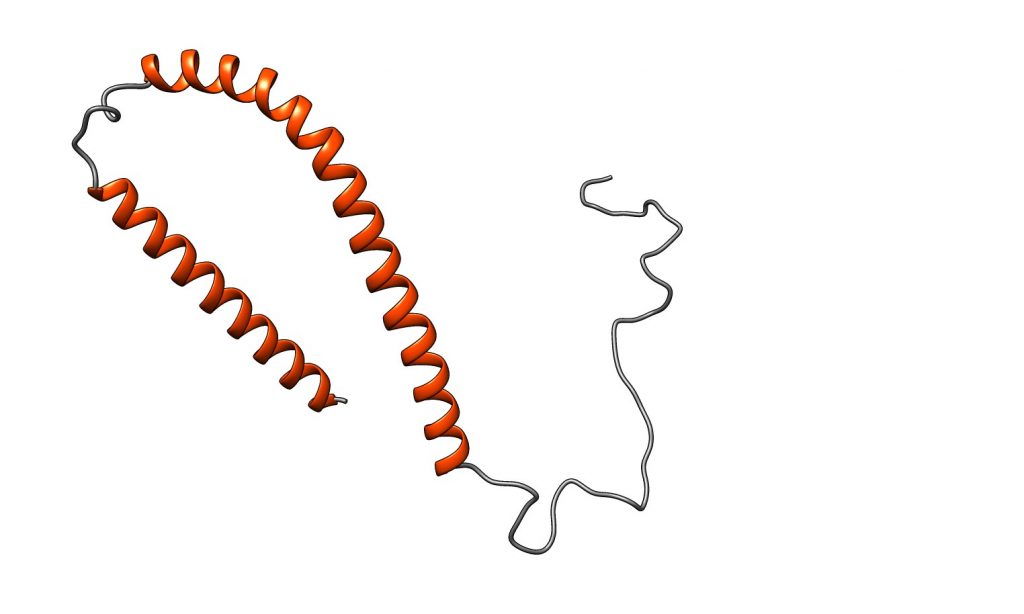
Figura IX: Estructura de la proteína Tau no mutada. Imagen obtenida con Chimera. Código PDB: 2MZ7. PDB DOI: 10.2210/pdb2MZ7/pdb
El gen que codifica para la proteína Tau, localizado en el gen 17, contiene 16 exones y en el cerebro adulto sufre diferentes tipos de empalme alternativo en los exones 2, 3 y 10 originando finalmente 6 isoformas de Tau. Estas isoformas contienen entre 3-4 repeticiones peptídicas de 31-32 residuos en la región carboxilo terminal que se corresponde con el dominio de unión a los microtúbulos. Así mismo, el estado de fosforilación del gen varía durante el desarrollo y es lo que conduce a un cambio en el empalme del ARN.
Figura X: Regiones donde se puede fosforilar a Tau (señaladas con flechas negras). Las regiones señaladas en rojo son aquellas que se encuentran anormalmente fosforiladas en el Alzheimer, las regiones verdes aquellas en las cuales Tau está fosforilada de manera normal y en azul las fosforilaciones encontradas de Tau en el Alzheimer y en condiciones normales. Por tanto, las regiones señaladas en rojo serían aquellas que no deberían encontrarse fosforiladas y las que conllevan a una agregación de Tau en el Alzheimer. Luna-Muñoz, et al (2012)
Tau en el embrión aparece fosforilada, pero en adultos está muy poco fosforilada. Se puede fosforilar por más de 80 sitios Ser/Thr y 5 de Tyr, gracias a GSK-3, PKA, la cuales son Ser/Thrk o Fyn (TyrK). En la enfermedad de Alzheimer, está hiperfosforilada.
Además, cuando Tau está fosforilada cambia de localización y, en lugar de localizarse preferentemente en los axones, se dirige hacia las dendritas y se lleva a Fyn secuestrada consigo. Fyn fosforila a receptores de glutamato (Glu) (receptores NMDA sobre todo) lo que producirá una muerte neuronal por sobreexcitación.
De esta manera, y dado la importancia de la fosforilación en este gen, se ha podido observar que en los procesos de neurodegeneración Tau sufre una fosforilación anormal irreversible que impide su función normal y su agregación. Como consecuencia, no puede unirse correctamente a los microtúbulos y todo ello junto con la formación de las fibrillas por su agregación, impiden que la neurona pueda transmitir señales eléctricas. Además todo ello favorece un mal transporte axonal de nutrientes, lo cual contribuye a la degeneración sináptica y a la muerte neuronal.
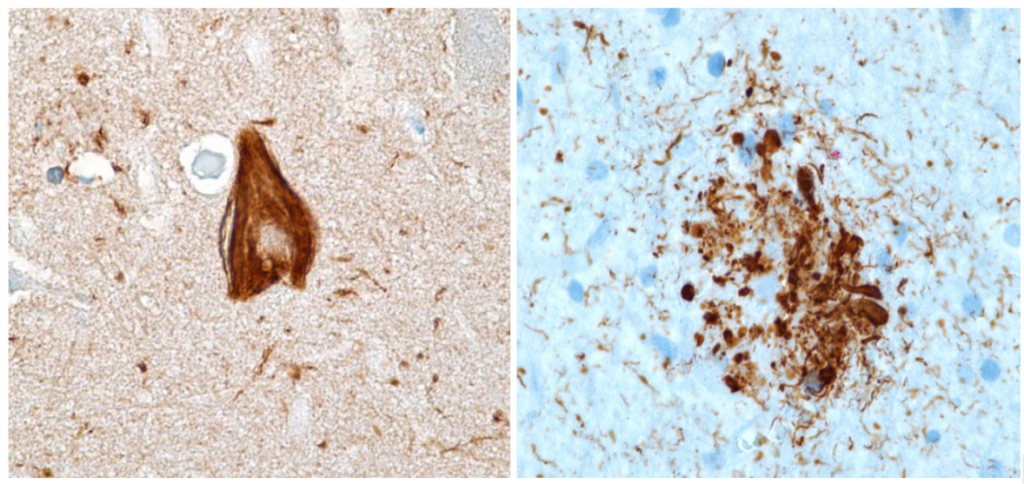
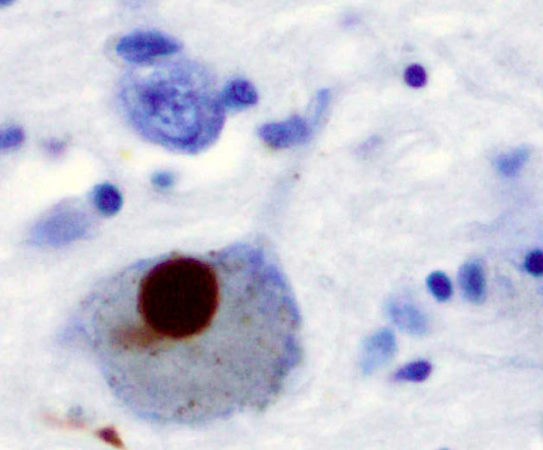
Figura XI: Tinción inmunohistoquimica de ovillos neurofibrilares (marcado en marrón). Castellani, et al (2010)
Además, parece ser que, una vez Tau está fosforilada y ha perdido su estructura, se empieza a transmitir de unas neuronas a otras, induciendo el cambio conformacional patológico en proteínas Tau normales por contacto (como los priones). Gracias a ello, la enfermedad puede propagarse y afectar a diferentes estructuras.
4.-¿Qué es la ApoE?
La ApoE es una proteína de 299 aminoácidos, que está dentro del grupo de las apolipoproteínas, las cuales están implicadas en el metabolismo de los lípidos (forman las lipoproteínas), siendo la ApoE la única que encontramos en el cerebro.
En la ApoE hay varios dominios de unión a destacar, como por ejemplo el de unión a lípidos, al receptor, a heparina o la región bisagra. Todos ellos con gran importancia en las funciones de la ApoE como veremos a continuación.
Sin embargo, el interés reciente hacia la ApoE no es por su papel en el metabolismo de los lípidos, sino por su relación con el péptido Aβ y la proteína Tau, ambos implicados en el inicio y avance de la enfermedad de Alzheimer.
4.1- Relación ApoE – péptido Aβ
La ApoE es sintetizada por astrocitos y la microglía, debido a la alta importancia de los lípidos para el desarrollo y mantenimiento de las neuronas, pero, ¿cuál es su función dentro del metabolismo de lípidos?¿y que nexo tiene esto con el péptido Aβ y la enfermedad de Alzheimer?
FUNCIÓN DE LA ApoE
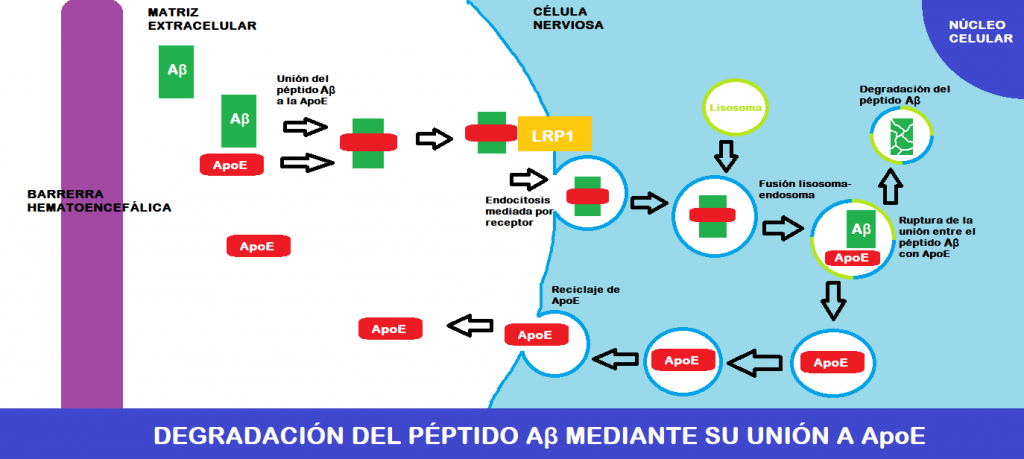
La ApoE se encuentra circulando por la matriz extracelular nerviosa, y es ahí dónde se une al colesterol, que proviene del interior de las células nerviosas (sale a través de transportadores de la familia ABC), y ambos forman lipoproteína discoidal. Esta se une a recetores celulares para lipoproteínas (LRP1 principalmente) y entra en las células por endocitosis mediada por receptor. Ya en el interior, esta lipoproteína entra en el metabolismo de los lípidos.
Sin embargo, lo más interesante de esto es que el péptido Aβ es capaz de unirse a la ApoE circulante (a su dominio de unión a heparina concretamente), formando una molécula que es capaz de entrar en las células al igual que la lipoproteína que hemos mencionado antes (endocitosis mediada por LRP1).
Una vez dentro, el endosoma donde está la molécula ApoE-péptido Aβ se fusiona con lisosomas, y esto hace que por aumento del pH y acción de proteasas lisosomales ApoE y el péptido Aβ se separen. El destino de cada uno es bien distinto:
ApoE queda en una vesícula que se fusionará con la membrana plasmática, permitiendo que salga al exterior y se recicle.
El péptido Aβ es sustrato para proteasas lisosomales, por lo que acaba siendo degradado.
FIGURA XII: Modelo explicativo sencillo de la degradación del péptido Aβ a través de su unión a ApoE. Imagen creada en Paint
Pero yendo más allá, lo que más ha despertado el interés científico de todo esto y lo que está relacionado con el caso concreto de Chris Hemsworth son las distintas isoformas de ApoE que aparecen en las poblaciones, cada una de ellas con un papel distinto en la predisposición a sufrir Alzheimer.
4.2- Isoformas de ApoE y su importancia en el Alzheimer
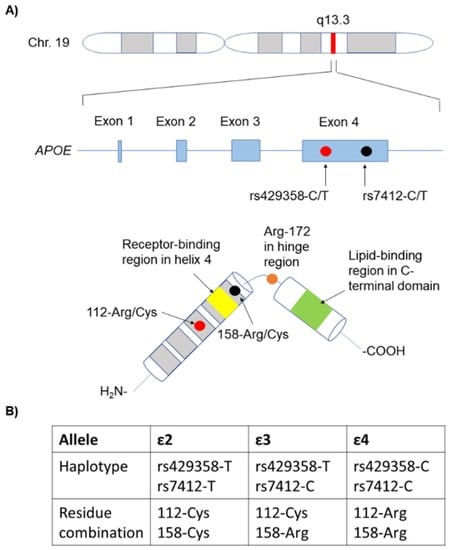
Hay 3 isoformas de la ApoE, las cuales sólo se diferencian en los residuos 112 y 158:
ApoE2: posee cisteínas (en 112 y 158). Se la considera «protectora» frente a la enfermedad de Alzherimer. Se cree que su eliminación del péptido Aβ es más eficaz.
ApoE3: posee cisteína (en 112) y arginina (en 158). Es la isoforma mayoritaria en la población y se considera que su papel es neutro frente a la enfermedad de Alzheimer.
ApoE4: posee argininas (en 112 y 158). Su presencia aumenta la probabilidad de sufrir la enfermedad de Alzheimer hasta 4 veces si aparece en heterocigosis y hasta 10 veces si aparece en homocigosis.
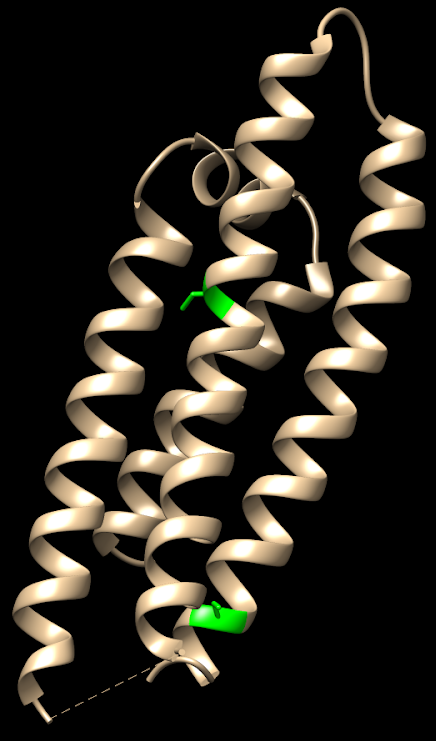
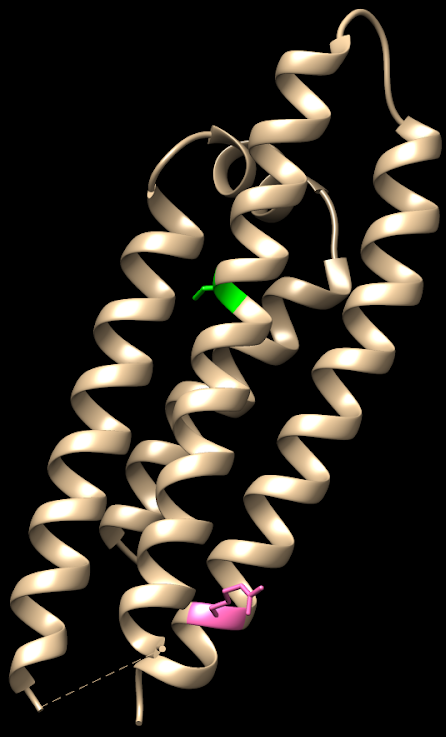
FIGURA XIII: Distintas isoformas de ApoE con sus diferencias estructurales. Fernandez, et al (2011)FIGURA XIV: ApoE2: (2 Cys en verde, en posiciones 112 y 158). Imagen obtenida con Chimera. Código PDB: 1NFO. PDB DOI: 10.2210/pdb1NFO/pdbFIGURA XV: ApoE3: (Cys en verde, en posición 112 y Arg en rosa en posición 158). Imagen obtenida con Chimera. Código PDB: 1NFN. PDB DOI: 10.2210/pdb1NFN/pdbFIGURA XVI: ApoE4: (2 Arg en rosa, en posiciones 112 y 158). Imagen obtenida con Chimera. Código PBD: 1B68. PDB DOI: 10.2210/pdb1B68/pdb
Pero, ¿por qué ApoE4 aumenta tanto la probabilidad de desarrollar Alzheimer?, ¿en qué afecta el cambio de tan solo 2 aminoácidos a una proteína que posee 299? Ahora mismo te lo explicamos
4.3- ApoE4
Su única diferencia estructural frente a la isoforma protectora es el cambio de dos cisteínas (aminoácidos polares sin carga), por dos argininas (aminoácidos polares con carga positiva). En principio parece un cambio insignificante, pero este pequeño cambio hace que la ApoE4 tenga una conformación algo más «cerrada» que el resto de ApoE, afectando más de lo que creemos en su función.
La conformación que adopta ApoE4 hace que se una más deficientemente a sus receptores, por lo que ApoE4 se encuentra durante más tiempo circulante por la matriz extracelular. Al disminuir la endocitosis mediada por receptor, disminuye la proteólisis intracelular y para compensarlo, aumenta la proteólisis extracelular.
Esta proteólisis favorece la acumulación del péptido Aβ, su unión con otros péptidos Aβ (formando oligómeros Aβ) y el inicio y desarrollo de la formación de placas amiloides y de la enfermedad de Alzheimer. De hecho se han descubierto restos de esta ApoE en las placas amiloides.
Pero esto no es todo, ya que recientemente se ha visto que ApoE también interactúa con la proteína Tau, una proteína muy importante en el desarrollo de la enfermedad de Alzheimer y cuya hiperfosforilación explica muchos de los signos clínicos de la enfermedad.
4.4- Relación ApoE – proteína Tau
Se ha visto que ApoE está relacionada con la acumulación de Tau y Tau fosforilada, especialmente ApoE4 es quien favorece en mayor medida está acumulación, la cual deriva en la formación de los ovillos neurofibrilares. Para explicar está relación de manera más detallada vamos a presentaros un experimento realizado con ratones en 2017.
En este experimento se quiso conocer más acerca del papel de ApoE en la neurodegeneración a través de Tau. Para ello se utilizaron ratones que sobreexpresaban Tau humana (ratones P301S) y las distintas isoformas de ApoE, creando distintos grupos de ratones:
Tau-ApoE2 (TE2): expresaban Tau y ApoE2.
Tau-ApoE3 (TE3): expresaban Tau y ApoE3.
Tau ApoE4 (TE4): expresaban Tau y ApoE4. Es el grupo de ratones en el que mayor neurodegeración se observó a los 9 meses.
Sin Tau y con ApoE (KI): no se observó neurodegeneración a los 9 meses. Esto confirmaba que ApoE por sí sola no ocasiona neurodegeneración.
Con Tau y sin ApoE (TEK0): se observó neurodegeneración a los 9 meses, pero en menor nivel que en cualquiera de los grupos con Tau y las distintas formas de ApoE.
Con esto se confirmó que ApoE (especialmente ApoE4) juega un papel importante en la neurodegeneración a través de Tau. Además en este experimento se observó que ApoE4 no influye en la síntesis de Tau, sino que la acumulación de Tau se debe a que ApoE4 afecta a su degradación por autofagia.
Por último se vio que cuando había patología de Tau (hiperfosforilación y acumulación), ApoE4 provoca neuroinflamación, promoviendo la activación de la microglía y la expresión de genes A1 en astrocitos.
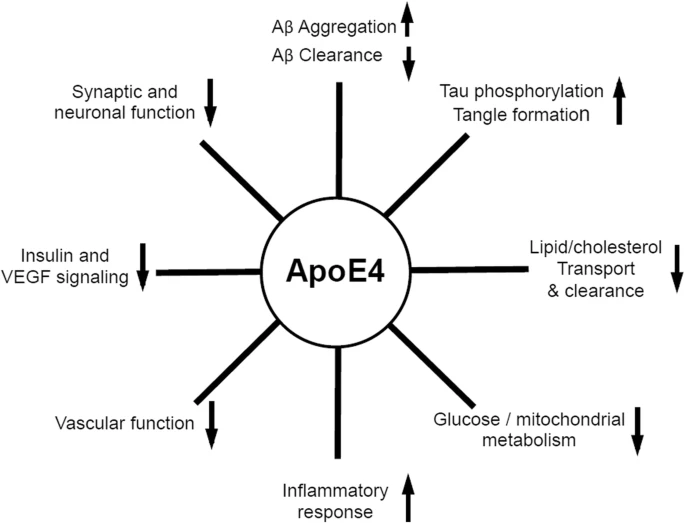
FIGURA XVII: Resumen de los efectos de ApoE4 a nivel cerebral, metabólico, vascular y hormonal. ApoE4 favorecería la fosforilación de Tau, los procesos inflamatorios y la agregación de Aβ. Por el contrario, disminuye la eliminación de Aβ, la función neuronal sináptica y vascular, la señalización de la insulina y VEGF (factor de crecimiento endotelial vascular), el metabolismo mitocondrial y el transporte y aclaramiento de lípidos y colesterol. Safieh et al (2019)
4.5- Genes implicados
En la prueba genética que se realizó el actor Chris Hemsworth, se analizo el gen que da lugar a la síntesis de la ya mencionada ApoE. El gen de la ApoE está formado por 4 exones y se encuentra en el cromosoma 19.
Las mutaciones de mayor interés en este gen son las que afectan a los codones que dan lugar a los aminoácidos 112 y 158, y por tanto a las distintas isoformas ya vistas (ApoE2, ApoE3 o ApoE4) y su influencia en la probabilidad de desarrollar la enfermedad de Alzheimer.
FIGURA XVIII: Representación del gen de la ApoE y su lugar en el cromosoma. Dibujo de la proteína ApoE, incluyendo sus principales dominios y tabla donde se compara el genotipo y aminoácidos que distinguen a las tres isoformas. Abondio, et al (2019)
5.Referencias
Breijyeh, Z., & Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules (Basel, Switzerland), 25(24), 5789. https://doi.org/10.3390/molecules25245789
Lane, C. A., Hardy, J., & Schott, J. M. (2018). Alzheimer’s disease. European journal of neurology, 25(1), 59–70. (2020) https://doi.org/10.1111/ene.13439
Lin, Y. T., Seo, J., Gao, F., Feldman, H. M., Wen, H. L., Penney, J., Cam, H. P., Gjoneska, E., Raja, W. K., Cheng, J., Rueda, R., Kritskiy, O., Abdurrob, F., Peng, Z., Milo, B., Yu, C. J., Elmsaouri, S., Dey, D., Ko, T., Yankner, B. A., … Tsai, L. H. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron, 98(6), 1141–1154.e7. (2018) https://doi.org/10.1016/j.neuron.2018.05.008
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., … & Holtzman, D. M. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature, 549(7673), 523-527. (2017)
Spinney, L. The forgetting gene: for decades, most researchers ignored the leading genetic risk factor for Alzheimer’s disease. That is set to change. Nature, 510(7503), 26-29. (2014) https://doi.org/10.1038/510026a
Figura III: Lane, C. A., Hardy, J., & Schott, J. M. Alzheimer’s disease. European journal of neurology, 25(1), 59–70. (2018) https://doi.org/10.1111/ene.13439
Figura IV: Breijyeh, Z., & Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules (Basel, Switzerland), 25(24), 5789. (2020)
Figura VIII: Duran-Aniotz, C., Moreno-Gonzalez, I., & Morales, R. Agregados amiloides: rol en desórdenes de conformación proteica. Revista médica de Chile, 141(4), 495-505. (2013)
Figura X: Luna-Muñoz, J., Zamudio, S., De La Cruz, F., Minjarez-Vega, B., & Mena, R. Acción protectora de la proteína tau en la enfermedad de Alzheimer. Revista Mexicana de Neurociencia, 13(3), 160-167. (2012)
Figura XI: Castellani, R. J., Rolston, R. K., & Smith, M. A. Alzheimer disease. Disease-a-month : DM, 56(9), 484–546 (2010)
Figura XIII: Fernandez, Celia & Hamby, Mary & McReynolds, Morgan & Ray, William. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Frontiers in Aging Neuroscience. (2011) 11. 10.3389/fnagi.2019.00014
Figura XVII: Safieh, M., Korczyn, A. D., & Michaelson, D. M. ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC medicine, 17(1), 1-17. (2019)
Figura XVIII: bondio, P., Sazzini, M., Garagnani, P., Boattini, A., Monti, D., Franceschi, C., Luiselli, D., et al. The Genetic Variability of APOE in Different Human Populations and Its Implications for Longevity. Genes, 10(3), 222. (2019)
Virus oncolíticos como nueva terapia frente al cáncer
escrito por natpmv_3C | 10 enero, 2023
Realizado por Natalia López Escobar y Pablo Martín Valenzuela.
Biología molecular. 3º Biología Sanitaria. Grupo C.
1. Introducción
Actualmente, existen diversas terapias frente al cáncer, por un lado, las tradicionales, donde encontraríamos la quimioterapia, la cirugía y la radioterapia; y por el otro, las de nueva incorporación, donde nos encontraríamos la terapia dirigida, la inmunoterapia y la terapia hormonal láser entre otras.
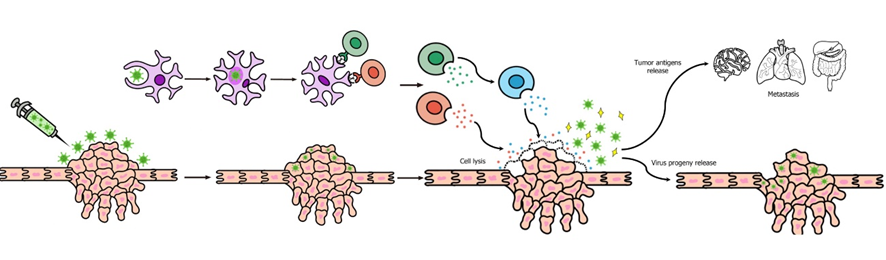
Una de las líneas de investigación más recientes frente al cáncer es el uso de virus oncolíticos. Estos virus son modificados genéticamente para reconocer al cáncer e infectarlo. Gracias a esto, no sólo conseguimos que las células del tumor infectadas mueran, si no que permite, además, una activación del sistema inmune del hospedador. Al lisar a las células tumorales, se liberan antígenos que serán reconocidos por células del sistema inmune que activarán la respuesta inmune.
1.1. El cáncer
El cáncer es un conjunto de enfermedades que se presentan cuando las células se multiplican sin control y se diseminan a los tejidos que los rodean. Las características que deben cumplir las células para ser consideradas células tumorales fueron descritas en 2011 por Hanahah y Weinberg (1 y 2). Estas características son:
Autosuficiencia de señales de crecimiento, es decir, no necesitan señales externas para crecer.
Insensibilidad a señales antiproliferativas.
Evasión de la apoptosis.
Adaptación metabólica.
Inmortalización, mediante el alargamiento de los telomeros
Capacidad de invasión y angiogénesis, es decir, capacidad de crear nuevos vasos sanguíneos.
Capacidad de colonización de otros tejidos (metástasis).
Evasión de la respuesta inmune
Catherine Sánchez, 2013. Conociendo y comprendiendo la célula cancerosa: Fisiopatología del cáncer
1.2. Los virus oncolíticos
La idea de que los virus pueden ser utilizados contra el cáncer no es novedosa, proviene de mediados del siglo XX, cuando se observaron, en pacientes con linfomas y leucemias, remisiones del tumor, coincidentes con infecciones por virus, como el de la hepatitis o el del sarampión (3). Se empezó entonces a probar la infección de pacientes oncológicos con virus. No tuvo la eficacia esperada y además, se encontraron muchos efectos secundarios causados por los virus, de modo que se detuvo la investigación (4).
Ahora, gracias a los avances de la ingeniería genética, se han podido desarrollar virus oncolíticos más seguros y específicos frente a determinados tipos de tumores.
2. Mecanismos moleculares de acción
Los virus oncolíticos son capaces de infectar células anormales a través de dianas celulares específicas: Transcriptasa inversa de telomerasa humana, antígeno específico de próstata, ciclooxigenasa-20, her2/neu…
La Transcriptasa inversa de telomerasa humana o hTERT, es una subunidad catalítica de la enzima Telomerasa. La telomerasa es una polimerasa ribonucleoproteica, que mantiene los extremos de los telomeros. No puede ser considerado un protooncogén, ya que su mutación por sí sola no induce el crecimiento. Si que es importante su papel en la inmortalización de las células tumorales. La mutación en el promotor de hTERT confiere una mayor agresividad al melanoma (5)
HER2/neu: es un tipo de HER (Human EGF Receptor). Es un receptor con actividad Tyr quinasa, que tiene como ligando EGF (Epidermal Growth Factor). HER2 tiene un peculiaridad, ya que presenta la capacidad de activarse sin necesidad de ligando. Se ha visto su sobreexpresión hasta en el 30% de los cánceres de mama. (6 y 7)
Una vez hemos visto ejemplos de algunas dianas que pueden usar los virus para reconocer a las células tumorales, podemos ver los mecanismos que producen la muerte del tumor. La infección viral provoca, en primer lugar, la lisis de células tumorales. Las células dendríticas, reconocen antígenos virales y estimulan la producción de Interferon de tipo I, factor de necrosis tumoral alfa. (TNF-α) y citoquinas como la interleucina 2 (IL-2). El TNF-α regula la expresión del complejo de histocompatibilidad, e influye positivamente en la acción de la enzima caspasa y contribuye a la apoptosis celular en algunos tumores. Además, está molécula está relacionada con la activación de los linfocitos T citotóxicos y las células NK. Por lo tanto, conseguimos la muerte de las células tumorales mediante dos modos: por un lado, la lisis celular provocada por el ciclo de infección del virus. (8)
Santos Apolonio et al. Oncolytic virus therapy in cancer.
Una de las principales ventajas que supone el uso de virus oncolíticos es que podría inducir regresión en casos de metástasis (que representan la mayor parte de las muertes por cáncer) ya que, al provocar la lisis celular, salen nuevas partículas virales que pueden viajar hacia zonas lejanas donde haya metástasis. Pero el mecanismo más importante son las nuevas respuestas inflamatorias, que se producen cuando se lisan las células tumorales y salen antígenos al exterior. Estas nuevas respuestas inflamatorias, unidas a la memoria inmune celular, pueden provocar la regresión de las metástasis. (8)
Uno de los virus oncolíticos más prometedores es el CTV-m7, el cual incrementa la acción citotóxica sobre el tumor y es capaz de lisar células metastásicas. Se ha probado su uso en cánceres de próstata y ha demostrado efectividad (9).
Hay un único virus oncolítico aprobado por la FDA, es el T-VEC (Imlygic®), que es el virus del herpes simple (VHS), modificado para atacar a las células cancerígenas del melanoma.
3. Virus de la Enfermedad de Newcastle como nueva aproximación terapéutica para el glioblastoma
3.1. Introducción
Vamos a poner un ejemplo de un estudio que se realizó sobre el virus de la enfermedad de Newcastle, para ver si es adecuado para usarlo como virus oncolítico y como terapia para el glioblastoma.
3.1.1. Glioblastoma (GBM)
El glioblastoma es el tumor cerebral más común en el SNC, siendo muy agresivo debido a su invasividad y alta proliferación. Las personas que lo padecen tienen una esperanza de vida muy corta una vez que se diagnostica, a pesar de la mejora de los tratamientos y establecimiento de terapias.
Este tumor, compuesto por células madre de glioma (GSCs), presenta resistencia a diferentes tratamientos contra el cáncer, como la quimio o la radioterapia, ya que estas células son capaces de autorrenovarse y diferenciarse (10). Las GSCs se cree que también son las causantes de la recurrencia del glioblastoma.
Los rasgos más característicos de este cáncer son la proliferación microvascular y la necrosis, es decir, se agrupan en capas y las células presentan la zona central con necrosis (11).
3.1.2. Virus de la Enfermedad de Newcastle (NDV)
Es un virus aviar, con propiedades oncolíticas e inmunoestimuladoras, por lo que su estudio en viroterapia y ensayos clínicos cada vez es mayor.
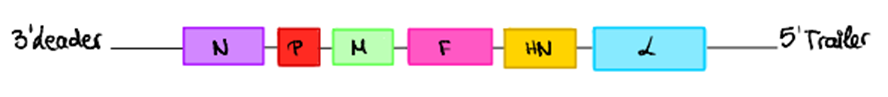
El genoma de este virus consiste en una molécula de ARN monocatenario, con polaridad negativa y formada por dos regiones en los extremos, leader en 3’ y tráiler en 5’, no codificantes; y seis genes que codifican 6 tipos de proteínas diferentes (12).
6 genes para 6 tipos de proteínas
Infecta células y se replica en ellas, destruyéndolas. Esto lo hace más rápido en las células cancerosas humanas, de ahí el interés en su estudio como tratamiento para el cáncer. Presenta dos cepas: las cepas líticas, que dañan la membrana de la célula; y las cepas no líticas que bloquea el metabolismo de la célula. Las cepas líticas son las que se estudian para el cáncer, ya que son capaces de eliminar directamente las células cancerosas; pero las dos cepas se usan en vacunas que ayudan al sistema inmune a combatir el cáncer (13).
Virus de la enfermedad de Newcastle
Los cultivos que se realizaron con GSCs y rNDV muestran como este virus afecta a la viabilidad de las células del tumor, induciendo apoptosis.
3.2. Glioblastoma: aspectos moleculares y patología
Las vías de señalización, moléculas y genes más comúnmente afectadas en el GBM, que hacen que sea resistentes a los tratamientos convencionales (14), son:
Receptores tirosina/quinasa (RTK): se encuentran en la membrana plasmática. Se autofosforilan en presencia de ligando para activarse. Se encargan de activar vías de transducción que continúan con vías de transcripción de genes que regulan el ciclo celular.
Vía de PI3K/AKT/mTOR: PI3K activa a AKT y este activa a mTOR, relacionado con la supervivencia y el ciclo celular.
Señalización de RAS/MAPK: genes transcritos por vías de traducción llevadas a cabo por segundos mensajeros (oncogenes o genes supresores de tumores), que participan en la proliferación celular. RAS es una GTPasa que actúa en la transducción de señal de RTK. Cuando se activan RTK, se activa RAS, que a su vez activa la vía de transducción de las MAPK. Las mutaciones en RAS la activan permanentemente, activando también permanentemente la vía de las MAPK. Esto induce una transcripción activa de genes relacionados con el ciclo celular.
P53 y retinoblastoma (RB): implicadas en regulación del ciclo celular. P53 es un gen supresor que se encarga de inducir apoptosis cuando el ADN está dado. Si p53 está mutado, se sigue con el ciclo celular y el daño en el ADN. También inhibe a mTOR, relacionado con el ciclo celular. El retinoblastoma está relacionado con la mutación de pRb, que hace que no se una a E2F y se siga con el ciclo celular.
Gen EGFR: es el gen del receptor del factor de crecimiento epitelial (GFR). Si está alterado, se hace independiente de EGF, por lo que se activa a muy bajas concentraciones de ligando.
3.3. NDV como agente oncolítico
En 1965 observó por 1ª vez que NDV presentaba un efecto antitumoral y baja neuroafinidad. Este potencial oncolítico que presenta el virus se debe a su propia capacidad de replicarse bastante mejor (unas 104 veces mejor) en las células tumorales que en las células normales, y además, sin afectar a las células sanas. Además, al ser un virus aviar, sus cepas virulentas provocan solo síntomas leves.
3.3.1. Mecanismo de oncólisis de NDV
NDV se asocia principalmente a la inducción de la apoptosis. La apoptosis es un tipo de muerte celular programada que ocurre en todos los tipos celulares. Además, también puede provocar necroptosis, que es un tipo de muerte celular que tiene características tanto de necrosis (por la morfología de las células) como de apoptosis (por lo de programada). También puede inducir la muerte celular por autofagia.
La infección por este virus induce la activación de la respuesta inmune, favoreciendo su efecto oncolítico. Las células tumorales infectadas presentan Ag virales, haciendo que las células de alrededor liberen citoquinas, que activan a macrófagos, NK y o monocitos, provocando la respuesta inmune innata; o haciendo que se activen las células presentadoras de Ag, que activan a los linfocitos T citotóxicos, que activarán la respuesta inmune adaptativa. Todo esto activa el estado de actividad inmunológica antitumoral causando la muerte celular inmunogénica de las células tumorales (15).
El genoma de este virus es muy fácil de modificar, por lo que la técnica de genética inversa es útil para obtener virus recombinantes, teniendo como objetivo aumentar su eficacia antitumoral.
Se han estudiado sus propiedades oncolíticas, dando en algunos casos reducción parcial y en otros total del tumor.
3.4. Resultados
La tesis concluye que el NDV induce cambios en la viabilidad de las GSCs, demostrando la capacidad oncolítica del virus en diferentes tipos de líneas celulares tumorales, incluidas las líneas tumorales de glioma. Además, interfiere en el crecimiento celular de las GSCs, y provoca la inducción de la apoptosis de las diferentes líneas celulares.
En cuanto a os xenotransplantes, también se observó que se reduce el tamaño de los tumores xenotransplantados en ratones Nude. Finalmente se demostró que en los ratones inmunodeprimidos, el virus causa 100% de mortalidad, siendo seguro solo para los ratones inmunocompetentes. Lo que puede suponer una importante limitación en el uso farmacológico del virus de la enfermedad de Newcastle.
4. Conclusión
Nos ha parecido un trabajo interesante y además hemos aprendido muchas cosas que no sabíamos y que nos han gustado mucho. Creemos que la investigación y el estudio de los virus como terapia para el cáncer es algo muy importante y que podría funcionar muy bien para solucionar el problema que provoca esta enfermedad. Si es verdad que aún queda mucho por avanzar, pero creemos firmemente, que de aquí a unos años esta nueva terapia será una opción más para combatir el cáncer.
5. Bibliografía
1. The Hallmarks of CancerHanahan D, Weinberg RCell (2000) 100(1) 57-70
3. Studies in Hodgkin’s syndrome; the association of viral hepatitis and Hodgkin’s disease; a preliminary report. HOSTER H, ZANES R, VON HAAM E. Cancer research (1949) 9(8) 473-80
4. Clinical studies of viruses as antineoplastic agents with particular reference to Egypt 101 virus. SOUTHAM C, MOORE A Cancer (1952) 5(5) 1025-34
5. Telomerase reverse transcriptase promoter mutations in primary cutaneous melanoma Heidenreich B, Nagore E, Rachakonda P, Garcia-Casado Z, Requena C, Traves V, Becker J, Soufir N, Hemminki K, Kumar R. Nature Communications (2014) 5(1) 3401
6. Homeostasis celular: crecimiento celular y cáncer – Bioquímica médica Michie, Alison M.; Paunovic, Verica; Harnett, Margaret M.; Bioquímica médica, Capítulo 28, 397-415
8. Santos Apolonio J, Lima de Souza Gonçalves V, Cordeiro Santos ML, Silva Luz M, Silva Souza JV, Rocha Pinheiro SL, de Souza WR, Sande Loureiro M, de Melo FF. Oncolytic virus therapy in cancer: A current review. World J Virol 2021; 10(5): 229-255
9. Therapy of prostate cancer using a novel cancer terminator virus and a small molecule BH-3 mimetic Sarkar S, Quinn B, Shen X, Dash R, Das S, Emdad L, Klibanov AWang X, Pellecchia M, Sarkar D, Fisher P Fisher POncotarget (2015) 6(13) 10712-10727
10. Piper K, DePledge L, Karsy M, Cobbs C. Glioma Stem Cells as Immunotherapeutic Targets: Advancements and Challenges.
11. Ohgaki and Kleihues, 2013; Perry and Wesseling, 2016; Urbanska et al., 2014
12. Marcos et al., 2005; Triosanti et al., 2018
13. Csatary LK, Eckhardt S, Bukosza I, Czegledi F, Fenyvesi C, Gergely P, Bodey B, Csatary CM. Attenuated veterinary virus vaccine for the treatment of cancer. Cancer Detect Prev. 1993;17(6):619-27.
13. Csatary LK, Moss RW, Beuth J, Töröcsik B, Szeberenyi J, Bakacs T. Beneficial treatment of patients with advanced cancer using a Newcastle disease virus vaccine (MTH-68/H). Anticancer Res. 1999 Jan-Feb;19(1B):635-8.
14. Crespo et al., 2015; Szopa et al., 2017
15. Matveeva et al., 2015; Zamarin and Palese, 2012ª
16. Virus de la Enfermedad de Newcastle como nueva aproximación terapéutica para el Glioblastoma, Rubio S (2018) 24-43
Los tratamientos conocidos hasta el momento contra la enfermedad del Alzheimer
escrito por A2_3D | 10 enero, 2023
Por Andrea Rufat Verdú y Ana Verdugo Abril. Grado en Biología Sanitaria, Universidad de Alcalá
Alzheimer y su histopatología
La enfermedad de Alzheimer (EA) es un trastorno neurológico que provoca la muerte de las células nerviosas del cerebro. A medida que avanza la enfermedad, se van deteriorando las capacidades cognitivas, entre ellas la capacidad para tomar decisiones y llevar a cabo las tareas cotidianas. Además, pueden surgir modificaciones de la personalidad, así como conductas problemáticas. En sus etapas avanzadas, la enfermedad de Alzheimer conduce a la demencia y finalmente a la muerte. [1] Siendo esta enfermedad una de las principales causas de muerte a nivel mundial. Además, se espera que esto aumente en los próximos años por lo que sería interesante conseguir un diagnóstico temprano para la enfermedad, así como, un tratamiento eficaz.
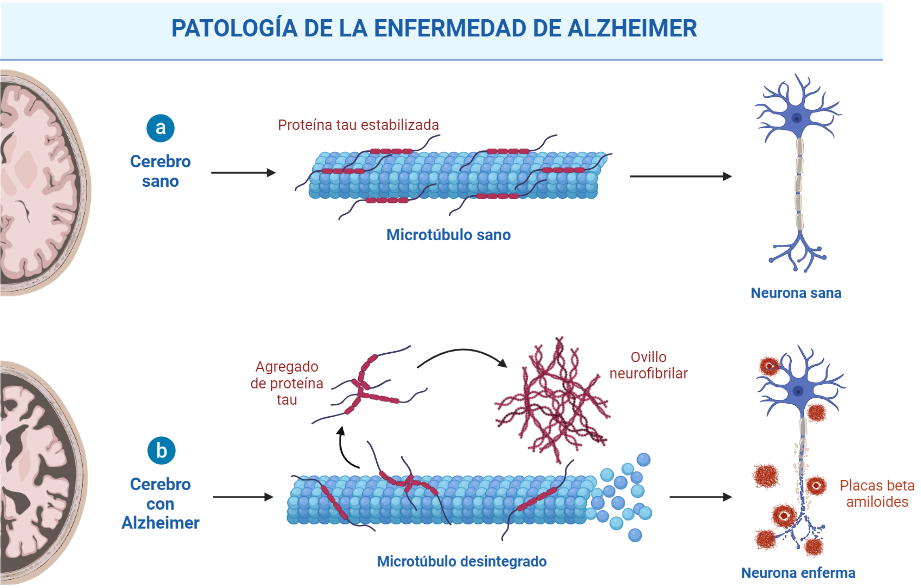
A nivel neuropatológico, la EA se caracteriza principalmente por la acumulación del péptido β-amiloide agregado en forma de placa (placas amiloides o placas de Aβ),y por la presencia de ovillos neurofibrilares (NFTs) que contienen a la proteína tau hiperfosforilada y agregada [2].
Las placas β-amiloides son depósitos extracelulares de proteínas, principalmente el péptido β-amiloide en su forma insoluble, mientras que los ovillos neurofibrilares son depósitos intraneuronales de la proteína asociada a microtúbulos Tau. Pese a que ambas son manifestaciones de la enfermedad del Alzheimer, la primera es característica también de otras patologías, como la arteriosclerosis, y en general del proceso de envejecimiento. [3]
Existen dos tipos de EA, que se diferencian en la edad de inicio, en la causa principal que provoca su aparición e incidencia. Sin embargo, comparten los mismos síntomas y lesiones histopatológicas.
Así encontramos la EA familiar o de inicio temprano, que afecta a individuos menores de 65 años, se asocia con la herencia mendeliana y representa alrededor del 5% de los casos de EA; y la EA esporádica o de inicio tardío, sin un modo de transmisión consistente que afecta a las personas mayores de 65 años y representa el mayor número de casos entre las personas mayores (90-95% de los casos de la EA). [2]
Figura 1. Comparación de un cerebro sano al de un paciente con Alzheimer Created with BioRender.com
2.Intervenciones terapéuticas
Los tratamientos disponibles hasta el momento para la EA pueden lograr una mejoría sintomática y en la calidad de vida de los pacientes, pero ninguno consigue revertir, frenar o curar la fatal progresión de la enfermedad [4].
2.1 Tratamientos farmacológicos
En su tratamiento se encuentran los inhibidores de la enzima acetilcolinesterasa (IACE): donepezilo, rivastigmina y galantamina y los inhibidores de glutamato como el namenda (memantina), además de plantas como Melissa officinalis y Gingko biloba como técnicas de la Medicina Natural China. [5] También se ha estudiado el posible uso de la Huperzina A, un alcaloide natural que atraviesa la barrera hematoencefálica con un posible papel como neuroprotector [6].
Los IACE inhiben la acción de una enzima que destruye la acetilcolina, un químico cerebral implicado en la memoria y otros procesos cognitivos y afectivos, por lo que su consecuencia será el aumento de la acetilcolina. Se ha observado que la enfermedad de Alzheimer afecta desde muy temprano a las neuronas que producen acetilcolina, de ahí que una de las primeras estrategias terapéuticas ha sido crear fármacos que impidan la degradación de la acetilcolina. [5] Estos medicamentos protegen contra el estrés oxidativo y la toxicidad amiloide,
pero son costosos y pueden dañar las membranas neuronales [6].
Por otro lado, los inhibidores de glutamato actúan sobre este neurotransmisor que se produce en grandes cantidades por las células dañadas por Alzheimer de forma que se adhiere a receptores NMDA que aceleran el daño celular. [5]
También se pueden usar como tratamiento combinaciones de medicamentos como Namzaric que contiene tanto donepezilo como memantina.
Asimismo, se ha investigado acerca del uso de los receptores muscarínicos M1 como blancos terapéuticos en el tratamiento de la enfermedad. Por ejemplo, la xanomelina es un agonista tanto de receptores M1 como M4 que atraviesa la barrera hematoencefálica. Se ha demostrado que mejora la función cognitiva, sin embargo como cualquier fármaco tiene efectos secundarios indeseables, en concreto, sobre el sistema gastrointestinal y cardiovascular. [6]
2.2 Otros remedios: antioxidantes, antiinflamatorios…
En la literatura científica se ha asociado la neurotoxicidad del péptido ß-amiloide a la generación de radicales libres desencadenando así un estado de estrés oxidativo con daños celulares como la peroxidación lipídica. [7] El cerebro es un órganoparticularmente vulnerable a este tipo de daños por estrés oxidativo, debido a su alto consumo de oxígeno, o bajos niveles de antioxidantes, entre otros. [8] Por ello, no se descarta el uso de antioxidantes como tratamiento o incluso, el hábito saludable de una buena dieta mediterránea, puesto que pueden prevenir la degeneración neuronal eliminando especies reactivas de oxígeno (ROS) o previniendo su formación. Algunos son la vitamina E, la selegilina o la melatonina. [6]
Cabe mencionar el uso de antiinflamatorios para la inflamación generada por las placas seniles, o el de quelantes de hierro como la desferroxamina. Aunque ambos palian los efectos de la EA, pueden causar reacciones adversas como toxicidad en la retina o en el hígado. [6]
La curcumina, sustancia presente en la cúrcuma, también tiene un papel importante en la prevención y el tratamiento de esta enfermedad. Cuenta con propiedades antioxidantes, lipofílicas e incluso antiinflamatorias que permiten la mejora de las funciones cognitivas en los pacientes con Alzheimer. [9]
En la patogénesis de la EA se observa que el péptido Αβ puede interrumpir los canales de calcio en la membrana, provocando una pérdida del flujo (o influjo) que conlleva al desequilibrio del ion y así a altas concentraciones de calcio intracelular. Las concentraciones de calcio demasiado altas o bajas tienen efectos tóxicos, como lo es la alteración sobre la producción y transmisión de neurotransmisores en las células nerviosas. Finalmente esto conlleva a un proceso de muerte celular. Es por eso que los antagonistas del calcio también han sido utilizados como intervención terapéutica. Un ejemplo es el del nimodipino, que inhibe el influjo de calcio y mejora la circulación sanguínea cerebral. [6]
2.3 Inmunoterapias
Como se ha mencionado con anterioridad, el péptido Αβ amiloide tiene un papel clave en esta enfermedad ya que es neurotóxico, altera la función sináptica y produce neurodegeneración. [4] Por lo tanto, las estrategias dirigidas a Aβ podrían frenar eficazmente la progresión de la EA. En la actualidad, los mecanismos de acción de los fármacos anti-Aβ incluyen principalmente la reducción de la producción de Aβ, la prevención de la agregación de Aβ y la promoción de la eliminación de Aβ. [10]
Las inmunoterapias anti-Aβ más elaboradas son las vacunas y los anticuerpos exógenos, conocidas como inmunoterapia activa y pasiva, respectivamente. La inmunización activa estimula el sistema inmunitario mediante la administración de Aβ o sus fragmentos, lo que desencadena una respuesta inmunitaria para producir anticuerpos endógenos contra Aβ. Sin embargo, estas vacunas presentan una baja reactividad y la aparición de reacciones adversas dependientes de células T, por lo que en la actualidad se desarrolla la inmunoterapia pasiva utilizando anticuerpos monoclonales humanizados o inmunoglobulinas policlonales para promover la eliminación de Aβ [10].
En cuanto a inmunización activa se han desarrollado varias vacunas como AN1792, amilomotida y UB-311.
Aducanumab, donanemab, lecanemab, solanezumab, crenezumab y gantenerumab son anticuerpos monoclonales humanizados que se están estudiando como tratamientos de la enfermedad de Alzheimer.
Recientemente ha sido aprobado el anticuerpo monoclonal Lecanemaben Estados Unidos. Es el primer fármaco que realmente modifica el curso de la enfermedad, pues reduce hasta un 27% el empeoramiento de los síntomas del Alzheimer después de administrarse durante 18 meses [11].
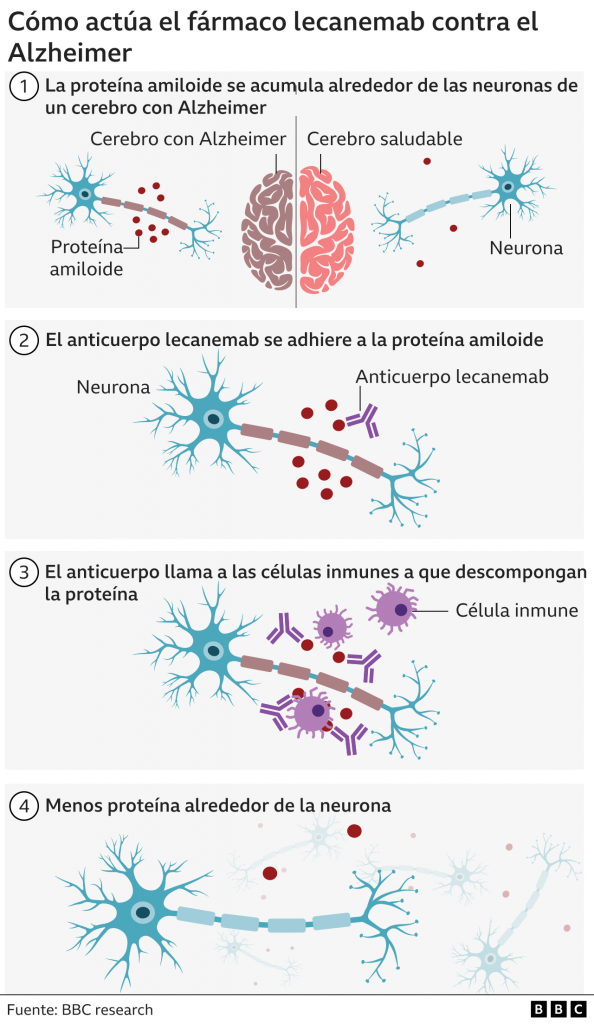
Los péptidos Aβ existen en varios estados conformacionales, incluidos monómeros solubles, agregados solubles de tamaño creciente y fibrillas y placas insolubles. Los agregados de Aβ solubles, como las protofibrillas de Aβ, son más tóxicos que los monómeros o las fibrillas insolubles. El modo de acción del Lecanemab consiste en unirse a las protofibrillas beta amiloides, por tanto, es capaz de reducir los niveles de beta amiloide patógeno (Aβ) y prevenir el depósito de Aβ. [12]
Figura 2. Mecanismo de acción del Lecanemab Tomada de: BBC News Mundo (2022) «Alzheimer: el medicamento aclamado como un avance trascendental en la lucha contra la enfermedad», BBC News Mundo, 30 noviembre. Disponible en: https://www.bbc.com/mundo/noticias-63806450.
Como ya se ha explicado, los ovillos neurofibrilares están formados por la proteína tau anormalmente fosforilada. Esta es una proteína citoplasmática que puede estabilizar los microtúbulos a través de la unión a la tubulina durante su polimerización en condiciones normales. Sin embargo, en la EA está hiperfosforilada, lo que implica que tenga una capacidad reducida para unirse a los microtúbulos y que, en ocasiones, cause la formación de ovillos neurofibrilares y la generación de agregados. Al igual que sucede con los péptidos Αβ amiloides, también existen estrategiasanti-tau que previenen la fosforilación anormal de tau, inhiben la agregación de tau y promueven la eliminación de agregados de tau. Actualmente, la mayoría de los agentes anti-tau en ensayos clínicos son inmunoterapias. [10]
Aβ y la proteína tau fosforilada son reconocidos por receptores en la superficie de la microglía, lo que promueve la liberación de factores inflamatorios en los cerebros con EA. Los factores inflamatorios, a su vez, aumentan la formación de depósitos de Aβ y ovillos neurofibrilares, creando así un círculo vicioso que exacerba el proceso de la enfermedad. Existen genes como TREM2 que se sobreexpresan en la microglía y que se pueden utilizar como blancos en tratamientos de inmunoterapia como es el caso del anticuerpo monoclonal AL002. [10]
3. Conclusiones
Es imprescindible el estudio de la neuropatología del Alzheimer con el fin de valorar posibles dianas farmacológicas que nos permitan desarrollar futuros fármacos eficaces contra la enfermedad.
A día de hoy no se conoce ningún fármaco aprobado capaz de mejorar la EA al 100%. Sin embargo, muchos pueden retrasar el progreso de la enfermedad, como lo son aquellos aprobados por la FDA (U.S Food and Drug Administration). Incluyendo en este grupo al Lecanemab como el último fármaco aprobado por dicha organización, capaz de reducir hasta un 27% el empeoramiento de la enfermedad del Alzheimer.
La EA no se trata únicamente con la administración de medicinas, sino que también es importante que las personas que la padezcan adopten en su vida cotidiana hábitos saludables, como una dieta mediterránea o la incorporación de especias como la cúrcuma gracias a los efectos de la curcumina. Porque pese a que no tengan un gran efecto sobre el progreso de la enfermedad, son capaces de colaborar en su mejoría.
La histopatología de la enfermedad de Alzheimer se debe principalmente a la formación de placas amiloides y ovillos neurofibrilares, los cuales son las principales dianas de los tratamientos de esta enfermedad. Sin embargo, la patogénesis de la EA también puede ser tratada consiguiendo un aumento de acetilcolina, disminuyendo el exceso de glutamato y fomentando la actividad antiinflamatoria de la microglía y suprimiendo la proinflamatoria.
Cabe destacar que la inmunoterapia actualmente está en pleno desarrollo como una de las formas más efectivas para conseguir un tratamiento para la cura del Alzheimer. Por otro lado, es necesario seguir con la investigación en este campo ya que muchos de los anticuerpos monoclonales y vacunas que se han conseguido han fracasado en sus últimas fases de ensayo.
No solo es necesario encontrar un buen tratamiento para la EA, sino que también hay que seguir investigando para contar con un diagnóstico temprano, ya que estos fármacos resultan más efectivos con una temprana administración.
4. Bibliografía
[1]: Del Huerto Paredes, N. M., Nissen, M. D., Parquet, C. A. & Romano, M. F., 2007. ENFERMEDAD DE ALZHEIMER. Revista de Posgrado de la VIa Cátedra de Medicina, Issue 175, pp. 9-12. [2]: Lara Ureña, N. (2020) Papel de HIF1 y PHD3 en la microglía de la enfermedad de Alzheimer. Universidad de Sevilla.
[3]: Heras Garvín, A. (2015) Microglía e hipoxia: Implicaciones en la enfermedad de Alzheimer. Universidad de Sevilla.
[4]: Rojas Delgado, K., Salazar Nassar, J. y Torrealba Acosta, G. (2019) “Alzheimer e Inmunoterapia: revisión de tres anticuerpos monoclonales humanizados dirigidos contra el Aβ amiloide (bapineuzumab, solaneuzumab y aducanumab)”, Revista Médica de Costa Rica, 84(627), pp. 2–7.
[5]: Gómez Tejeda, J. J., Hernández Pérez, C. y Iparraguirre Tamayo, A. E. (2020) “Tratamientos paliativos en la Enfermedad de Alzheimer”, 16 de Abril, 59(275) p. 727.
[6]: Cabrera, M.J.A. et al. (2014) «Patogenia y tratamientos actuales de la enfermedad de Alzheimer», Revista Cubana de Farmacia, 48(3), pp. 508-518.
[7]: Manzano-León, N. y Mas-Oliva, J. (2006) «Estrés oxidativo, péptido β-amiloide y enfermedad de Alzheimer», Gaceta Medica De Mexico, 142(3), pp. 229-238.
[8]: Bello-Medina, P. C. et al. (2022) “Estrés oxidativo, respuesta inmune, plasticidad sináptica y cognición en modelos transgénicos de la enfermedad de Alzheimer”, Neurología (English Edition), 37(8), pp. 682–690
[9]: Mishra, S. y Palanivelu, K. (2008) «The effect of curcumin (turmeric) on Alzheimer′s disease: An overview», Ann Indian Acad Neurol, 11(1), pp. 13-19.
[10]: Song, C., Shi, J., Zhang, P. et al. (2022) «Immunotherapy for Alzheimer’s disease: targeting β-amyloid and beyond», Translational Neurodegeneration, 11(18).
[12]: Lecanemab: Uses, Interactions, Mechanism of Action | DrugBank Online [en línea], (sin fecha). DrugBank Online | Database for Drug and Drug Target Info. Disponible en: https://go.drugbank.com/drugs/DB14580
Veneno: nuestro aliado
escrito por lorea_3B | 10 enero, 2023
Por Lorea Ayape, 3º Biología Sanitaria. Universidad de Alcalá.
Introducción
Desde la introducción de la insulina hace casi un siglo, se han comercializado más de 80 fármacos peptídicos para el tratamiento de una amplia gama de enfermedades, como la diabetes, el cáncer, la osteoporosis, la esclerosis múltiple, la infección por VIH y el dolor crónico (Muttenthaler et al., 2021). Los grandes avances en biología molecular y química peptídica siguen haciendo progresar este campo, así como la venómica integrada, una estrategia emergente que crea nuevas vías para el descubrimiento de fármacos peptídicos (Muttenthaler et al., 2021).
A continuación, explicaremos un ejemplo de cómo los venenos, esas sustancias dañinas que presentan ciertos animales de los que muchos huimos despavoridos, pueden convertirse en nuestros aliados con ayuda de la ciencia, pues contienen determinados péptidos que podemos usar en nuestro beneficio. Para ello, le propongo al lector que visualice a nuestra protagonista: la tarántula, ese animal grande y peludo de 8 patas que tan rápido se mueve con, aparentemente, enormes ganas de morder a su víctima humana cuando nos encontramos frente a él. No obstante, las tarántulas tienen poca importancia clínica debido a que no son muy agresivas y evitan el contacto con el entorno humano (Murray, Rosenthal and Pfaller, 2021). Sin más dilación, comencemos a tratar esta curiosidad científica y, quizás, incluso les terminemos cogiendo un poquito de cariño a las arañas.
Tarántula. Foto: Pixabay
Necesidad de un nuevo analgésico
Recientemente, se han descubierto determinados componentes del veneno de tarántulas capaces de bloquear la ruta por la que se envían las señales de dolor al cerebro.
El estudio de Klint et al. ha sido realizado debido a la gran necesidad clínica que tenemos de crear analgésicos eficaces para tratar el dolor crónico, puesto que la mayoría de los fármacos disponibles actualmente tienen una eficacia limitada y efectos secundarios que limitan la dosis (Klint et al., 2015). Por su parte, el dolor crónico es un importante problema de salud en todo el mundo que afecta a una media del 15% de las personas adultas (Gaskin and Richard, 2012). Además, tan solo en Estados Unidos, la carga económica anual que supone el dolor crónico es de unos 600.000 millones de dólares, lo que supera el coste económico en conjunto del cáncer, la diabetes y los accidentes cerebrovasculares (Gaskin and Richard, 2012).
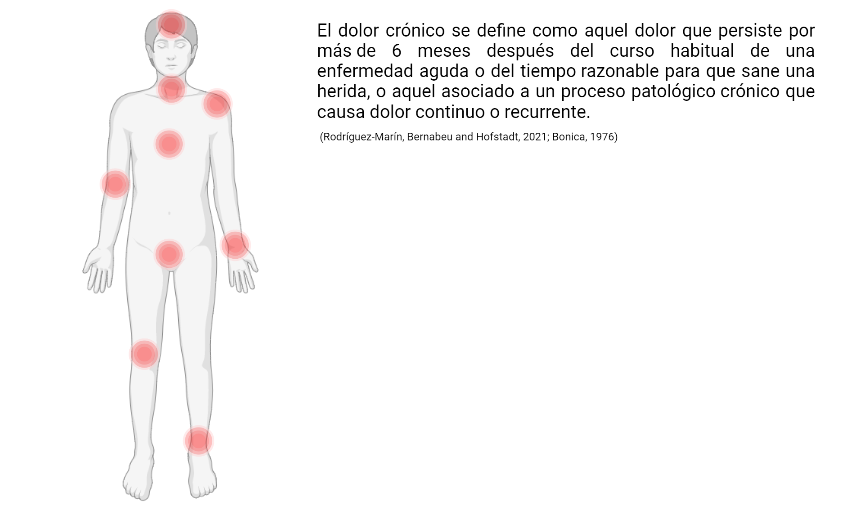
En definitiva, la prevención en dolor crónico es muy importante al ser un problema muy extendido, producir una inestimable cantidad de sufrimiento y constituir un coste económico enorme (Rodríguez-Marín, Bernabeu and Hofstadt, 2021).
Representación de múltiples puntos donde puede afectar el dolor crónico y su definición. Imagen creada por Lorea Ayape con BioRender.
Fisiología de los canales de sodio dependientes de voltaje
La comunicación celular en el sistema nervioso se basa en fenómenos de señalización eléctrica y química mediados por canales iónicos (Boron, 2017). En las células con dicha propiedad es posible desencadenar un impulso eléctrico denominado potencial de acción, que actúa como una señal capaz de propagarse a grandes distancias a lo largo de las fibras nerviosas o musculares. La conducción de los potenciales de acción permite que se pueda transmitir información a través de nervios desde los órganos hacia el cerebro (Boron, 2017). En el ejemplo que tratamos, las señales que se envían al cerebro viajan mediante la vía del dolor.
Por otro lado, debemos conocer los canales de sodio dependientes de voltaje (NaV), unas proteínas situadas en la membrana de estas células especiales que permiten el paso de los iones sodio a su través. Si no hay canales de sodio, no se producen potenciales de acción y, por tanto, tampoco se transmiten las señales.
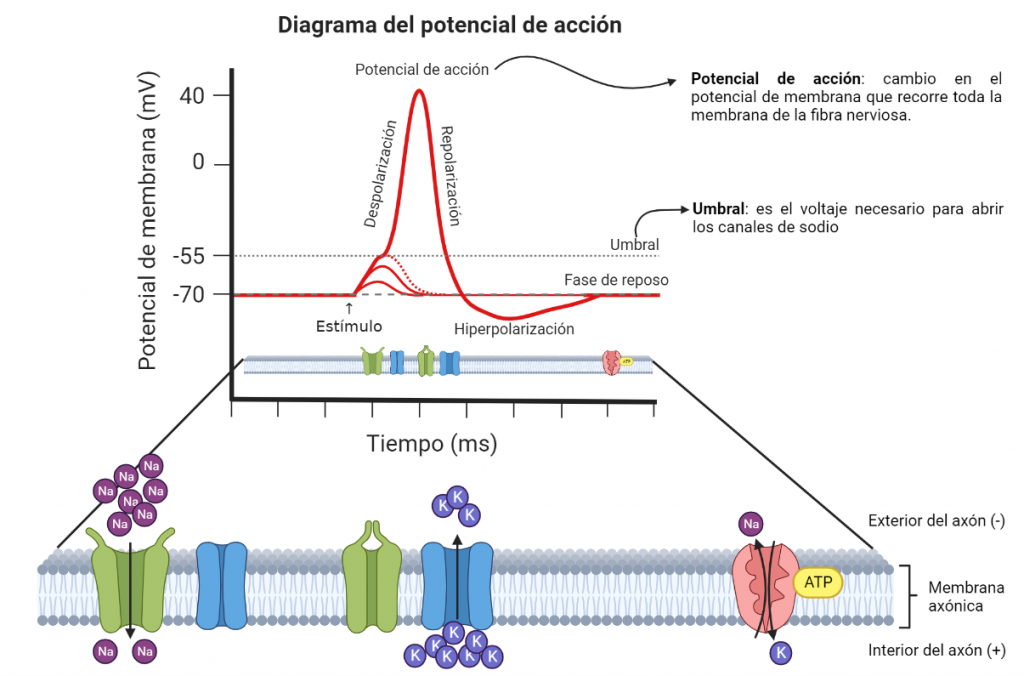
Representación de transmisión del impulso nervioso: Se alcanza el umbral, por lo que se abren los canales de sodio sensibles al voltaje, de manera que entra sodio (Na+) en la neurona. A continuación, se cierran los canales de sodio y se abren los de potasio (K+), saliendo éste desde el interior de la neurona. Después se inactivan los canales de potasio y se alcanza el potencial de reposo. Finalmente, se activa la bomba sodio-potasio con gasto de ATP (energía) para restablecer los niveles iniciales de los iones. Diagrama elaborado a partir de un modelo de BioRender.
En concreto, el hNaV1.7 es un canal de sodio presente en las neuronas nociceptivas. Estudios anteriores ya han demostrado que las mutaciones de pérdida de función en el gen que da lugar al hNaV1.7 generan una insensibilidad a todas las formas del dolor (Cox et al., 2006).
Estos hallazgos sugieren que el hNaV1.7 podría ser una buena diana en la investigación de nuevos fármacos para el tratamiento del dolor (Boron, 2017). Es por todo ello que Klint et al. se han centrado en los canales de sodio, específicamente en el hNaV1.7.
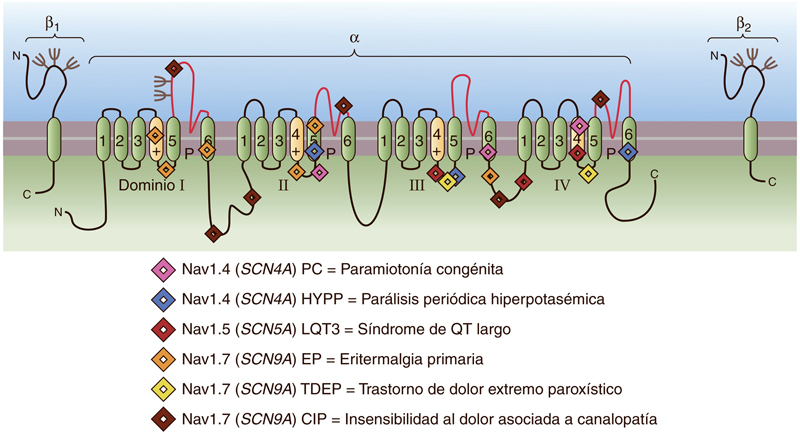
Mutaciones del canal de Na+ en enfermedades genéticas humanas. Los símbolos muestran lugares en una subunidad α del canal de Na+ genérico con los 4 dominios (Dominio I-IV), cada uno con 6 segmentos transmembrana (Drenth and Waxman, 2007). C indica el extremo C-terminal de la cadena peptídica, mientras que N indica el extremo N-terminal de la cadena peptídica. Se representan algunos ejemplos de las localizaciones de mutaciones pertenecientes a tres genes de canales de Na+ (SCN4A, SCN5A y SCN9A) (Boron, 2017). Todas estas mutaciones patológicas dan lugar a cambios o deleciones de aminoácidos, salvo en el caso de la insensibilidad al dolor, que se debe a mutaciones que ocasionan la falta de expresión funcional del canal NaV1.7 (Boron, 2017). Imagen obtenida de Boron, W. F. (2017) ‘Excitabilidad eléctrica y potenciales de acción’, in Fisiología médica. third, pp. 172–189.
Relación entre las arañas y los analgésicos
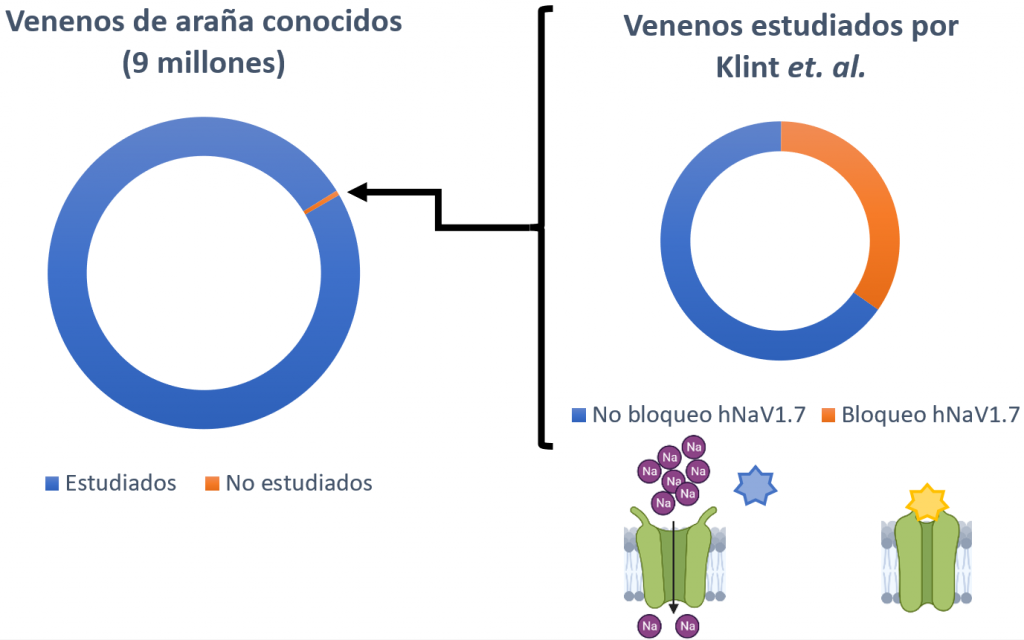
Las arañas son el grupo que más animales venenosos alberga. A su vez, existe un total de 9 millones de péptidos de veneno de araña, de los cuales solo se ha explorado un 0,01% (Klint et al., 2015); por lo tanto, estos venenos suponen una gran fuente para buscar nuevos moduladores del canal hNaV1.7.
En total se analizaron los venenos de 206 especies de arañas, de los cuales un 40% presentaban algún compuesto capaz de bloquear al canal hNaV1.7 (Klint et al., 2015). Finalmente, la estructura del Hd1a (un péptido de la araña Haplopelma doriae) demostró que se trataba del mejor candidato a posible analgésico, puesto que presenta una gran estabilidad química, térmica y biológica (Klint et al., 2015). Además, inhibió el hNaV1.7 con un alto nivel de selectividad sobre todos los demás subtipos, excepto el hNaV1.1 (Klint et al., 2015).
Gráfico de los venenos de araña estudiados por Klint et al. Elaborado por Lorea Ayape mediante Word y BioRender.
Otros canales de sodio como diana de analgésicos
A partir de una serie de venenos que activan neuronas sensoriales de ratón, Osteen et al. aislaron dos péptidos (Hm1a y Hm1b) del veneno de la tarántula Heteroscodra maculata que activaban tan sólo a unas pocas neuronas sensoriales (Osteen et al., 2016). Después comprobaron que el efecto de dichos péptidos era bloqueado mediante la tetrodotoxina (TTX), la cual actúa inhibiendo a los canales de sodio activados por voltaje (NaV) (Osteen et al., 2016). Esto llevó al descubrimiento de que los péptidos aislados son agonistas de los canales NaV1.1 y, a su vez, de que éstos están implicados en la señalización del dolor, lo cual se desconocía hasta entonces (Osteen et al., 2016).
También se demostró que el NaV1.1 regula la excitabilidad de unas fibras nerviosas que transmiten señales de dolor a la médula espinal, con lo que este canal iónico también se convirtió en una posible diana analgésica (Osteen et al., 2016). De hecho, se ha comprobado que los inhibidores del NaV1.1 reducen la hipersensibilidad mecánica en varios modelos de dolor visceral crónico (Salvatierra et al., 2018).
Es decir, en este caso los péptidos de araña mencionadas no se usarían como analgésico contra NaV1.1 (como veíamos en el caso anterior), puesto que realmente contribuyen a que dichos canales produzcan su efecto. No obstante, Hm1a y Hm1b permitieron descubrir que el NaV1.1 está relacionado con la señalización del dolor, posicionándolo así como un objetivo más contra el que aplicar analgésicos.
Conclusiones
El proceso de elaboración de un nuevo fármaco requiere muchísimos años, no solo por las fases que debe superar dicho medicamento hasta aparecer en las farmacias, sino también por el trabajo previo que implica detectar la diana correcta y el compuesto capaz de dirigirse hasta ella afectándola específicamente, como hemos podido comprobar.
Por su parte, todavía quedan millones de péptidos de veneno sin ser explorados, debido a la lentitud del método tradicional de descubrimiento, en el que las fracciones crudas de veneno se analizan frente a dianas conocidas, seguidas de purificación y secuenciación para dilucidar la secuencia de una diana (Muttenthaler et al., 2021).
En esta entrada del blog nos hemos centrado en algunos estudios con péptidos de arañas, pero los escorpiones y los caracoles cono también proporcionan una de las más ricas diversidades químicas (Muttenthaler et al., 2021). Afortunadamente, los nuevos avances tecnológicos están sentando las bases de un potente enfoque denominado «venómica integrada» (Muttenthaler et al., 2021).
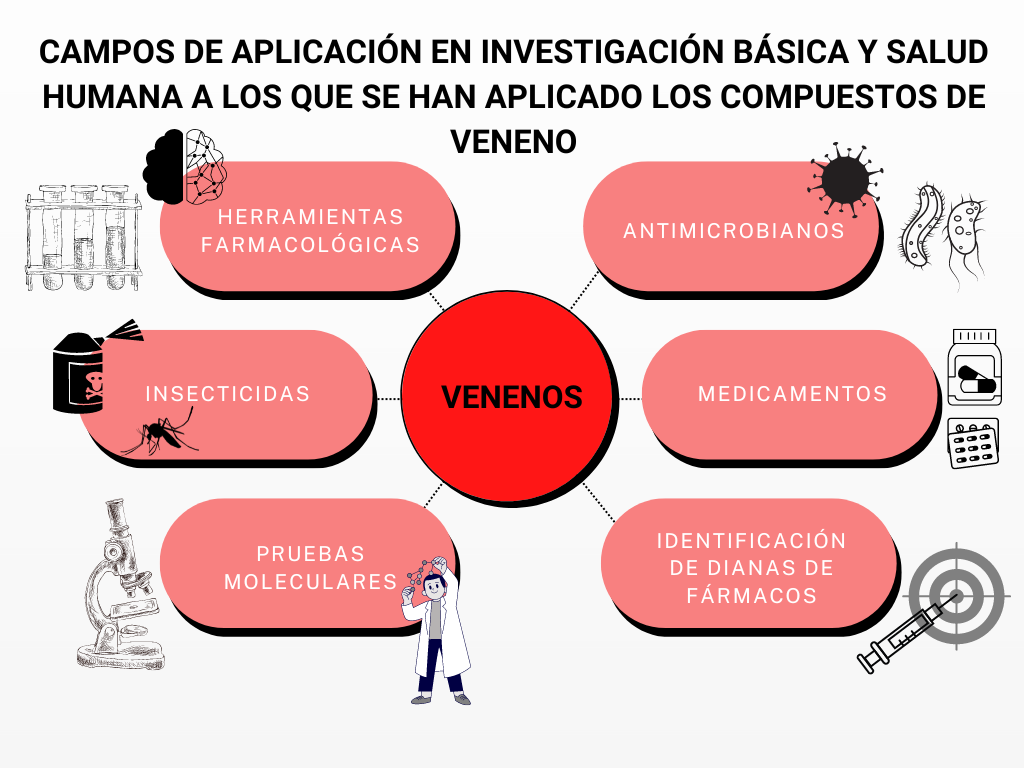
Los compuestos de veneno pueden utilizarse como herramientas farmacológicas, terapéuticas, insecticidas o para combatir vectores de enfermedades humanas u organismos patógenos (Herzig et al., 2020). Imagen creada por Lorea Ayape con Canva.
Referencias
Bonica, J. J. (1976) ‘Organization and function of a multidisciplinary pain clinic’, in Pain. Springer, pp. 11–20.
Boron, W. F. (2017) ‘Excitabilidad eléctrica y potenciales de acción’, in Fisiología médica. third, pp. 172–189.
Cox, J. J. et al. (2006) ‘An SCN9A channelopathy causes congenital inability to experience pain’, Nature, 444, pp. 894–898.
Drenth, J. P. H. and Waxman, S. G. (2007) ‘Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders’, Journal of Clinical Investigation, 117(12), pp. 3603–3609. doi: 10.1172/JCI33297.
Gaskin, D. J. and Richard, P. (2012) ‘The economic costs of pain in the United States’, Journal of Pain, 13(8), pp. 715–724. doi: 10.1016/j.jpain.2012.03.009.
Herzig, V. et al. (2020) ‘Animal toxins — Nature’s evolutionary-refined toolkit for basic research and drug discovery’, Biochemical Pharmacology, 181(May), p. 114096. doi: 10.1016/j.bcp.2020.114096.
Klint, J. K. et al. (2015) ‘Seven novel modulators of the analgesic target NaV1.7 uncovered using a high-throughput venom-based discovery approach’, British Journal of Pharmacology, 172(10), pp. 2445–2458. doi: 10.1111/bph.13081.
Li, T. and Chen, J. (2018) ‘Voltage-Gated Sodium Channels in Drug Discovery’, Ion Channels in Health and Sickness. doi: 10.5772/intechopen.78256.
Murray, P. R. ., Rosenthal, K. S. . and Pfaller, M. A. (2021) ‘Artrópodos’, in Elsevier (ed.) Microbiología médica. ninth, pp. 791–807.
Muttenthaler, M. et al. (2021) ‘Trends in peptide drug discovery’, Nature Reviews Drug Discovery, 20(4), pp. 309–325. doi: 10.1038/s41573-020-00135-8.
Osteen, J. D. et al. (2016) ‘Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain’, Nature, 534(7608), pp. 494–499. doi: 10.1038/nature17976.
Rodríguez-Marín, J., Bernabeu, P. and Hofstadt, C. J. van-der (2021) ‘Dolor crónico, enfermedades crónicas y enfermedades terminales’, in Psicología médica. second, pp. 391–412.
Salvatierra, J. et al. (2018) ‘NaV1.1 inhibition can reduce visceral hypersensitivity.’, JCI insight, 3(11). doi: 10.1172/jci.insight.121000.
Progeria: El curioso caso de Benjamin Button.
escrito por marinatomas_3b | 10 enero, 2023
Por Marina Casado Grandía y Tomás Cantis López (3ºB de Biología Sanitaria, UAH).
Si has visto El curioso caso de Benjamin Button, seguramente te haya impresionado la condición que sufría este niño, que nació en el cuerpo de una persona de 80 años, y conforme iba «creciendo», rejuvencía. Pues bien, esta película está inspirada en una enfermedad real, llamada progeria o síndrome de Hutchinson-Gilford. Aquí te contamos sus causas, mecanismos moleculares, y sus posibles tratamientos.
Es sabido que los núcleos de las células eucariotas son muy importantes y cumplen muchísimas funciones diferentes. Esto se debe en parte a las laminas nucleares, redes filamentosas compuestas por proteínas situadas en la cara interna de la membrana nuclear. Esta estructura cumple funciones mecanoquímicas, de organización estructural para la cromatina, de regulación epigenética y señalización de muchas rutas, así como de regulación de la expresión génica.
La lamina nuclear está formada por una gran cantidad de proteínas filamentosas de tipo V (filamentos intermedios), llamadas laminas. Estas proteínas están constituidas por una región central de hélice α o coiled coil rodeada por 2 regiones globulares, los extremos C y N terminal. El extremo N terminal es algo más grande, por lo que recibe el nombre de cabeza, mientras que el extremo C terminal es la cola. En esta cola, compuesta por láminas β, presentan además una secuencia señal de direccionamiento hacia el núcleo (llamada NLS, de Nuclear Localization Sequence), con una estructura altamente conservada similar a la de una inmunoglobulina G. Generalmente, esta secuencia está formada por una caja CaaX (donde “C” es una cisteína, “a” es un aminoácido alifático, y “X” es un aminoácido cualquiera). (Vidak, S. et al., 2016).
Para su ensamblaje, las laminas primero se encajan individualmente entre sí para formar dímeros, en los que los motivos centrales (coiled coil) se enrollan mutuamente. Entonces interaccionan las cabezas de un dímero con las colas de otro dímero, de forma que se generan polímeros. Por último se forma el filamento o red, en el que varios polímeros se posicionan paralelamente. (2)
Modelo de ensamblaje de las laminas nucleares: las laminas se ensamblan entre sí para formar dímeros, los cuales se unen para formar polímeros, que finalmente generan la red que constituye la lamina nuclear. Imagen tomada de (Cooper G. et al, 2006).
Se han descrito 2 grandes grupos de estas proteínas:
Laminas A y C: están codificadas por el mismo gen, LMNA, cuyo ARN mensajero sufre 2 vías de splicing para dar lugar a los dos tipos de laminas (A y C). Intervienen en la rigidez de los núcleos, y mutaciones en este tipo de laminas provocan las enfermedades progeroides.
Laminas B: están codificadas por los genes LMNB1 (que produce las laminas B1) y LMNB2 (que produce las laminas B2 y B3). Todas ellas intervienen en la elasticidad nuclear. (Vidak, S. et al., 2016).
Procesamiento de las laminas A
Este proceso de maduración de la pre-lamina A (proteína precursora de la lamina A madura, en el núcleo) es muy importante, ya que en la mayoría de casos, la progeria está causada por mutaciones que afectan en estos pasos.
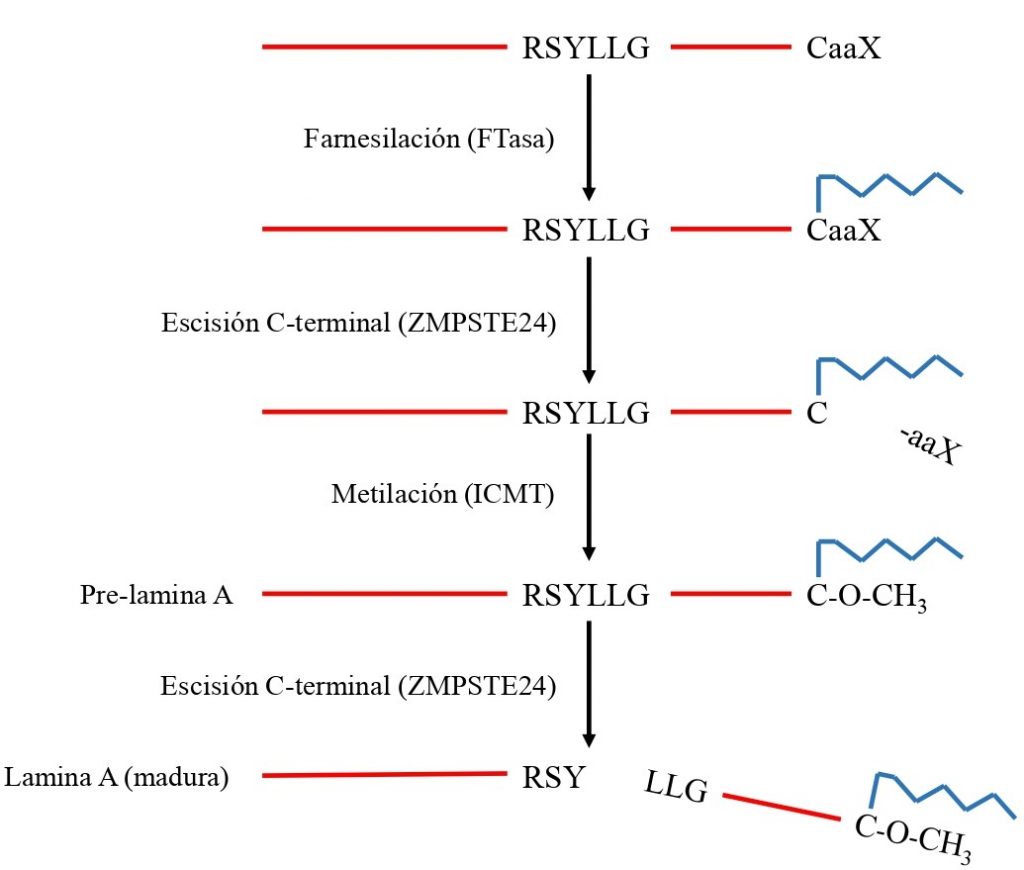
Al transcribirse el gen LMNA, se obtiene un transcrito de mRNA que es traducido para producir la pre-lamina A. Esta proteína sufre una serie de modificaciones:
Primero es farnesilada (se añade un grupo farnesilo, una estructura que facilita su inserción en la membrana) por una enzima FTasa (farnesiltransferasa) en el residuo de cisteína de la caja CaaX.
Una proteasa (FACE1/ZMPSTE24) elimina los 3 aminoácidos del final (aaX), quedando en el extremo C-terminal la cisteína unida al farnesilo. Este farnesilo que queda en la cisteína puede interaccionar fuertemente con la membrana nuclear, por su naturaleza lipídica.
Después, la cisteína es carboxilada (se añade un grupo metilo) por una enzima llamada ICMT (isoprenil-cisteína.carboxil-metiltransferasa).
Por último, la misma proteasa de antes elimina 15 aminoácidos del extremo C terminal, incluida la cisteína (carboximetilada y farnesilada).
Procesamiento de las laminas A: la pre-lamina A se farnesila mediante una FTasa, sufre una escisión en su extremo C-terminal (por una proteasa ZMPSTE24), es metilada en el residuo de cisteína de la caja CaaX (por una enzima ICMT), y finalmente es escindida de nuevo por la misma proteasa. El resultado es una lamina A madura, preparada para insertarse en la membrana nuclear interna.
Las laminopatías son aquellas enfermedades que afectan a la lamina nuclear, causadas por mutaciones en los genes que codifican las laminas. Estos trastornos afectan a muchos tejidos, pero suelen estar asociados al envejecimiento prematuro, y algunos ejemplos de estas enfermedades son el síndrome de Hutchinson-Gilford (progeria) o el síndrome de Charcot-Mary-Tooth 2B1, que afecta a los nervios periféricos. (Méndez-López, I., 2012).
El envejecimiento se puede definir como la degeneración de las funciones celulares y tisulares, que fisiológicamente, está asociada al tiempo. Los principales eventos que caracterizan el envejecimiento son 9:
Inestabilidad genómica y defectos en la arquitectura nuclear (fallos en el ADN).
Acortamiento de los telómeros, los extremos de los cromosomas.
Alteraciones epigenéticas y remodelación de la cromatina.
Pérdida de la proteostasis (regulación de las proteínas).
Disminución en la sensibilidad a los nutrientes.
Disfunción mitocondrial (déficit energético).
Senescencia (envejecimiento) celular.
Agotamiento de las células madre.
Alteración en la comunicación entre células.
(Vidak, S. et al., 2016).
1. SIGNOS Y SÍNTOMAS
En el momento del nacimiento, los pacientes con progeria no muestran ninguna característica diferencial que haga sospechar de la enfermedad, pero cumplido el año de edad, aparecen los primeros síntomas. Estos comienzan con retardo en el crecimiento, pérdida de pelo (alopecia) y de grasa subcutánea (que puede conducir a una resistencia a la insulina y sensibilidad al frío), ojos y venas craneales prominentes, piel envejecida, rigidez articular, baja densidad ósea, esclerodermatitis (piel gruesa y endurecida), displasia (fallos en el desarrollo) mandibular, macrocefalia, etc.
A medida que van creciendo, otros síntomas son osteoporosis, aterosclerosis (endurecimiento de las arterias) y enfermedades cardiovasculares. Entre los 14 y los 20 años de edad, se produce la muerte de estas personas, por infarto de miocardio, fallos cardiacos o aterosclerosis progresiva (que conduce a hemorragias letales).
(Ullrich, N. J. et al., 2015), (Goldman, R. et al., 2004).
Principales signos y síntomas en pacientes con progeria: macrocefalia, alopecia, ojos prominentes, nariz puntiaguda, displasia mandibular, enfermedades cardiovasculares prematuras, esclerodermatitis, afecciones articulares, etc. Estas características aparecen a partir del primer año de vida y se agravan a medida que avanza la vida del paciente. Imagen tomada de (Kreienkamp, R. et al., 2020).
2. GENÉTICA Y PATOGÉNESIS MOLECULAR DE LA PROGERIA
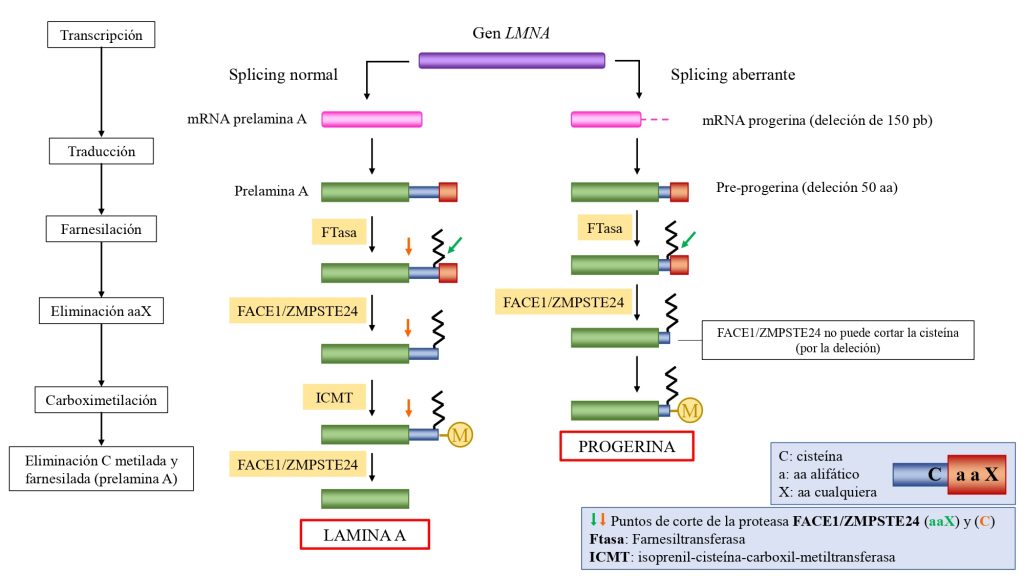
La progeria es causada por una mutación autosómica (en cromosomas no sexuales) y heterocigótica (es suficiente una copia del gen mutada) en el gen LMNA. Con frecuencia esta mutación es el cambio de citosina a timina en la posición 1824 del gen, la cual activa una proteína que produce un corte de 150 pares de bases en el mRNA de la pre-lamina A.
Esta deleción provoca que en la proteína resultante (pre-lamina A) presente una deleción de 50 aminoácidos, incluidoel sitio de reconocimiento para el segundo corte de la enzima. Con ello, no se pueden eliminar los últimos 18 aminoácidos, con la la cola y el farnesilo en la cisteína. (Ullrich, N. J. et al., 2015).
De esta manera, la pre-lamina A en su forma inmadura, acumulándose en estas células como progerina.
Procesamiento molecular de la lamina A normal (izquierda) y de la progerina o lamina A mutante (derecha). En el caso de la progerina, la deleción de 50 aminoácidos en la proteína precursora (pre-progerina) contiene la cisteína terminal (el sitio de reconocimiento de la proteasa ZMPSTE24), con lo que no puede ser escindida. Esto provoca que el grupo farnesilo no sea eliminado, y la progerina se inserta con mucha afinidad en la membrana nuclear interna, generando el fenotipo HGPS.
Esta progerina siempre se encuentra en la membrana nuclear (y no en el nucleoplasma), de forma que la lamina se ve engrosada y se producen cambios en la morfología del núcleo. La progerina, además, produce una disminución en los niveles de laminas B1, y de las proteínas HP1α y LAP2α, de gran importancia en numerosos procesos.
La progerina se expresa únicamente en tejidos de origen mesenquimal (piel, hueso, músculo esquelético, tejido adiposo, corazón y arterias), y como consecuencia de ello, aparecen los siguientes efectos:
Núcleos lobulados y con laminas engrosadas.
Disminución y pérdida de heterocromatina periférica.
Acumulación de daños en el ADN.
Aberraciones teloméricas.
Disfunción mitocondrial.
Es por ello que el organismo de estas personas sufre procesos senescentes prematuros, que conducen a la muerte del individuo antes de los 20 años de edad. (Vidak, S. et al., 2016).
3. DIAGNÓSTICO DE LA PROGERIA
La enfermedad se puede diagnosticar por el reconocimiento del fenotipo (exploración clínica). El análisis genético del gen LMNA puede servir para detectar mutaciones y confirmar el diagnóstico. (Ullrich, N. J. et al., 2015).
4. DIANAS TERAPÉUTICAS
Aunque la progeria no tiene cura, hay ciertos tratamientos que permiten ralentizar la evolución tan rápida de la enfermedad. Entre ellos, destacan:
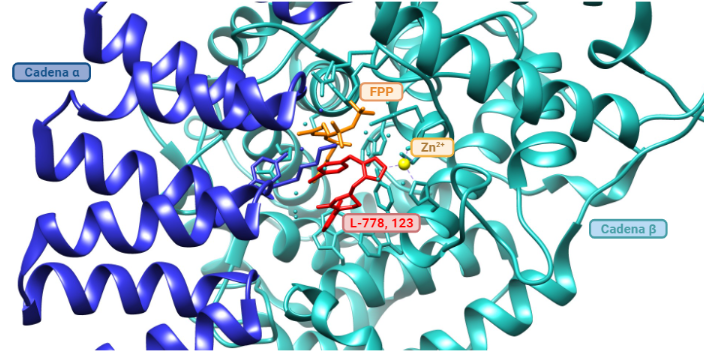
Inhibidores de la farnesiltransferasa (FTIs): Los FTIs son moléculas que se unen al farnesilo del centro activo de la farnesiltransferasa de manera reversible, inhibiendo así la farnesilación de la progerina y su posicionamiento en la membrana nuclear. Se ha probado in vivo en ratones y se han obtenido buenos resultados en los fenotipos como una mejor formación de los huesos, aumento de peso y con todo, alargamiento de la esperanza de vida. Aún así, la farnesilación no es la única forma de prenilación (adición de moléculas hidrofóbicas), sino que las proteínas también se pueden geranil-geranilar (lo que explica la eficacia moderada de los FTIs). Con ello, se remodeló la terapia a inhibir farnesil-pirofosfatasa sintasa (que genera el farnesil pirofosfato) con aminobifosfonato, o la HMG-CoA reductasa (que interviene en rutas sintéticas de isoprenoides) con estatinas. (Ullrich, N. J. et al., 2015), (Vidak, S. et al., 2016). Un ejemplo de FTI es el L-778, 123. (Reid, T. S. et al., 2004).
Modelo de inhibidor de la farnesiltransferasa (L-778, 123) en el centro activo de la enzima, representado en rojo. En azul oscuro se muestra la subunidad α de la farnesiltransferasa, y en azul claro, la subunidad β. FPP hace referencia a farnesil difosfato, el lípido a incorporar a las proteínas u otras moléculas. En el centro activo de la FTasa también hay un átomo de zinc (que actúa como cofactor). Imagen obtenida mediante Chimera y BioRender, a partir de la entrada de PDB 1SA4 (Reid, T. S. et al., 2004).
Activación de la macroautofagia: la autofagia lleva proteínas defectuosas y organelos dañados a lisosomas para su degradación, y a veces está hiper-estimulada (por ejemplo, en condiciones de estrés o ayuno), con el fin de obtener aminoácidos. La rapamicina es un inhibidor de la vía de mTOR, que aumenta la autofagia, y con ello la vida media de las células HGPS (al eliminar la progerina de estas células). También tiene un efecto de reducción de los fenotipos más comunes de la progeria (núcleo lobulado, niveles de LAP-2α y daño en el ADN). Los FTIs también pueden actuar en esta vía, ya que al inhibir la farnesilación de la proteína Rheb GTPasa, activadora de la vía mTOR, también se reducen los niveles de progerina. (Vidak, S. et al., 2016).
Sulforafano: es un antioxidante obtenido de crucíferas (plantas como el brócoli, la col, etc.) que estimula la actividad proteosomal y la autofagia en cultivos de fibroblastos. Con ello provoca la eliminación de la progerina, reduciéndose ciertos efectos de la enfermedad.
Oligonucleótidos morfolinos: son polímeros de hasta 75 nucleótidos que bloquean esteáricamente el sitio de splicing en el exón 11 del pre-mRNA de la lamina A, revirtiendo algunos síntomas o fenotipos, y aumentando la vida de células HGPS.
Resveratrol: activa la deacetilasa SIRT1, y ha mostrado efectos beneficiosos en algunos modelos de ratón, aunque no se conoce con exactitud su mecanismo de acción. Se cree que en presencia de progerina o laminas A, SIRT1 se asocia en menor medida a la matriz nuclear y disminuye su actividad deacetilasa. Con el resveratrol se ha visto que ocurre lo contrario, lo que en última instancia produce una disminución en la pérdida de masa corporal (aumentando la vida de los ratones).
La progeria es una enfermedad que se encuadra en aquellas consideradas «raras», ya que afecta a un número muy reducido de personas en todo el mundo. Sin embargo, a lo largo de los últimos años ha llamado mucho la atención de muchos investigadores e investigadoras, que tratan de comprender mejor los procesos moleculares que la producen, así como idear nuevos tratamientos que permitan aumentar la esperanza de vida de estas personas. Entendiendo este tipo de enfermedades, se podrá en un futuro mejorar o revertir otros problemas relacionados con el envejecimiento fisiológico, lo que también recibe mucha atención últimamente.
(1) Vidak, S., & Foisner, R. (2016). Molecular insights into the premature aging disease progeria. Histochemistry and Cell Biology, 145(4), 401–417. https://doi.org/10.1007/s00418-016-1411-1
(2) Cooper G., Hausman R. El núcleo. En: La Célula. 4ª edición. Marbán. 323-340. (2006).
(3) Méndez-López, I. (2012). Laminopatías. Enfermedades de la lámina nuclear. Medicina Clínica, 138(5), 208–214. https://doi.org/10.1016/j.medcli.2011.03.032
(4) Kreienkamp, R., & Gonzalo, S. (2020). Metabolic Dysfunction in Hutchinson–Gilford Progeria Syndrome. Cells, 9(2), 395. https://doi.org/10.3390/cells9020395
(5) Goldman, R. D., Shumaker, D. K., Erdos, M. R., Eriksson, M., Goldman, A. E., Gordon, L. B., Gruenbaum, Y., Khuon, S., Mendez, M., Varga, R., & Collins, F. S. (2004). Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome. Proceedings of the National Academy of Sciences, 101(24), 8963–8968. https://doi.org/10.1073/pnas.0402943101
(6) Ullrich, N. J., & Gordon, L. B. (2015). Hutchinson–Gilford progeria syndrome (pp. 249–264). https://doi.org/10.1016/B978-0-444-62702-5.00018-4
(7) Reid, T. S., Long, S. B., & Beese, L. S. (2004). Crystallographic Analysis Reveals that Anticancer Clinical Candidate L-778,123 Inhibits Protein Farnesyltransferase and Geranylgeranyltransferase-I by Different Binding Modes. Biochemistry, 43(28), 9000–9008. https://doi.org/10.1021/bi049280b
VIH: Ciclo biológico, patogenia y tratamiento (ART y sistema CRISPR-Cas9)
escrito por Grupo 22 | 10 enero, 2023
Jorge Maldonado Torres, Carlos Medina Sánchez y Daniel Martín Ruíz
Biología Sanitaria, Universidad de Alcalá
El VIH o virus de la inmunodeficiencia humana es muy conocido por la mayoría de la población debido a que ocasiona el síndrome de la inmunodeficiencia humana (SIDA). En un primer momento, el SIDA fue relacionado con drogadictos y gays, gente que en aquella época era tratada como los desechos de la sociedad. Esta situación es recreada muy bien en el siguiente video, donde se cuenta la historia de cómo un linfocito infectado por VIH es repudiado por el resto pero finalmente los antirretrovirales hacen efecto y permite que este siga bailando junto a otro por el torrente.
Origen
El VIH es un retrovirus perteneciente a la familia de los lentivirus. Este tipo de virus proviene de un salto inter-especie del virus de la inmunodeficiencia del simio (SIV). El VIH-2 (menos patogénico y transmisible) proviene de la variedad SIVsm (Sooty mangabey) y el VIH-1 (causante de la pandemia) a su vez lo podemos dividir en diferentes grupos: el VIH-1 de los grupos M y N proviene del SIVcpz (chimpancé) (1), mientras que el VIH-1 de los grupos O y P proviene del SIVgor (gorila) (2).
Dentro del VIH-1 grupo M encontramos diferentes subtipos a partir de los cuales mediante recombinación entre ellos se puede formar formas recombinantes circulantes. En Europa el subtipo que predomina es el subtipo B (3).
Figura 1. Origen de los tipos de VIH.
Morfología y ciclo biológico
El VIH es un virus esférico con una envoltura lipídica, donde se localizan las espículas, matriz y nucleocápside, donde encontramos dos cadenas de RNA. El genoma viral está compuesto por 9 genes (Gag, Pol, Env, Tat, Rev, Nef, Vif, Vpr, Vpu), cuya estructura aparece en la Figura 2 y cuyas funciones aparecen en la Tabla 1.
Gen
Proteína
Función
gag
p24
Proteína de la cápside.
gag
p17
Proteína de la matriz, favorece el anclaje en la membrana y dirige el complejo de pre-integración hacia el núcleo.
gag
p7
Proteína de la nucleocápside, responsable del reconocimiento y la incorporación del RNA al virión.
gag
p6
Interviene en la incorporación de la proteína Vpr al virión en formación.
pol
Transcriptasa inversa (TI)
Sintetiza DNA usando como molde RNA.
pol
Integrasa (IN)
Integra el material genómico viral en el genoma del huésped.
pol
Proteasa (PR)
Escinde precursores como los del gen gag, interviniendo en la maduración.
env
gp120
Proteína externa de la espícula de la membrana, relacionada con la gp41. Interviene en la fijación a células CD4.
env
gp41
Proteínas transmembrana de la espícula, relacionada con gp120. Interviene en la fijación a células CD4.
tat
Tat
Proteína reguladora Activador de la transcripción.
rev
Rev
Proteína reguladora Factor de exportación de los RNAm al citosol para la expresión de proteínas.
nef
Nef
Proteína accesoria Interfiere en la activación de linfocitos T, induce la regulación negativa de células CD4 y del complejo de histocompatibilidad clase I (MHC I). Interviene en la evasión ante la respuesta inmune (4).
vif
Vif
Proteína accesoria Interfiere anulando el efecto antiviral de la proteína A3G (5), favoreciendo la maduración y la infectividad de la partícula vírica.
vpr
Vpr
Proteína accesoria Interviene en la entrada al núcleo del complejo de pre-integración.
vpu
Vpu
Proteína accesoria Interfiere anulando el efecto antiviral de la proteína Tetherina (6), favoreciendo la liberación de viriones y promoviendo la degradación de CD4 en el retículo endoplásmico.
Tabla 1. Genes, proteínas y función del VIH. (7)(8)
Figura 2. Estructura del genoma del VIH. (9)
Para que se pueda llevar a cabo la infección y transmisión del VIH es necesario su integración en la célula del huésped. Primero es necesario la interacción con receptores CD4 (localizados en linfocitos T, linfocitos B, monocitos …) y posteriormente con correceptores CCR5 y CXCR4 (receptores con 7 dominios transmembrana), de tal forma que las partículas virales pueden tener tropismo por receptores CCR5, CXCR4 o ambos.
En un primer lugar, la proteína gp120, localizada en la zona exterior de las espículas de la membrana del virión interacciona con el receptor CD4, provocando un cambio de conformación en gp120 que deja expuesto a su dominio V3, para que se una a los correceptores (receptores de quimiocinas). Estos cambios a su vez provocan un cambio conformacional en la proteína gp41, que deja expuesto el péptido de fusión (N-terminal), el cual se ancla a la membrana plasmática provocando el acercamiento y finalmente la fusión de ambas membranas.
Figura 3. Fusión de la membrana viral y la membrana celular (10).
En el siguiente video representa el final del proceso mediado por gp41.
Este proceso está favorecido por las interacciones que se producen entre el VIH y las lectinas DC-SIGN y L-SIGN, localizadas en la superficie de las células dendríticas, provocando el aumento de la infección de los linfocitos circulantes. De esta forma, en las prolongaciones de las células dendríticas se facilita la infección de linfocitos CD4 (10).
Figura 4. Facilitación de la infección de linfocitos por parte de células dendríticas. (10)
Tras la fusión de las membranas se produce la entrada de la nucleocápside viral, para a continuación descapsidar el genoma viral. Este proceso es inhibido por la proteína celular TRIM5α (específica de cada especie), que provoca que cada retrovirus deba generar variantes de la proteína de la cápside para evitar la acción de estas proteínas celulares (11)(12).
Figura 5. Mecanismo de acción de TRIMα.
Una vez que tenemos libre el RNA viral en el citoplasma, se produce la retrotranscripción, proceso en el cual interviene la transcriptasa inversa (TI). Para la retrotranscripción será necesario unos niveles de nucleótidos y factores celulares, lo cual implica que el linfocito esté activo, porque en caso de que se encontrase en reposo se produciría una retrotranscripción incompleta dando lugar a un DNA retrotranscrito parcial que se degradaría por nucleasas celulares. Por el contrario, en linfocitos activados, una vez que tenemos el DNA transcrito este se va a unir a factores virales (Vpr) y celulares formando el complejo de pre-integración localizado en el citoplasma.
Este complejo será transportado al núcleo para ser integrado mediante la acción de la integrasa en cualquier parte del genoma de la célula huésped (es más frecuente en secuencias intrónicas de genes que estén activados). Sin embargo, no todo el DNA viral será integrado, quedando un porcentaje que será susceptible de integración.
Una vez que ya se haya integrado el genoma viral, se pueden dar diferentes escenarios, en los que el VIH puede permanecer latente, replicarse de forma masiva o de forma controlada.
Para que se inicie la replicación es necesario que se produzca la transcripción, proceso que únicamente depende de factores celulares, entre los que podemos destacar el NF-κB (13) el cual no se encuentra activado en linfocitos en reposo y sí en linfocitos activados.
Una vez que se inicia la síntesis, la proteína Tat aumenta la tasa de transcripción del RNA viral (es necesario para transcripción completa del RNA viral), el mRNA viral es transportado al citosol para ser procesado en transcritos de menor tamaño. Ambos procesos (procesamiento y transporte) están regulados por la proteína Rev (localizada en el núcleo), y si no hay Rev el mRNA viral se acumula en el núcleo. De esta forma obtenemos diferentes proteínas virales que serán procesadas para su posterior ensamblaje dando lugar a las partículas maduras. En este proceso es importante remarcar el papel del las proteínas Vif y Vpu.
Vif va a producir la degradación proteosomal de APOBEC3G (A3G) impidiendo su entrada en los viriones en formación. La acción que tiene APOBEC3G es producir la desaminación de citosina en la primera hebra retrotranscrita por la transcriptasa inversa, provocando mutaciones en el genoma viral que provocan su degradación o una hipermutación y posterior degradación. De esta forma los efectos de su acción se ven sobre los viriones (14).
Figura 6. Mecanismo de acción de APOBEC3G y Vif.
Vpu actúa inhibiendo la expresión de Tetherina, de tal forma que permite y facilita la liberación de viriones de la célula al espacio extracelular, ya que la Tetherina actúa secuestrando los viriones recién formados en la membrana celular.
Finalmente se llevará a cabo la maduración final en el proceso de gemación mediante la acción de la proteasa viral sobre poliproteínas. Cabe destacar que el precursor gp160 (da lugar a gp120 y gp41) es procesado por la proteasa celular.
Figura 7. Ciclo biológico del VIH. (10)
La transmisión del VIH principalmente se produce vía parenteral, vía sexual o vertical (transmisión madre-hijo).
Patogenia
El VIH una vez que se introduce en un organismo va a provocar principalmente un debilitamiento del sistema inmunitario, el cual estará muy relacionado con la disminución en el número de linfocitos T CD4+ (10). Esta disminución de linfocitos CD4 puede deberse a diferentes motivos que aparecen representados en la Figura 8.
Figura 8. Esquema de las causas de linfocitopenia CD4.
La alteración en la homeostasis esta ocasionada por una acumulación de los linfocitos alrededor de prolongaciones de células dendríticas de los órganos linfoides donde se localizan las partículas víricas. A su vez la replicación vírica provoca un bloqueo en la generación de nuevos linfocitos por parte del timo y la médula ósea, dificultando la sustitución de los linfocitos destruidos por el VIH.
En la destrucción directa por efecto citopático, es importante destacar que la destrucción es preferentemente en aquellos linfocitos activados (tienen altos niveles de CCR5, altos niveles de nucleótidos, ATP… En definitiva, factores que son necesario para la replicación del virus).
En el sistema GALT (tejido linfoide asociado al intestino) hay un predominio de linfocitos CD4 activos (debido a que están en continuo contacto con partículas antigénicas). Esto provoca una gran destrucción del sistema GALT que es irreversible. A su vez, la destrucción de linfocitos activados también explica que se destruyan linfocitos de memoria, lo cual agrava la situación inmunológica. Finalmente, el proceso de activación y reconocimiento del VIH provoca la activación de linfocitos que reconocen y atacan las células infectadas, esto ocasiona que las células que iban a matar al VIH se activen siendo infectadas y destruidas. De esta forma, el VIH elimina de manera específica los linfocitos de memoria contra él, facilitando el escape viral ante la respuesta inmune.
Por otro lado, la destrucción indirecta de los linfocitos esta mediada principalmente por dos mecanismos. En primer lugar los linfocitos infectados por el VIH presentan péptidos virales en sus moléculas de HLA clase I siendo objetivos de linfocitos T CD8 citotóxicos.
En segundo lugar, se produce apoptosis, esta se puede dar por dos vías: extrínseca (unión a la membrana plasmática de citocinas de la familia del factor de necrosis tumoral alfa (TNF-alfa)) e intrínseca (alterando la permeabilidad mitocondrial). El VIH puede inducir dicha apoptosis mediante síntesis de citocinas por macrófagos y linfocitos, aumento de la expresión de ligandos citotóxicos o mediante el efecto tóxico de proteína virales. Por otro lado, se ha visto que la proteína Tat puede tener un papel antiapoptótico en linfocitos infectados.
Finalmente, la hiperactivación y el agotamiento vienen ocasionados por una sobreproducción de linfocitos que se puede ver en los linfocitos CD8 citotóxicos producidos frente al VIH, ya que carecen de la capacidad citolítica requerida, debido al agotamiento por la sobrecarga antigénica. A su vez, es fácil pensar que la activación constante del sistema inmune viene ocasionada por la carga viral. Sin embargo, se ha visto que en pacientes con carga viral indetectable y con tratamiento antirretroviral se sigue produciendo esa activación, esto es debido, a la activación de virus endógenos (SIDA) y al aumento de la translocación de productos bacterianos debido al daño producido en el sistema GALT.
Sumado al descenso de linfocitos CD4 ocasionado por la infección del virus es necesario destacar la inflamación crónica que se produce debido a la secreción de citocinas proinflamatorias secretadas por la activación desmesurada que sufre el sistema inmune ocasionando daño endotelial y tisular sistémico.
Figura 9. Esquema de la Patogenia del VIH.
Esta acción esta favorecida por los diferentes mecanismos que tiene el VIH para evadir la respuesta inmune, los cuales están representados en la Figura 10.
Figura 10. Mecanismos de evasión de la respuesta inmune por parte del VIH.
La infección por VIH a su vez se puede dividir en diferentes etapas según avanza el curso de la enfermedad (10).
Una primera etapa o infección reciente. Si se tiene en cuenta que la forma de contacto con el virus ha sido por la vía sexual, el primer contacto que tiene el virus es con las células dendríticas y los linfocitos localizados en la vagina y el recto (sistema GALT), y de ahí se va a producir una diseminación a linfocitos circulantes y ganglios (la estancia en las mucosas es relativamente corta pronto se puede observar una viremia en sangre). A partir de la infección hay un lapso de 4-12 semanas durante el cual no hay respuesta del sistema inmune, lo que ocasiona un gran avance del virus que destruye gran cantidad de linfocitos CD4, provocando la destrucción de gran parte del sistema GALT, que ya no se recuperará y que facilitará la translocación microbiana.
Una segunda etapa o infección crónica, la cual empezaría a las 12 semanas de la infección, donde se se empiezan a generar anticuerpos y linfocitos CD8 citotóxicos. Esto produce que haya un control de la viremia, pero a su vez favorece que se empiecen a producir variantes para escapar de la respuesta inmune. Durante esta fase, el sistema inmune se va debilitando provocando la aparición de alteraciones en la activación y maduración linfocitos CD8 y CD4.
Finalmente, hay una tercera etapa correspondiente a un estadio avanzado, que se caracteriza por la aparición de enfermedades oportunistas a causa de una bajada en el número de linfocitos CD4 y una elevación de la carga viral. En esta fase se ha podido observar un aumento en partículas virales con tropismo por los correceptores CXCR4.
Por lo tanto esta última fase se correspondería con la aparición del síndrome de la inmunodeficiencia humana (SIDA).
Figura 11. Evolución virológica e inmunológica de la infección por VIH. (15)
Tratamiento mediante ART
Inhibidores de la transcriptasa inversa análogos de los nucleósidos y nucleótidos (ITIAN)
Son fármacos que inhiben transcriptasa inversa del VIH mediante una inhibición competitiva actuando como análogos. Su target farmacológico se localizará en el centro catalítico (situado en el subdominio de la palma en la subunidad p66). Se dividen en dos clases:
Análogos de nucleósidos: Abacavir, emtricitabina, lamivudina y zidovudina.
Análogos de nucleótidos: Fumarato de disoproxilo de tenofovir (16).
Análogos de nucleósidos
Actuarán como análogos estructurales de diferentes nucleósidos, pero diferirán de ellos, sobre todo en el radical del C 3´ de la desoxirribosa. Serán fosforilados por las mismas enzimas celulares que los nucleósidos con los que competirán.
Estos análogos de nucleósidos trifosforilados serán incorporados en el DNA viral perdiendo 2 fosfatos y quedando incorporados como análogos de nucleósidos monofosfato. Por tanto, se dará una competición entre estos fármacos y los nucleósidos trifosforilados para incorporarse en el DNA viral.
Las diferencias que poseen estos fármacos en el C 3´de la desoxirribosa impedirán la formación del enlace fosfodiéster con el siguiente nucleótido, deteniendo la elongación de la cadena prematuramente lo cual provoca la inhibición de la transcriptasa inversa (17).
Figura 12. Mecanismo de acción de la zidovudina.
Abacavir (sulfato de abacavir, ABC)
Este fármaco será un análogo estructural del desoxirribonucleósido de guanina. Diferirá de este en que el C 3´ de la desoxirribosa no poseerá un grupo hidroxilo, por lo que formará un doble enlace, además de diferentes variaciones en la guanina.
Figura 13. Comparación abacavir vs desoxiguanosina.
Emtricitabina (FTC)
Este fármaco será un análogo estructural de la desoxicitidina. Diferirá de este en que el C 3´ de la desoxirribosa será sustituido por un átomo de azufre, además de diferentes variaciones en la citosina como un radical de flúor.
Figura 14. Comparación emtricitabina vs desoxicitidina.
Lamivudina (3TC)
Este fármaco será un análogo estructural del desoxicitidina. Poseerá la misma diferencia que posee la emtricitabina (átomo de azufre) pero no poseerá un átomo de flúor en la citosina.
Figura 15. Comparación lamivudina vs desoxicitidina.
Zidovudina (azidotimidina, AZT, ZDV)
Este fármaco será un análogo estructural de la timidina (desoxirribonucleósido de timina). Diferirá de este en que el C 3´de la desoxirribosa no poseerá un grupo hidroxilo, sino un grupo azida.
Figura 16. Comparación zidovudina vs desoxitimidina.
Análogos de nucleótidos: fumarato de disoproxilo de tenofovir (tenofovir DF, TDF)
Este compuesto en un profármaco que sufrirá diversas hidrolisis diestéricas hasta transformarse en tenofovir.
El tenofovir será un análogo estructural del desoxirribonucleótido de adenina (desoxiadenosina monofosfato). A diferencia de los análogos de nucleósidos, este compuesto solo sufrirá dos fosforilaciones por las mismas enzimas celulares que fosforilan el nucleótido de adenina.
El tenofovir difosfato será incorporado al DNA viral perdiendo 2 fosfatos y quedando incorporado simplemente como tenofovir. Por tanto, se dará una competición entre el tenofovir difosfato y la desoxiadenosina trifosfato por la incorporación.
El tenofovir poseerá una estructura química que diferirá de desoxiadenosina monofosfato ya que no poseerá una desoxirribosa como conector, sino que poseerá un metilisopropileter. Al no poseer la desoxirribosa, no poseerá su C 3´ y, por tanto, impedirá la formación del enlace fosfodiéster con el siguiente nucleótido. Esto supondrá la detención de la elongación de la cadena prematuramente provocando la inhibición de la transcriptasa inversa (16)(17).
Figura 17. Mecanismo de acción del Tenofovir.
Inhibidores de la transcriptasa inversa no análogos de nucleósidos (ITINN)
Estos fármacos inhiben la actividad polimerasa de la transcriptasa inversa del VIH-1. No necesitarán ser fosforilados ni se incorporarán a la cadena de DNA viral, como los anteriores, sino que realizarán una inhibición no competitiva uniéndose a la propia enzima.
Se dividen en dos generaciones:
Primera generación: Efavirenz (EFV) y nevirapina (NVP): precisan pocas mutaciones para desarrollar resistencia.
Segunda generación: Etravirina (ETR) y rilpivirina (RPV) utilizados contra cepas del VIH-1 resistentes a la primera generación (18).
Además de estas dos generaciones, en los últimos años, surge un fármaco de nueva generación llamado doravirina (DOR).
Se unirán en la transcriptasa inversa en un bolsillo hidrofóbico situado a 10-15 Å del centro catalítico de la actividad polimerasa y a 60 Å del sitio activo de la actividad ribonucleasa H.
Estos fármacos actúan contra el VIH-1 y no contra el VIH-2 debido a que los residuos 181 y 188 en la RT-VIH-1 son tirosina mientras que en la RT-VIH-2 son isoleucina y leucina, respectivamente. Esto provoca que no se pueda dar la unión del fármaco (19).
Los fármacos de primera generación (EFV y NVP) se unen a los residuos de tirosina (Y181 y Y188) localizados en la región de la palma. Por otro lado, los fármacos de segunda generación (ETR y RPV) se unen a otros puntos como un residuo de triptófano (W229), sitio en el que se producen menos mutaciones. Debido a esto, los fármacos de primera generación sufrirán más resistencia que los de segunda generación (18).
La unión a estos puntos provoca cambios en la conformación que provocan la inhibición de la catálisis del RNA viral y la formación de DNA viral disminuyendo la replicación del VIH-1.
Figura 18. Unión de NVP a la subunidad p66 de la RT-VIH-1. (Imagen realizada con Chimera y BioRender)
Inhibidores de la proteasa (IP)
La proteasa se encarga de realizar la hidrolisis del enlace peptídico de las poliproteínas virales gag y gag-pol. En concreto, se tiene como consenso que en estas poliproteínas se da la hidrolisis del enlace peptídico entre fenialanina (posición 1) y prolina (posición 1´).
Estos fármacos inhibirán la proteasa del VIH y los clasificaremos según su modo de acción:
Actuando como peptidomiméticos y compitiendo con las poliproteínas virales en la unión al centro activo (Asp25-Asp25´): Saquinavir (SQV), ritonavir (RTV), fosamprenavir (FPV), atazanavir (ATV) y darunavir (DRV).
Uniéndose a otra zona del centro activo (Ile50-Ile50´): Tipranavir (TPV).
Los diferentes sustituyentes que tendrán formarán interacciones con aminoácidos de la estructura de la proteasa produciendo más afinidad.
Cabe destacar que el ritonavir actualmente se utiliza como potenciador farmacocinético ya que inhibe el citocromo CYP3A4, el cual se encarga del metabolismo de estos fármacos. Por ello, este se administrará junto con otro IP (20).
Figura 19. Interacción de DRV con el centro catalítico de la proteasa del VIH. (Imagen realizada con Chimera y BioRender)
Inhibidores de la integrasa
El dolutegravir (DTG) y el raltegravir (RAL) son fármacos antirretrovirales que actúan inhibiendo la integrasa. Pertenecen al grupo de los INSTI (inhibidores de la transferencia de cadenas de la integrasa).
La integrasa posee dos cationes metálicos divalentes (Mg 2+) en su sitio activo, en el cual se unirá al DNA del hospedador. Estos dos átomos metálicos serán el target farmacológico.
Se unen a los dos cationes divalentes en el sitio activo cuando la integrasa forma el complejo de preintegración. Esto provoca que la 3-desoxiadenina, que se encuentra en el extremo 3´del DNA viral, se desplace impidiendo la transferencia e integración del DNA viral al DNA del hospedador (21).
Figura 20. Unión de DTG y RAL a la integrasa del VIH.
Inhibidores de la fusión
La proteína de fusión gp41 de los retrovirus posee dos regiones de repetición: HR1 y HR2. La HR1 forma una estructura de enrollamiento trimérica y HR2 se pliega dentro de los surcos hidrofóbicos de HR1 para formar una horquilla. En esto consiste el zapping de gp41. Esto provoca el acercamiento de las dos membranas y la formación de un poro de fusión.
Figura 21. Zapping de gp41. (Imagen extraída de esta página)
Poseemos un único fármaco que inhiba este proceso el cual es la enfuvirtida. Este previene este proceso uniéndose a la región HR1 previniendo el zapping de gp41. La secuencia de aminoácidos de este fármaco es similar a la secuencia de HR2 provocando que realice el pliegue que realizaría HR2.
Se cree que esta acción se realiza después de que se una la proteína gp41 en la membrana de la célula y antes de que se realice el pliegue de HR2. Esto provoca la inhibición del zapping de gp41 y los sucesivos cambios conformacionales para que se dé la fusión (22).
Antagonistas de CCR5
Poseemos un único fármaco, el maraviroc (MVC). Este es un inhibidor alostérico del correceptor CCR5, que inhibe la entrada de retrovirus a la célula.
Es una imidazopiridina que se une a una región transmembrana de CCR5 (dominios transmembrana 2, 3, 6 y 7). Esto provoca una serie de cambios conformacionales, especialmente en el dominio ELC2, lo cual impide que la región V3 de la glucoproteína gp120 de la envuelta interaccione con el receptor (23). Esto impide que se dieran cambios conformacionales posteriores para que se insertara gp41 en la membrana celular provocando la fusión.
Figura 22. Unión de maraviroc a CCR5. (Imagen realizada con Chimera y BioRender)
Inhibidores de la fijación: Fostemsavir
Fostemsavir es un profármaco del temsavir, el cual impedirá la fijación del VIH. Se unirá directamente a la proteína gp120 e inhibe la interacción entre el receptor CD4 celular y el virus. Por tanto, también inhibirá los procesos posteriores a la fijación impidiendo la entrada a linfocitos T CD4+ (24).
Figura 23. Mecanismo de acción del fostemsavir [Rukobia].
Inhibidores posfijación: Ibalizumab-uiyk
Ibalizumab es un anticuerpo IgG monoclonal humanizado el cual bloquea la entrada del virus a los linfocitos T4 sin deteriorar el sistema inmune. Es el primer anticuerpo monoclonal para el tratamiento del VIH y el primer inhibidor posfijación.
Se une al dominio 2 del receptor CD4 en la superficie opuesta tanto en el sitio de unión del complejo mayor de histocompatibilidad de clase II como en el sitio de unión de g120 (25).
Figura 24. Mecanismo de acción del Ibalizumab. (Imagen realizada con BioRender)
Tratamiento mediante CRISPR-Cas9
¿Qué es el sistema CRISPR-Cas9?
El sistema CRISPR-Cas9 ha evolucionado en procariotas como un sistema inmune de memoria que confiere a estos microorganismos resistencia a una segunda entrada de material genético exógeno, como aquel que puede ser introducido a través de fagos o plásmidos.
En el genoma bacteriano existe una región, el locus CRISPR (siglas de “Clustered regularly interspaced short palindromes repeats”, o “Agrupaciones de repeticiones palindrómicas cortas regularmente interespaciadas”), que porta porciones de RNA exógeno (de unos 20pb), perteneciente a fagos u otros elementos genéticos, insertadas por la propia bacteria cuando esta sobrevive a una infección. Estas porciones de RNA, denominadas “protoespaciadores”, se encuentran intercaladas entre otras secuencias cortas que se repiten.
Además, en este locus están también las secuencias que codifican para otros elementos formadores del sistema CRISPR-Cas9, como la enzima Cas9 (“CRISPR asociated protein 9”, una nucleasa) o el RNA transactivador de CRISPR (TracrRNA). La defensa frente un nuevo DNA “conocido” entrante por parte de la bacteria se basa en la complementariedad entre este y el protoespaciador específico (el fragmento de RNA que constituye ese protoespaciador recibe el nombre de crRNA) que se corresponde con ese material genético, además de la interacción de estos con el TracrRNA y la nucleasa Cas9, que desarrolla la acción de degradación del nuevo material genético infectante.
Figura 25. Esquema del sistema CRISPR-Cas9 nativo de bacterias.
Este sistema, nativo de bacterias, ha servido de base para desarrollar en los últimos años un sistema de edición genética que puede ser usado para manipular y escindir partes concretas del DNA humano causantes de graves enfermedades, así como para prevenir y tratar diferentes enfermedades infecciosas como las causadas por el VIH, entre muchos otros agentes infectantes (26).
La maquinaria que permite desarrollar este método de edición génica consiste en una enzima Cas9 unida a una porción de RNA denominado RNA guía (gRNA), formado por la conjunción de un TracsRNA y un crRNA que cuenta con un protoespaciador sintetizado artificialmente y de manera intencionada para que sea complementario de la región del DNA celular (secuencia diana) que se quiere modificar o escindir.
Además de la secuencia diana, para la unión de la proteína Cas9 (y para el correcto emparejamiento con el protoespaciador) es necesaria una secuencia PAM, de apenas 3 nucleótidos, situada junto a ella. Los dominios HNH (Histidina-Asparagina-Histidina) y RuvC de la nucleasa Cas9 participan en la unión de la enzima a la secuencia diana del DNA celular (aquellas con la que interacciona el protoespaciador) y a la hebra complementaria a la secuencia diana, respectivamente (9)(27).
Figura 26. Interacción y procesamiento del DNA diana por el sistema CRISPR-Cas9. (9)Figura 27. Estructura tridimensional de la proteína Cas9 e interacción y acción sobre el DNA diana. (27)
Una vez se ha producido el corte en el DNA celular, el proceso puede continuar con la reparación de las hebras mediante recombinación homóloga o no homóloga. A través de la primera, gracias a un DNA donador, se puede introducir nueva información genética.
Sin embargo, si el proceso desarrollado por el sistema CRISPR-Cas9 no se acompaña por ningún DNA donador y se produce la recombinación no homóloga (unión de los extremos no homólogos formados en el DNA tras el corte), el resultado será un knock-out del gen afectado, un gen no funcional, pues simplemente se ha anulado la función del gen y no se ha introducido nueva información genética.
Figura 28. Edición genética tras la acción del sistema CRISPR-Cas9 gracias a la presencia de un DNA donador y a la recombinación homóloga.Figura 29. Generación de un gen no funcional por la reparación de la rotura generada por sistema CRISPR-Cas9 a través de recombinación no homóloga (en ausencia de DNA donador).
DNA provirus en latencia en linfocitos T CD4+ como diana del sistema CRISPR-Cas9 para el tratamiento por VIH
Por lo general, la terapia antirretroviral (ART) para tratar el SIDA es capaz de controlar la viremia de manera efectiva, pero no consigue eliminar el virus presente en estado de latencia en los linfocitos T infectados. Durante el estado de latencia, las células infectadas por el VIH apenas generan productos víricos, lo que les permite evitar el ataque del sistema inmune del hospedador y, por tanto, permanecer en él durante larguísimos periodos de tiempo.
En estos linfocitos T CD4+ de memoria (cuando se encuentran en un estado de descanso (“a memory resting state”), algo normal tras producirse la infección de estas células por parte del virus), las copias inactivas del provirus (se denomina así al DNA viral resultante de la retrotranscripcción que se encuentra integrado en el genoma celular ) pueden reactivarse cuando lo hicieran las células en las que se encuentran y causar un nuevo rebrote si el tratamiento antirretroviral se interrumpe (28).
Debido a que se cree que estas células son los principales “almacenes” de DNA viral en estado de latencia (29) se han convertido en el objetivo de los principales tratamientos contra el SIDA basados en la utilización del sistema CRISPR-Cas9.
Escisión del provirus del genoma de linfocitos T CD4+ de memoria
En la siguiente tabla aparecen algunas secuencias diana del provirus sobre las que se puede actuar a través del sistema CRISPR-Cas9, así como los métodos de introducción de los elementos necesarios (gRNA y nucleasa Cas9) para desarrollar la técnica (9).
Tabla 2. Secuencias diana del provirus para el sistema CRISPR-Cas9, nucleasa Cas9 específica empleada y métodos de liberación de los componentes del sistema empleados en diferentes estudios. (9)(30)(31)(32)(33)(34)(35)(36)(37)
En diferentes estudios, se consideró la posibilidad de provocar la eliminación de las células hospedadoras del virus en estado de latencia a través de su reactivación, pues esto provocaría una respuesta inmune contra ellas que conllevaría también la eliminación del virus (28). Sin embargo, debido a diversos inconvenientes, consideraron mejores como cura métodos que eliminasen directamente el genoma del virus de las células hospedadoras, como es el caso del método de edición genética CRISPR-Cas9.
En su estudio, Kaminski et al. (28) utilizaron el algoritmo de secuenciación CREST (clipping reveals structure) (38) para la localización de alteraciones estructurales en el genoma de células de la línea de células Jurkat 2D10. Se encontraron cuatro puntos de rotura en el DNA de la célula hospedadora en los cromosomas 1 y 16, concretamente en las regiones p13.2 y p13.3, respectivamente.
Mediante una amplificación de corto alcance se encontró un fragmento de 504 nucleótidos correspondiente a la unión de las regiones 5’ LTR (Long Terminal Repeat) y 3’ LTR del genoma viral tras la acción del sistema CRISPR-Cas9 con un gRNA denominado B. Esta secuencia era 7 nucleótidos más larga que la secuencia de 497 nucleótidos amplificada en las células de control, pues se había producido, además, una inserción de un fragmento de 7 nucleótidos.
La acción del sistema CRISPR-Cas9 / gRNA B permitió eliminar el genoma viral, pues no se pudo secuenciar la región de 257 pares de bases correspondientes al elemento de respuesta Rev (RRE), que se encuentra en el centro de este.
Figura 30. A) Análisis de los amplicones de DNA de las regiones LTR y RRE sin y con tratamiento con sistema CRISPR-Cas9. B) Estructura de la región del genoma viral entre las regiones 5′ y 3′ escindida por el sistema CRISPR-Cas9. Se indican también los 7 nucleótidos añadidos y la región RRE que se pierde. (28)
Examinaron mediante PCR de alto rango las posiciones en las que se inserta el genoma viral dentro de los cromosomas 1 y 16. En ambos, se obtuvieron tres tipos distintos de amplicones: uno de larga longitud (de 6130 pb para el cromosoma 1 y de 5467 pb para el 16), correspondiente al genoma integrado del VIH-1 más su secuencia de ADN flanqueante derivado del cromosoma; uno de tamaño medio (de 909 pb y de 759 pb, respectivamente), que se corresponde con el mismo fragmento pero tras escindir por completo el genoma viral; y otro mucho menor (de 264 pb y 110 pb), que se trata del fragmento de DNA correspondiente del cromosoma homólogo, que no ha sufrido modificación alguna.
Figura 31. Puntos de integración del genoma del VIH en los cromosomas 1 (C) y 16 (D). Además, de cada uno de ellos, se observa la longitud del genoma completo del virus integrado antes del tratamiento con el sistema CRISPR-Cas9, tras su acción y la secuencia diana en el cromosoma homólogo (en la que no se ha producido integración del genoma viral), todos ellos secuenciados mediante PCR de largo alcance. (28)
Revisiones posteriores del método determinaron que el genoma viral escindido es mayoritariamente degradado a continuación, por lo que el riesgo de transposición o reintegración en el genoma del huésped es bajo (28).
Escape viral al tratamiento y como consecuencia del tratamiento
Sin embargo, según otros estudios, como se recoge en la publicación de Das, Binda and Berkhout (39), es posible que el virus escape a la inhibición mediante mutaciones efectuadas en la región del genoma viral que es diana del corte efectuado por Cas9 que impidan su acción pero permitan la replicación viral. Entre estas mutaciones se encontraron indels (inserciones o deleciones) en regiones poco conservadas (promotores de la región LTR) y sustituciones e inserciones en regiones conservadas, como dominios que codifican para proteínas, lo que constituye un patrón de mutaciones diferentes al que ocurre para escapar de los fármacos ART.
El origen de esas mutaciones puede encontrarse, además de en el proceso de retrotranscripción desarrollado para integrar el genoma viral en el genoma de la célula hospedadora, en el proceso de modificación del genoma proviral desarrollado por parte del propio sistema CRISPR-Cas9.
Infectividad por VIH de células T tratadas con el sistema CRISPR-Cas9
En el mismo estudio (28) se probó cual era la reacción a la reinfección de clones de células T de las que se erradicó el genoma viral mediante el sistema con CRISPR-Cas9. Estas células contaban con diferentes componentes del sistema (unas con la enzima Cas9 en distintos niveles de concentración, otras con el gran y otras con ambos).
Los resultados fueron la resistencia a la reinfección por parte de aquellas células que contaban con ambos elementos (mayor resistencia en las que la expresión de Cas9 era mayor), pero no en las que solamente poseían uno de los dos elementos del sistema.
Otras dianas para el tratamiento del SIDA mediante terapia génica: Correceptores CCR5 y CXCR4
Además de tener el genoma proviral como diana para el tratamiento genético del VIH, el sistema CRISPR-Cas9 puede ser utilizado para bloquear la entrada del virus en los linfocitos T CD4+ a través de la modificación génica de los correceptores de membrana que empleados por el VIH para acceder al interior celular (9).
Simulando la resistencia al VIH que aparecen de manera natural en personas que presentan deleciones en gen que codifica para el correceptor CCR5 (40), el método CRISPR-Cas9 puede ser utilizado (como corroboran múltiples estudios, recogidos en el artículo de Xiao et al.) con la intención de editar los genes de los correceptores CCR5 y CXCR4 para evitar la infección por VIH sin que se pierda la función propia.
Ventajas y limitaciones de la utilización del sistema CRISPR-Cas9 como tratamiento contra el SIDA
La utilización del sistema de edición genética CRISPR-Cas9 se ha antepuesto a otros, como el uso de nucleasas con dedos de zinc (ZFNs) o de nucleasas que actúan como activadores transcripcionales (TALENs), debido al alto coste y tiempo requerido por los mismos. Entre los motivos que llevan al sistema CRISPR-Cas a ser el más ampliamente utilizado están su precisión, su sencillez o su alta eficacia (9).
Por otro lado, uno de los mayores inconvenientes que aparecen a la hora de utilizar el sistema CRISPR-Cas9 es el método de “aporte” o “liberación” (delivery) de los elementos que lo componen en las células infectadas. Entre los métodos comúnmente utilizados se encuentran los vectores adenovirales, vectores de AAV (virus adenoasociados, que requieren la coinfección con adenovirus) o los vectores lentivirales (41), en función de los requerimientos de expresión o el tamaño del material genético que se quiere introducir en el interior celular. También se puede llevar a cabo la transfección directa de los componentes del sistema (procedentes de, por ejemplo, Streptococcus pyogenes o Staphylococcus aureus) tras electroporación (42) o utilizando vesículas generadas artificialmente para permitir el transporte hasta el interior celular (43).
Además, como se comentó anteriormente, otra desventaja de este método es su alta especificidad (su precisión se le puede volver en contra), pues le da al virus la posibilidad de escapar y hacerse resistente al sistema mediante simples mutaciones en su secuencia.
Bibliografía
1. Delgado R. Virological characteristics of HIV. Enferm Infecc Microbiol Clin. 2011;29(1):58–65.
2. D’Arc M, Ayouba A, Esteban A, Learn GH, Boué V, Liegeois F, et al. Origin of the HIV-1 group O epidemic in western lowland gorillas. Proc Natl Acad Sci U S A. 2015;112(11):E1343–52.
3. Albert Hernández M, San Miguel Hernández Á. Epidemiology, detection of resistance and tropism of HIV-1: update. Rev Med Lab. 2020;1(1):10–20.
4. Salmen S, Gabaldon-figueira JC, Teran-angel G. Nef- VIH-1 como principal orquestador de la disfunción celular durante la inmunopatogenia de la infección por el VIH-1 ( Nef- HIV-1 as the main orchestrator of the cellular dysfunction in the immunopathogenesis of the HIV-1 infection ) Introducción. Av en Biomed. 2016;4(3):126–37.
5. Goila-Gaur R, Strebel K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008;5:1–16.
6. Neil SJD, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451(7177):425–30.
7. Noé REA, Guadalupe HMP, Israel NG, Ricardo GPC, Denise CAF. Caracteristicas estructurales y funcionales del Virus de la Inmunodeficiencia Humana. Vol. 33, Enfermedades Infecciosas y Microbiologia. 2013. p. 163–73.
8. Alcamí J. The HIV replication cycle. Established therapeutic targets and potential targets. Enferm Infecc Microbiol Clin [Internet]. 2008;26(SUPPL. 12):3–10. Available from: http://dx.doi.org/10.1016/S0213-005X(08)76566-2
9. Xiao Q, Guo D, Chen S. Application of CRISPR/Cas9-based gene editing in HIV-1/AIDS therapy. Front Cell Infect Microbiol. 2019;9(MAR):1–15.
10. Alcamí J, Coiras M. Inmunopatogenia de la infección por el virus de la inmunodeficiencia humana. Enferm Infecc Microbiol Clin. 2011;29(3):216–26.
11. Lukic Z, Campbell EM. The cell biology of TRIM5α. Curr HIV/AIDS Rep. 2012;9(1):73–80.
12. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature [Internet]. 2004 Feb;427(6977):848–53. Available from: https://pubmed.ncbi.nlm.nih.gov/14985764/
13. Lozano De León-Naranjo F. Infección por el VIH (I). Med. 2014;11(49):2893–901.
14. Imahashi M, Nakashima M, Iwatani Y. Antiviral mechanism and biochemical basis of the human APOBEC3 family. Front Microbiol. 2012;3(JUL):1–7.
15. Simon V, Ho DD, Abdool Karim Q. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet. 2006;368(9534):489–504.
16. Ribera E, Tuset M, Martín M, Del Cacho E. Characteristics of antiretroviral drugs. Enferm Infecc Microbiol Clin. 2011;29(5):362–91.
17. McTavish D, Campoli-Richards D, Sorkin EM. Carvedilol: A Review of its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Efficacy. Drugs. 1993;45(2):232–58.
18. Portilla J, Estrada V. Mecanismo de acción y farmacocinética de rilpivirina. Enferm Infecc Microbiol Clin [Internet]. 2013;31(SUPPL.2):2–5. Available from: http://dx.doi.org/10.1016/S0213-005X(13)70136-8
19. Wang Y, De Clercq E, Li G. Current and emerging non-nucleoside reverse transcriptase inhibitors (NNRTIs) for HIV-1 treatment. Expert Opin Drug Metab Toxicol [Internet]. 2019;15(10):813–29. Available from: https://doi.org/10.1080/17425255.2019.1673367
20. Mahmoud SS, Hussain ZM, O’Shea P, Schaubach KR, Iv NJD, Rappaport TS, et al. الابتزاز الإلكتروني.. جرائم تتغذى على طفرة «التواصل الاجتماعي»No Title. CNR-ISTI Tech Rep [Internet]. 2015;3(2):356–69. Available from: https://www.metis2020.com/wp-content/uploads/METIS_D1.4_v3.pdf%0Ahttps://www.metis2020.com/documents/deliverables/index.html%0Ahttps://www.metis2020.com/metis-deliverables-d1-4-d2-4-d3-3-d4-3-d6-5-and-d7-3-were-completed-in-february-2015/index.html%0Ahttp
21. Ribera E, Podzamczer D. Mecanismo de acción, farmacología e interacciones de dolutegravir. Enferm Infecc Microbiol Clin [Internet]. 2015;33(S1):2–8. Available from: http://dx.doi.org/10.1016/S0213-005X(15)30002-1
22. Adams J. Drug evaluation. Aust Prescr. 1996;19(1):5.
23. Soriano V, Poveda E. Maraviroc: Farmacocinética, interacciones y mecanismo de acción. Enferm Infecc Microbiol Clin [Internet]. 2008;26(SUPPL. 11):12–6. Available from: http://dx.doi.org/10.1016/S0213-005X(08)76558-3
24. Markham A. Fostemsavir: First Approval. Drugs [Internet]. 2020;80(14):1485–90. Available from: https://doi.org/10.1007/s40265-020-01386-w
25. Markham A. Ibalizumab: First Global Approval. Drugs [Internet]. 2018;78(7):781–5. Available from: https://doi.org/10.1007/s40265-018-0907-5
26. McCarthy MW. Harnessing the potential of CRISPR-based platforms to advance the field of hospital medicine. Expert Rev Anti Infect Ther [Internet]. 2020;18(8):799–805. Available from: https://doi.org/10.1080/14787210.2020.1761333
27. Ricci CG, Chen JS, Miao Y, Jinek M, Doudna JA, McCammon JA, et al. Deciphering Off-Target Effects in CRISPR-Cas9 through Accelerated Molecular Dynamics. ACS Cent Sci. 2019;5(4):651–62.
28. Kaminski R, Chen Y, Fischer T, Tedaldi E, Napoli A, Zhang Y, et al. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci Rep [Internet]. 2016;6(December 2015):1–15. Available from: http://dx.doi.org/10.1038/srep22555
29. Bruner KM, Hosmane NN, Siliciano RF. Towards an HIV-1 cure: Measuring the latent reservoir. Trends Microbiol [Internet]. 2015;23(4):192–203. Available from: http://dx.doi.org/10.1016/j.tim.2015.01.013
30. Ebina H, Misawa N, Kanemura Y, Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep. 2013;3:1–7.
31. Hu W, Kaminski R, Yang F, Zhang Y, Cosentino L, Li F, et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A. 2014;111(31):11461–6.
32. Liao HK, Gu Y, Diaz A, Marlett J, Takahashi Y, Li M, et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat Commun. 2015;6:1–10.
33. Zhu W, Lei R, Le Duff Y, Li J, Guo F, Wainberg MA, et al. The CRISPR/Cas9 system inactivates latent HIV-1 proviral DNA. Retrovirology. 2015;12(1):1–7.
34. Wang Z, Pan Q, Gendron P, Zhu W, Guo F, Cen S, et al. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell Rep [Internet]. 2016;15(3):481–9. Available from: http://dx.doi.org/10.1016/j.celrep.2016.03.042
35. Lebbink RJ, De Jong DCM, Wolters F, Kruse EM, Van Ham PM, Wiertz EJHJ, et al. A combinational CRISPR/Cas9 gene-editing approach can halt HIV replication and prevent viral escape. Sci Rep [Internet]. 2017;7(February):1–10. Available from: http://dx.doi.org/10.1038/srep41968
36. Yin C, Zhang T, Qu X, Zhang Y, Putatunda R, Xiao X, et al. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol Ther [Internet]. 2017;25(5):1168–86. Available from: http://dx.doi.org/10.1016/j.ymthe.2017.03.012
37. Wang Q, Liu S, Liu Z, Ke Z, Li C, Yu X, et al. Genome scale screening identification of SaCas9/gRNAs for targeting HIV-1 provirus and suppression of HIV-1 infection. Virus Res [Internet]. 2018;250:21–30. Available from: https://doi.org/10.1016/j.virusres.2018.04.002
38. Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods. 2011;8(8):652–4.
39. Das AT, Binda CS, Berkhout B. Elimination of infectious HIV DNA by CRISPR–Cas9. Curr Opin Virol [Internet]. 2019;38:81–8. Available from: https://doi.org/10.1016/j.coviro.2019.07.001
40. Biti, R., Ffrench, R., Young, J. et al. HIV-1 infection in an individual homozygous for the CCR5 deletion allele. Nat Med 3, 252–253 (1997). Available from: https://doi.org/10.1038/nm0397-252
41. Deng Q, Chen Z, Shi L, Lin H. Developmental progress of CRISPR/Cas9 and its therapeutic applications for HIV-1 infection. Rev Med Virol. 2018;28(5):1–7.
42. Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A. 2015;112(33):10437–42.
43. Campbell LA, Coke LM, Richie CT, Fortuno L V., Park AY, Harvey BK. Gesicle-Mediated Delivery of CRISPR/Cas9 Ribonucleoprotein Complex for Inactivating the HIV Provirus. Mol Ther [Internet]. 2019;27(1):151–63. Available from: https://doi.org/10.1016/j.ymthe.2018.10.002
El boom de los miRNA
escrito por grupo 29 | 10 enero, 2023
Ana Paola Cano Morris, María del Rosario García Sánchez, Luna Guerra Núñez. Biología Sanitaria, UAH.
Los microRNA son cadenas pequeñas de RNA no codificante formadas por unos 22 nucleótidos, que intervienen en la regulación de RNA. Al tener de diana a gran parte de los transcritos codificantes de proteínas, los miRNA están involucrados en casi todos los procesos patológicos y de desarrollo de organismos.
Algunos estudios plantean la hipótesis de que los miRNA provienen de una evolución de RNAs pequeños en eucariotas para suprimir material genético y transcritos no deseados.
¿Cómo se forman?
La mayoría de los miRNA son transcritos en el núcleo por la RNA polimerasa IIcomo intrones o exones de RNAs no codificantes, siendo por tanto, su expresión regulada por factores de transcripción o reguladores epigenéticos. Como resultado se generará un primiRNAde unas 1000 bases que deberá sufrir una maduración hasta ser un miRNA funcional.
En estas secuencias aparecen pequeñas horquillas de unos 33-35 pares de bases en cuyos extremos hay un bucle y segmentos de cadena simple 5′ y 3′ que unirán las diferentes horquillas.
Biogénesis de miRNA, creada con Biorender. [4]
En el comienzo de la maduración participa la proteína Drosha, una endonucleasa RNasa-III cuya acción es sobre RNA de cadena doble, es decir, actuará directamente sobre la horquilla formada anteriormente. También colabora en este punto el cofactor DGCR8, cuya actividad principal es aumentar la interacción del Microprocesador que forman con el primiRNA.
Es importante que el tamaño de la horquilla sea regular para una correcta maduración. Este Microprocesador realiza un corte sobre el RNA de doble cadena aproximadamente a 11 pares de bases de la unión basal (lugar en el que comienza la doble cadena) y a 22 de la unión apical (lugar donde se sitúa el loop terminal de la horquilla). Si como hemos dicho antes, la horquilla tiene un tamaño de entre 33-35 pares de bases, el Microprocesador realizará un corte en el sitio correcto y se formará un premiRNA; este corte no es perfecto, sino que deja en el extremo 3′ dos nucleótidos de más que quedan desapareados. Es importante que esta nueva molécula también tenga en su extremo 5′ un monofosfato; ambas estructuras permitirán la correcta interacción con el resto de proteínas. El Microprocesador es susceptible de modificaciones que regulen tanto su expresión como su función, pudiendo ser la formación de miRNAs regulada en múltiples puntos.
Complejo microprocesador.Click para versión interactiva. Conformado por DROSHA, que presenta dominios con acción RNAsa III (RIIIDa y RIIIDb) y dominios de unión a RNA bicatenario (dsRBD). La unión a dsRNA se ve reforzada por los dominios de DGCR8, factor que se ancla a DROSHA mediante sus C-terminal conservados e interactúa con el pri-miRNA mediante dos dominios dsRBD. Imagen creada con Biorender.
Una vez formado el premiRNA el resto de su maduración se realizará en el citoplasma. El exporte de la molécula es llevado a cabo por la proteína Exportina 5 (EXP5), que reconoce la estructura del RNA de doble cadena e interacciona con el saliente en su extremo 3′. Se forma un complejo en forma de canasta con la proteína RAN-GTP y el premiRNA que interacciona con el poro nuclear. El GTP es hidrolizado después de la translocación del complejo, de forma que este se desensambla y el premiRNA queda libre en el citoplasma. En algunos tumores la proteína EXP5 está truncada por una mutación en el gen que la codifica, impidiendo la maduración de miRNAs.
Complejo EXP5 ; RAN-GTP. Click para versión interactiva. La estructura recuerda a un guante de baseball, con una zona de interacción tipo túnel debajo, donde se sitúa el overhang 3´del pre-miRNA que le permite ser reconocido por EXP5. Imagen creada en Biorender.
En el siguiente paso de la maduración, interviene la proteína Dicer acompañada de la proteína TRBP que aumenta su eficacia. Dicer, al igual que Drosha, es una endonucleasa RNasa III cuya función es realizar un corte en el premiRNA cerca del loop terminal de la horquilla. Así se liberará una corta cadena de RNA duplexo de unos 22 pares de bases, que será el futuro miRNA. La proteína Dicer necesitará de los extremos 5′ fosforilado y 3′ desapareado para su correcta interacción y funcionamiento.
DICER-TRBP. Click para versión interactiva. El centro catalítico de DICER está formado por un dímero intramolecular de RIIIDs en el extremo C-terminal, el dominio helicasa en el extremo N-terminal facilita la interacción con lazo terminal de pre-miRNA en su reconocimiento. El dominio PAZ se ancla al extremo terminal del pre-miRNA y se hipotetiza que el espacio entre PAZ y RIIIDs actúa a modo de «regla» al momento del corte. Imagen creada con Biorender.
La pequeña doble cadena de RNA se asocia a continuación con a proteína AGO, formando el complejo silenciador inducido por RNA (RISC). Será otra vez necesario que los extremos 5′ y 3′ estén correctamente formados para una apropiada interacción con la proteína. Para la carga del miRNA en la proteína AGO, es necesaria la formación un complejo intermedio denominado RLC compuesto por el miRNA, la proteína Dicer explicada antes y la nueva AGO. En humanos existen hasta 4 tipos de la proteína AGO, pero se unen indistintamente a los miRNAs para la formación del complejo.
Por el momento seguimos teniendo el miRNA como una cadena doble, por lo que debemos referirnos al complejo como pre-RISC. Por último, se deberá eliminar la cadena que no vaya a funcionar como guía en el complejo por medio de la endonucleasa C3PO, teniendo ya así el complejo RISC con un miRNA funcional. La selección de la cadena guía se realiza en función de la estabilidad termodinámica de los extremos de la cadena, normalmente es elegida la que posee un extremo 5′ menos estable; la cadena descartada será rápidamente degradada (será una pequeña cadena de RNA sin ningún tipo de protección). No siempre es elegida la misma cadena, ya que existe cierta preferencia hacia una de ellas en un 96-99% de los casos. La cadena de miRNA menos frecuente, aún siendo también activa, será menos efectiva y aparece señalada como miRNA*.
Se acaba de explicar el funcionamiento de la biogénesis de miRNA de forma canónica. Por el contrario, no es la única vía y existen otras vías alternativas. Destacaremos los mirtrons, los cuales se forman desde los intrones de mRNAs inmaduros y también forman una horquilla; producto del splicing que sufren los mRNA en su maduración. Estas secuencias de RNA serán similares a premiRNA y se introducirán en el proceso de síntesis de miRNA habiéndose saltado el procesamiento por Drosha y DGCR8.
¿Cómo funcionan?
Como se ha adelantado antes, la complementariedad de bases entre el complejo RISC y el mRNA a regular es crucial para que el miRNA pueda llevar a cabo su función. La regulación del producto codificado por el mRNA en cuestión puede llevarse a cabo por dos vías:
Degradación del mensajero
Represión de la traducción
Ambas dependerán en el grado de complementariedad al que se llegue entre el miRNA y el mRNA.
Modelo especulativo del funcionamiento de cada región del miRNA. Imagen creada con Biorender. [2]
Principalmente interactúan los nucleótidos de 2-8 del miRNA, altamente conservados, en el extremo 5’ (conocida como secuencia “seed”) con la región 3’UTR del mRNA, aproximadamente 15 nucleótidos después del codón de paro y junto a una secuencia rica en AU. Actualmente se sabe que la secuencia diana puede también encontrarse en la región codificante (ORF) o 5’UTR, aunque menos frecuente y eficaz que la unión canónica.
Que sean esos nucleótidos presentados al mRNA no es algo casual, viene determinado por la disposición del complejo RISC que consigue presentar este fragmento concreto de la cadena guía en forma de hélice A, lo que aumenta la afinidad y especificidad con el mRNA. Se ha estudiado que un segmento mayor supondría un problema para la unión, mientras que uno menor no tendría la especificidad suficiente para su buen funcionamiento.
El miRNA debe estar totalmente cubierto por la proteína AGO para evitar su degradación por RNasas. Solo se muestra la secuencia “seed” que se unirá específicamente al mRNA. En este punto ocurre la represión de la traducción. Existen varias hipótesis sobre este mecanismo:
Ralentización o paralización de los ribosomas en un punto posterior a la iniciación de la traducción.
Marcaje del polipéptido para su posterior degradación
Prevenir la interacción entre el promotor y su activador traduccional
Para que ocurra la degradación del mRNA es necesario alcanzar un grado mayor de complementariedad con el miRNA. Después de la unión típica del extremo 5’ hay un cambio conformacional del complejo que permite extender la interacción en las regiones central y 3’ de la cadena, después la proteína AGO vuelve a su conformación típica. Es en este momento cuando puede ocurrir la degradación, realizándose un corte entre los nucleótidos 10 y 11. Es la proteína AGO quien posee una actividad endonucleolítica capaz de realizar el corte. Los fragmentos del mRNA serán posteriormente degradados, mientras que el miRNA permanece intacto para poder seguir actuando.
En eucariotas, los complejos RISC se encuentran principalmente asociados a Cuerpos P, zonas electrodensas donde se acumulan enzimas implicadas en la degradación de mRNA, siendo así considerado un lugar para la regulación postranscripcional de mRNA.
Se sabe que existe una función cooperativa de los miRNA ya que aparecen múltiples sitios de unión a estas moléculas en los mRNA, multiplicando así las respuestas de cada sitio de unión trabajando por si mismo. De esta forma el mecanismo de acción es más sensible a cambios en los niveles de miRNA. Los miRNA que se unieran al mismo mRNA también podrían ser distintos, debiendo tener en cuenta todas las interacciones a la hora de determinar una patología o tratamiento.
Los miRNA normalmente no tienen un mensajero diana determinado, sino que pueden regular la expresión de varios genes. Esto es posible en primer lugar, gracias a la conservación de los sitios de unión en las regiones 3’UTR de modo que aparecen compartidos entre una familia de genes. Por otra parte, la complementariedad entre la región 3’UTR y el miRNA no es completa en la mayoría de casos, sino que solo se da en una pequeña secuencia de 7 nucleótidos, lo suficientemente corta como para poder estar en otros mRNA.
Transporte de miRNA
Tan importante como conocer su funcionamiento en el interior celular, es conocer su transmisión entre diferentes células. Dependiendo de los mecanismos de transporte intercelular que utilicen, su transmisión tendrá características diferentes, las cuales afectaran a los procesos fisiológicos y patológicos. En el caso de estos últimos, determinaran la capacidad de diseminación y extensión de la enfermedad en el organismo.
Las principales vías de comunicación intercelular son: la señalización dependiente de contacto célula-célula (uniones intercelulares), la señalización mediante moléculas solubles y la señalización mediante vesículas extracelulares.
En el caso de los miRNA se ha demostrado su transmisión mediante: microvesículas, HDL, lipoproteínas, riboproteínas, cuerpos apoptóticos, exosomas y gap junctions. Estos dos últimos procesos serán los que explicaremos en profundidad debido a la gran importancia que están teniendo en las últimas investigaciones sobre miRNA.
Gap-junctions
Son microdominios de la membrana plasmática de células adyacentes, en los cuales se encuentran conexinas (en los invertebrados se denominan inexinas) formando canales proteicos que permiten un intercambio rápido de sustancias entre ambas células mediante el transporte directo de moléculas de pequeño tamaño.
Diversos estudios han demostrado la existencia de un trasporte de miRNA desnudos, es decir, sin el complejo RISC. Sin embargo, aún se desconoce el mecanismo molecular de dicho transporte.
Transporte de miRNA a través de las conexinas de gap-junctions. Se transporta el miRNA duplexo, antes de la unión al complejo RISC. Imagen creada con Biorender. [7]
Exosomas:
Los exosomas fueron descritos por primera vez por Pan y Johnstone en 1983. Son vesículas extracelulares de entre 40 y 120 nm de diámetro constituidos por una bicapa lipídica. Pueden transportar moléculas como: lípidos, proteínas, RNA… Además, se han detectado en la mayoría de líquidos corporales, lo cual los convierte en potenciales biomarcadores para ciertas enfermedades.
La biogénesis de los exosomas se produce siguiendo el siguiente proceso: Primero se forman endosomas tempranos a partir de la invaginación de la membrana celular. Seguidamente, se generan invaginaciones de la membrana del endosoma, dando lugar a múltiples vesículas intraluminales (ILVs), lo que resulta en la formación de cuerpos multivesiculares (MVBs). El contenido es seleccionado e introducido en las ILVs, que cuando sean secretadas al exterior celular por exocitosis, pasarán a considerarse exosomas.
Biogénesis de exosomas. Imagen creada con Biorender. [10]
El mecanismo para la introducción de los miRNA en los exosomas aún no está completamente estudiado y descrito, siendo necesaria una mayor investigación en ese campo. Las cuatro vías que vamos a exponer a continuación han sido descritas en diversos estudios y actualmente son la principal hipótesis para explicar dicho proceso.
Vías de introducción de los miRNA en exosomas. Imagen creada con Biorender. [10]
MiRNA en el ámbito clínico
Fallos en la regulación de la expresión y la función de los miRNA, ya sea por alteraciones genómicas o cambios en su biogénesis, han mostrado ser factores importantes en el desarrollo y la progresión de múltiples enfermedades.
Los miRNA son por lo tanto, un área de estudio importante para futuras técnicas de diagnóstico y tratamiento.
MiRNA como biomarcadores
Los miRNA tienen el potencial de ser el biomarcador perfecto; se expresan de forma ubicua, se pueden aislar fácilmente y cuantificar con gran sensibilidad y especificidad en tejidos o fluidos. Además, son estables, tienen especificidad celular y relevancia fisiológica en estados patológicos.
Sin embargo, aún no se logra una estandarización en cuanto a su análisis, y la expresión y función de los miRNA ha presentado variabilidad en distintas poblaciones.
Los principales test de diagnostico en el mercado son útiles para distintos tipos de cáncer y enfermedades relacionadas con la edad, esto mediante el análisis de varios miRNA. Aun así, hay proyectos en desarrollo que se centran en los niveles de un único miRNA.
En los últimos años, muchos estudios han propuesto el uso clínico de los exosomas con miRNA como posibles biomarcadores para la detección y seguimiento de enfermedades en las que participan. Esto se debe a las siguientes características de los miRNAS exosomales:
No todos los miRNA son transportados por exosomas.
Algunos miRNA solo han sido detectados en exosomas en estados patológicos.
Se han detectado diferencias en los niveles de secreción de exosomas con miRNA, además de una diferente concentración de estos en su interior, tanto entre estado fisiológico normal y estado patológico, como en la evolución de la enfermedad.
Ante estos resultados, el departamento de Hematología/Oncología del Hospital Pediátrico Bambino Gesù ha propuesto el análisis de los miRNA exosomales mediante RT-qPCR para la detección del cáncer infantil y un mejor seguimiento del progreso de la enfermedad, además de su reacción a los diferentes tratamientos.
Diagnóstico con miRNA exosomal en pacientes oncológicos A partir de la extracción de sangre del paciente, se realiza un ultracentrifugado para la separación del plasma. Una vez obtenido la fracción con los exosomas, se realiza una RT-qPCR. Los datos se envían a un programa para su análisis, terminando en la identificación y cuantificación de los biomarcadores. [3]
MiRNA como terapia
Debido a sus patrones de expresión y su habilidad de unirse a varias dianas, usualmente en un mismo proceso biológico, los miRNA pueden potencialmente regular la expresión de múltiples elementos en rutas de señalización conocidas por estar afectadas en determinados estados patológicos.
Sin embargo, la baja especificidad de unión, aumenta el peligro de efectos secundarios importantes producto de la afectación de otras rutas fuera de la diana deseada.
Es indispensable así el desarrollo de herramientas que permitan una predicción más exacta de la unión miRNA – mRNA, validando su uso terapéutico.
MiRNA “mimics” y “AntagomiRs”
Distintas patologías se caracterizan por alteraciones en la expresión de miRNA; tanto sobreexpresión como infraexpresión. Es por ello que las terapias basadas en miRNA, buscan principalmente aumentar o reducir los niveles de un miRNA especifico, según sea el caso.
Actuación de las distintas moléculas terapéuticas en la biogénesis de miRNA. Imagen creada con Biorender. [1]
Infraexpresión de miRNA;
Para aumentar los niveles de un miRNA especifico, se utilizan “mimics” de miRNA. Son moléculas sintéticas de RNA que presentan la misma secuencia que el miRNA endógeno, pudiendo restablecer así los niveles y función normal de este.
También se utilizan “Short hairpin RNAs (shRNAs)”; moléculas sintéticas que son procesadas de forma endógena para dar miRNAs maduros. Los shRNAs han mostrado minimizar la toxicidad y permiten sintetizar varios miRNAs a partir de un solo promotor.
En cáncer, la mayoría de miRNAs se encuentran infraexpresados, muchos de estos regulan el ciclo celular y se consideran supresores de tumores, por lo que restablecer sus niveles puede complementar o potenciar la eficacia de otros tratamientos oncológicos.
Sobreexpresión de miRNA;
Para inhibir la función de un miRNA que se encuentra sobreexpresado, se utilizan “AntagomiRs”; moléculas sintéticas que se unen al miRNA endógeno, bloqueándolo y regulando su expresión.
Otra forma de inhibir la expresión de miRNAs, es utilizando sitios de unión competitivos. Estos sitios de unión suelen ser “Antisense oligonucleotides (ASOs)”. Estos ASOs se modifican para aumentar su resistencia a la degradación por nucleasas, su unión a proteínas plasmáticas, su complementariedad con el miRNA diana y su habilidad para activar el sistema inmune innato. Lo que tiene como resultado que los miRNAs tendrán mayor afinidad por estos ASOs sintéticos que por sus ARNm complementarios, viéndose inhibida su función.
Tabla sobre la relación entre miRNA, patologías y posibles tratamientos en fase de estudio clínico. Creada con Biorender. [1]
En la tabla se recogen datos de ensayos clínicos relacionados con el desarrollo de medicamentos basados en terapia mediante miRNA, exponiendo también su relación con algunas enfermedades. A continuación, vamos a exponer el funcionamiento de dos de ellos
Miravirsen: La creación de dicho medicamento se basó en el funcionamiento del miR-122 en la infección por el virus de la hepatitis C (HCV). Este utiliza los miRNAs de las células infectadas durante su ciclo replicativo, siendo capaz de modular su expresión para favorecer la continuidad del virus. MicroRNA-122 (miR-122) es un miRNA específico del hígado, el cual se une a la región 5´UTR del genoma del HCV promoviendo la estabilidad de su RNA, produciéndose también su acumulación. El complejo formado entre el miR-122 y el HCV, protege el genoma del virus de la degradación por exonucleasas celulares, además de evitar una respuesta inmune innata. Al introducir el AntimiR-122, se inhibe el miR-122, evitándose el proceso anteriormente explicado. En los ensayos clínicos se ha visto que los niveles de miR-122 en plasma se reducen en gran medida tras la administración del medicamento.
MRG-106 / Cobomarsen: La sobreexpresión de miR-155 está relacionada con el desarrollo algunos cánceres, debido a su papel en la proliferación y supervivencia celular y la inestabilidad del genoma de células malignizadas. Además, se han demostrado que algunos subtipos de este miRNA están implicados en el linfoma cutáneo de células T. La desregulacion de las vías JAK/STAT, NF-jB y PI3K/AKT tienen un papel muy importante en la patogénesis y progresión del linfoma cutáneo de células T. La activación de esas vías está relacionada con la sobreexpresión del miR-155. Se ha demostrado que el Cobomarsen o MRG-106 reduce la actividad de estas vías, siendo un potencial tratamiento de esta enfermedad.
Conclusión:
Desde el descubrimiento del primer microRNA su estudio se ha expandido considerablemente. Un mejor entendimiento del papel de los miRNA en el desarrollo y la enfermedad, los han convertido en herramientas y dianas atractivas para un enfoque terapéutico innovador. Es por esto que la comunidad científica tiene muchas esperanzas en el futuro desarrollo de tratamientos mediante medicina con miRNAs, ya que es notablemente más eficaz que la realizada con DNA. Sin embargo, la regulación mediada por miRNAs es una red muy compleja, de la cual todavía desconocemos gran parte de su funcionamiento y por lo tanto, aún ignoramos el verdadero alcance de su potencial, por lo que es necesaria una mayor investigación en este campo.
Red de asociaciones entre las diferentes enfermedades relacionadas con la regulación mediante miRNAs. Obtenida de HMDD v3.2.Click en la imagen para visitar el sitio web.
Bibliografía
[1] Bajan, S. and Hutvagner, G. (2020) ‘RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs’, Cells, 9(1), pp. 1–27. doi: 10.3390/cells9010137.
[2] Bartel, D. P. (2009) ‘MicroRNAs: Target Recognition and Regulatory Functions’, Cell, 136(2), pp. 215–233. doi: 10.1016/j.cell.2009.01.002.
[3] Galardi, A., Colletti, M., Di Paolo, V., Vitullo, P., Antonetti, L., Russo, I. and Di Giannatale, A. (2019) ‘Exosomal MiRNAs in pediatric cancers’, International Journal of Molecular Sciences, 20(18). doi: 10.3390/ijms20184600.
[4] Ha, M. and Kim, V. N. (2014) ‘Regulation of microRNA biogenesis’, Nature Reviews Molecular Cell Biology. Nature Publishing Group, 15(8), pp. 509–524. doi: 10.1038/nrm3838.
[5] Monaghan, M., Pandit, A., Bartel, D. P., Lee, R. and Feinbaum, R. (2008) ‘MicroRNAs : Genomics , Biogenesis , Mechanism , and Function Genomics : The miRNA Genes’, Advanced Drug Delivery Reviews, 116(4), pp. 197–208.
[6] Rupaimoole, R. and Slack, F. J. (2017) ‘MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases’, Nature Reviews Drug Discovery. Nature Publishing Group, 16(3), pp. 203–221. doi: 10.1038/nrd.2016.246.
[7] Saliminejad, K., Khorram Khorshid, H. R., Soleymani Fard, S. and Ghaffari, S. H. (2019) ‘An overview of microRNAs: Biology, functions, therapeutics, and analysis methods’, Journal of Cellular Physiology, 234(5), pp. 5451–5465. doi: 10.1002/jcp.27486.
[8] Seto, A. G., Beatty, X., Lynch, J. M., Hermreck, M., Tetzlaff, M., Duvic, M. and Jackson, A. L. (2018) ‘Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma’, British Journal of Haematology, 183(3), pp. 428–444. doi: 10.1111/bjh.15547.
[9] Van Der Ree, M. H., Van Der Meer, A. J., Van Nuenen, A. C., De Bruijne, J., Ottosen, S., Janssen, H. L., Kootstra, N. A. and Reesink, H. W. (2016) ‘Miravirsen dosing in chronic hepatitis C patients results in decreased microRNA-122 levels without affecting other microRNAs in plasma’, Alimentary Pharmacology and Therapeutics, 43(1), pp. 102–113. doi: 10.1111/apt.13432.
[10] Zhang, J., Li, S., Li, L., Li, M., Guo, C., Yao, J. and Mi, S. (2015) ‘Exosome and exosomal microRNA: Trafficking, sorting, and function’, Genomics, Proteomics and Bioinformatics, 13(1), pp. 17–24. doi: 10.1016/j.gpb.2015.02.001.
BRCA1 Y BRCA2: MUTACIONES Y CÁNCER
escrito por GRUPO11 | 10 enero, 2023
Noelia Ares Bóveda, Belén Asenjo Lajusticia e Irene Cañadilla González. Biología Sanitaria. Universidad de Alcalá.
Poco sabía Mary-Claire King en 1994 que su descubrimiento revolucionaría la biología molecular del cáncer: el gen BRCA1 que, junto con el gen BRCA2 descubierto un año después, abrió la puerta a la investigación de diferentes tipos de cáncer y su enfoque en clínica.
Estos dos genes juegan un papel fundamental en el mantenimiento de la estabilidad del genoma. Ante su pérdida de funcionalidad, falta o mutación, pueden conducir a fenómenos de tumorigénesis, además de otras patologías como la anemia de Fanconi.
Sin embargo, no toda mutación implica riesgo de aparición de tumores, y aquí subyace la complejidad de su estudio.
¿Cómo son estos genes y cómo funcionan? ¿En qué tipos de cáncer están implicados? ¿Qué aproximaciones terapéuticas existen a día de hoy?
FUNCIÓN Y ESTRUCTURA
BRCA1/2 (Breast Cancer Genes) son genes supresores de tumores que juegan un papel fundamental en el mantenimiento de la estabilidad genómica. Cuando se producen roturas en la doble hebra de DNA (DSB), ocasionando así una interrupción en la lectura del mismo, entran en juego estos genes: las proteínas que codifican llevan a cabo el proceso de recombinación homóloga (HR), uno de los mecanismos de reparación fundamentales que tienen lugar en la fase S del ciclo celular.
La inactividad de estos genes desencadenaría una recombinación homóloga defectiva que en último término podría desembocar en una predisposición al tumor, especialmente en la mama.
Figura 1: se ilustra el papel supresor de tumores de BRCA1/2 cuando se encuentra activo, mientras que ante su pérdida de funcionalidad se genera una predisposición a la formación de tumores. Created with BioRender.com.
A pesar de tener una función similar, BRCA1 y BRCA2 presentan diferencias en cuanto a su biología molecular, sus interacciones con proteínas y su relación con el cáncer.
BRCA1
El gen BRCA1 se sitúa en el brazo largo del cromosoma 17 (17q21), y fue descubierto e identificado en 1994. Este gen codifica la proteína BRCA1 supresora de tumores (1863 aminoácidos).
En la proteína BRCA1 encontramos tres clusters asociados al cáncer de mama (BCCR: “Breast Cancer Cluster” Region) y un único cluster asociado al cáncer de ovario (OCCR: “Ovarian Cancer Cluster” Region). Mutaciones en estas regiones aumentan la posibilidad del desarrollo de un cáncer en dichos tejidos específicos.
Figura 2: estructura de la proteína BRCA1 (dominios, clusters) y las proteínas con las que interacciona señaladas con un recuadro. Extraída de la referencia [1].
Esta proteína presenta en su extremo N-terminal un dominio RING rico en cisteína con el que interacciona con la proteína BARD1. Así forma un complejo BRCA1-BARD1 que tiene actividad ubiquitina ligasa. En caso de que haya una mutación en el dominio RING de la proteína BRCA1, esta no podrá unirse a BARD1 y por tanto, se perderá la función ubiquitina ligasa, pudiendo suponer una predisposición al desarrollo de cáncer.
En su extremo C-terminal presenta el dominio BRCT, que interacciona con la RNA polimerasa II relacionada con el proceso de transcripción. Además, este dominio interactúa con tres proteínas: Abraxas, BACH1, CtIP; estas uniones permitirán la formación de tres complejos distintos implicados en la reparación de DSBs:
La proteína BRCA1 se asocia a las Abraxas a través de RAP80, y cuando hay un daño en el DNA este complejo interacciona con las histonas dañadas.
El complejo BRCA1-BACH1 participa en la recombinación homóloga. La proteína BACH1 también es conocida como BRIP1 y tiene función helicasa.
La proteína CtIP unida a BRCA1 forma un complejo encargado de la resección de DSBs en pasos tempranos de la reparación.
Figura 3: esquema en el que se ilustran las interacciones de diferentes proteínas con BRCA1 y las funciones de los complejos mencionados. Created with BioRender.com.
BRCA2
El gen BRCA2 se encuentra en el cromosoma 13 (13q12), y fue descubierto e identificado en 1995. Este gen codifica la proteína BRCA2 supresora de tumores, menos conocida pero de mayor tamaño que la codificada por BRCA1 (3418 aminoácidos).
La proteína ácida pequeña DSS1 se une a una región de la proteína BRCA2, formando un complejo proteico que se asociará a las zonas dañadas de DNA.
La proteína BRCA2 presenta unas repeticiones BRC que permitirán la unión directa a RAD51, una enzima de recombinación necesaria en el proceso. El complejo formado por las dos proteínas juega un papel importante en el reconocimiento de DSBs y el inicio de la recombinación.
Los clusters ya mencionados localizados en la proteína BRCA1 también se encuentran en la proteína BRCA2.
Figura 4: esquema de la proteína BRCA2, se pueden observar los cluster (BCCR y OCCR). Extraído de referencia [2].
Además, se ha demostrado que el gen BRCA2 tiene un papel fundamental en la citocinesis, ya que mutaciones en este gen conllevan anomalías cromosómicas.
Figura 5: ilustra la unión de las proteínas RAD51 y DSS1 a la proteína BRCA2, así como las interacciones de sus estructuras moleculares (PDB: 1N0W y PDB: 1IYJ). Created with BioRender.com and Chimera.
CONEXIÓN ENTRE BRCA1 Y BRCA2: PALB2
Pero, ¿de qué manera se conectan las proteínas codificadas por BRCA1 y BRCA2? Esto es posible gracias a la proteína PALB2 (Partner And Localizer of BRCA2). La interacción entre las tres proteínas ocurre de la siguiente forma, tal y como se ilustra en el esquema:
El extremo N-terminal coiled-coil de PALB2 interacciona con el dominio coiled-coil de BRCA1.
El extremo N-terminal de BRCA2 se conecta con el extremo C-terminal de PALB2.
Esto daría lugar a la formación del complejo BRCA1-PALB2-BRCA2 el cual es esencial para la recombinación homóloga, así como la activación y mantenimiento del punto de control G2/M del ciclo celular.
PALB2 es susceptible a sufrir mutaciones que podrían conducir a la pérdida de estabilidad genómica y, en consecuencia, un posible desarrollo de cáncer.
Figura 6: se ilustran las regiones de la proteína PALB2 (Partner And Localizer of BRCA2) con las que interaccionan las proteínas BRCA1 y BRCA2, formando el complejo BRCA1-PALB2-BRCA2. Además, se puede observar la interacción ya mencionada de BRCA1 con la proteína BRIP1 (también conocida como BACH1). Esquema extraído de referencia [5].
RELACIÓN CON p53
Se cree que la proteína producto del gen BRCA1 lleva a cabo su función mediante la formación de grandes complejos. En estos complejos participan diversas proteínas, entre las cuales se encuentra la p53, que juega un papel crucial en el mantenimiento de la integridad genómica por su función supresora de tumores.
En muchos de los tipos de cáncer debidos a mutaciones de los genes BRCA, se ha visto que ha habido interrupciones en la relación entre algunos componentes de estos complejos, provocando que la función de los genes BRCA se vea alterada y por tanto se inhiba la supresión del tumor, dando como resultado la aparición de cáncer.
Figura 7: alteraciones en el complejo proteico en el que intervienen BRCA1/2 y p53 pueden conducir a la formación de un tumor, al inhibirse su supresión. Created with BioRender.com.
Por ejemplo, el complejo BRCA1-BARD1 está implicado en la activación de ciertos puntos de control del ciclo celular. Estas tareas complementarias de BRCA1 en los puntos de control y su rol estabilizador de p53 podrían ayudar en su función de mantenimiento de la estabilidad genómica.
Muchos estudios sugieren que en la formación del tumor existe una estrecha relación entre la pérdida de función de BRCA1/2 y la pérdida de función de la p53.
Se cree que la rotura de la molécula de DNA (debida a esa falta de funcionalidad de BRCA) hace que se activen puntos de control dependientes de p53 y/o apoptosis para evitar la tumorigénesis.
MUTACIONES Y CÁNCER
Cuando consultamos la página web del BRCA Exchange, una iniciativa internacional de la Global Alliance for Genomics and Health, encontramos a mes de febrero de 2021 un total de 40389 variantes de estos genes, clasificadas según su significado en clínica en cuanto a su patogenicidad (una escala de patogénico a benigno o sin significado clínico).
Esta cifra nos ilustra uno de los desafíos y la complejidad que supone el estudio de BRCA1 y BRCA2: no todas las mutaciones implican cáncer.
Las mutaciones en BRCA1/2 implicadas en tumorigénesis generalmente afectan a la mama y al ovario. Pero, ¿a qué se debe el tropismo por estos tejidos?
Ambos tienen gran susceptibilidad a sufrir estimulación hormonal por señales fuertes de crecimiento. La enzima aromatasa está implicada en la síntesis de estrógenos y está regulada negativamente por el gen BRCA1. Además, se ha observado un incremento de los niveles de estrógenos en sangre en personas portadoras de mutaciones en BRCA1, así como de progesterona. Por ello, actualmente se encuentra en estudio la correlación entre niveles hormonales y la predisposición a tumores cuando BRCA1 se encuentra mutado.
Además, en menor medida estas mutaciones están relacionadas con el cáncer de páncreas, entre otros.
CÁNCER DE MAMA
Tal y como nos sugiere su nombre, estos genes están asociados con el desarrollo de cáncer de mama, de manera que el riesgo de padecer este tipo de cáncer a lo largo de la vida cuando se produce una pérdida de funcionalidad de BRCA2 es del 45%, mientras que cuando se trata del gen BRCA1 alcanza el 57%.
Un aspecto a remarcar en el desarrollo del cáncer de mama es la importancia de la predisposición genética: cuando se estudian casos de esta enfermedad debidos a la herencia de una mutación, entre todos los genes de susceptibilidad al cáncer de mama descritos, la mayoría de estas mutaciones se han encontrado en BRCA1 y BRCA2.
A modo de curiosidad, en un estudio se ha observado que la expresión tanto de BRCA1 como de BRCA2 es menor en trabajadores a turnos (incluyen jornadas laborales nocturnas) que en trabajadores en turno de día y, además, cuanto mayor es el número de noches trabajadas por mes, menor es la expresión de los genes. Estos resultados llevaron a algunos autores a proponer que esta podría ser la causa de que la alteración del ritmo circadiano pueda conducir a un incremento del riesgo de desarrollo de cáncer de mama (tal y como han comprobado diferentes estudios), y a plantear que los genes BRCA podrían incluirse dentro de los conocidos como “clock-controlled genes”.
CÁNCER DE OVARIO
El ovario, junto a la mama, es uno de los órganos más afectados por las mutaciones en los genes BRCA. Se piensa que esto puede deberse al estrés oxidativo derivado de los ciclos menstruales, así como al papel de la regulación hormonal (especialmente por estrógenos).
Por ello, la alta predisposición en estos tejidos a sufrir daños en su DNA implica la importancia de los mecanismos de reparación en los que intervienen los genes BRCA. Las mutaciones en el gen BRCA1 suponen un aumento del 11% en el riesgo de sufrir cáncer de ovario y un 40% en el caso de pérdida de funcionalidad del gen BRCA2.
Figura 8: created with BioRender.com.
CÁNCER DE PÁNCREAS
Se trata del tercer tipo de cáncer más común asociado a la mutación de BRCA.
Se caracteriza por tener una alta letalidad, debido potencialmente a que presenta una gran resistencia hacia los tratamientos.
Sólo un pequeño porcentaje de pacientes con cáncer de páncreas presenta mutaciones germinales en BRCA1/2 (aproximadamente el 7%). Por tanto, el cáncer de páncreas causado por mutaciones en BRCA1/2 es poco común; y cabe destacar que presenta una “ventaja” ya que al poseer características biológicas únicas (diferentes a las del cáncer de páncreas común) se pueden crear tratamientos específicos.
Las mutaciones en BRCA1 o en BRCA2 aumentan de manera diferente el riesgo de padecer cáncer de páncreas: si es BRCA2 el afectado, el riesgo de padecer la enfermedad aumenta mucho más que si se trata de mutaciones de BRCA1.
TERAPIAS INNOVADORAS
INHIBIDORES DE PARP (PARPi)
Los inhibidores de PARP (PARPi) se han usado en tratamientos de cáncer en pacientes sensibles a quimioterapia con platino.
PARP (Poli-ADP-ribosa polimerasa) es una enzima con papel fundamental en el reclutamiento del complejo proteico encargado de restaurar los daños del DNA. Las proteínas BRCA, entre otras, forman parte de estos complejos.
Como ya se ha tratado, una mutación en los genes BRCA resultaría en la transcripción de las proteínas BRCA1 y BRCA2 con una pérdida de funcionalidad y por consecuente, una errónea reparación.
Figura 9: participación de PARP en la formación del complejo proteico. Created with BioRender.com.
Este tratamiento basado en el uso de inhibidores de PARP consiste en la “captura” (PARP-trapping) de esta enzima. Esto ocasiona la inhibición de los mecanismos de reparación, ya que en caso de que se produjese una mutación en BRCA1/2, la reparación errónea podría conducir a la formación de tumores. De esta forma, al no actuar los mecanismos de reparación, se activa la apoptosis celular y esas células morirían.
Figura 10: esquema ilustrativo de la función de PARPi. Created with BioRender.com.
TERAPIAS BASADAS EN G-CUADRUPLEXOS
Un enfoque terapéutico de gran interés actualmente para tumores en los que el gen BRCA2 se ve implicado se centra en los G-cuadruplexos, unas estructuras formadas por tres láminas de tétradas de guanina unidas por apareamientos de Hogsteen y estabilizadas por un catión metálico. Podemos encontrar estas estructuras al final de las secuencias teloméricas, interfiriendo con su replicación, y se ha observado que BRCA2, entre otras proteínas, interviene en la replicación de los telómeros (de forma que estos no se acortan).
En trabajos experimentales se ha demostrado que tratamientos de células con pérdida de funcionalidad de BRCA2 con compuestos estabilizadores de G-cuadruplexos disminuye la viabilidad de las mismas, conduciendo de alguna manera a un aumento de la letalidad específica al incrementar la fragilidad de los telómeros. Concretamente, se utilizó la piridostatina (PDS). Otro aspecto que genera interés de este trabajo es que también afecta a aquellas células con pérdida de funcionalidad de BRCA2 que muestran resistencia a los tratamientos con inhibidores de PARP ya mencionados.
Figura 11: en este gráfico se representa un descenso de la viabilidad de las células deficientes en BRCA2 (línea roja) cuando son tratadas con piridostatina (PDS), un estabilizador de G-cuadruplexos. Extraído de los resultados de la referencia [13].
Además, otro compuesto estabilizador de G-cuadruplexos conocido como CX-5461 entró en 2016 en fase I de ensayo clínico para el tratamiento de tumores relacionados con el gen BRCA.
Con BRCA1 y BRCA2 surgió una nueva manera de concebir el estudio del genoma y el cáncer: pocas décadas atrás existía cierto rechazo en el campo de la ciencia a vincular la aparición de tumores a cambios en el material genético; pero esto cambió con este descubrimiento que tuvo gran impacto en la investigación contra el cáncer.
REFERENCIAS
[1] Takaoka, M. and Miki, Y. (2018) ‘BRCA1 gene: function and deficiency’, International Journal of Clinical Oncology. Springer Japan, 23(1), pp. 36–44. doi: 10.1007/s10147-017-1182-2.
[2] Venkitaraman, A. R. (2019) ‘How do mutations affecting the breast cancer genes BRCA1 and BRCA2 cause cancer susceptibility?’, DNA Repair. Elsevier, 81(July), p. 102668. doi: 10.1016/j.dnarep.2019.102668.
[3] Semmler, L., Reiter-Brennan, C. and Klein, A. (2019) ‘BRCA1 and breast cancer: A review of the underlying mechanisms resulting in the tissue-specific tumorigenesis in mutation carriers’, Journal of Breast Cancer, 22(1), pp. 1–14. doi: 10.4048/jbc.2019.22.e6.
[4] Bracci, M., Ciarapica, V., Zabaleta, M. E., Tartaglione, M. F., Pirozzi, S., Giuliani, L., Piva, F., Valentino, M., Ledda, C., Rapisarda, V., Stevens, R. G. and Santarelli, L. (2019) ‘BRCA1 and BRCA2 gene expression: diurnal variability and influence of shift work’, Cancers, 11(8), pp. 1–15. doi: 10.3390/cancers11081146.
[5] Murphy, C. G. and Moynahan, M. E. (2010) ‘BRCA Gene Structure and Function in Tumor Suppression’, The Cancer Journal, 16(1), pp. 39–47. doi: 10.1097/ppo.0b013e3181cf0204.
[6] López-Urrutia, E., Salazar-Rojas, V., Brito-Elías, L., Coca-González, M., Silva-García, J., Sánchez-Marín, D., Campos-Parra, A. D. and Pérez-Plasencia, C. (2019) ‘BRCA mutations: is everything said?’, Breast Cancer Research and Treatment. Springer US, 173(1), pp. 49–54. doi: 10.1007/s10549-018-4986-5.
[7] Roy, R., Chun, J. and Powell, S. N. (2012) ‘BRCA1 and BRCA2: Different roles in a common pathway of genome protection’, Nature Reviews Cancer. Nature Publishing Group, 12(1), pp. 68–78. doi: 10.1038/nrc3181.
[8] Luo, G., Lu, Y., Jin, K., Cheng, H., Guo, M., Liu, Z., Long, J., Liu, C., Ni, Q. and Yu, X. (2015) ‘Pancreatic cancer: BRCA mutation and personalized treatment’, Expert Review of Anticancer Therapy, 15(10), pp. 1223–1231. doi: 10.1586/14737140.2015.1086271.
[9] E. Filippini, S. and Vega, A. (2013) ‘Breast cancer genes: beyond BRCA1 and BRCA2’, Frontiers in Bioscience, 18, pp. 1358–1372.
[10] Venkitaraman, A. R. (2014) ‘Cancer suppression by the chromosome custodians, BRCA1 and BRCA2’, Science, 343(6178), pp. 1470–1475. doi: 10.1126/science.1252230.
[11] Simhadri, S., Vincelli, G., Huo, Y., Misenko, S., Foo, T. K., Ahlskog, J., Sorensen, C., Oakley, G., Ganesan, S., Bunting, S., Xia, B. (2018) ‘PALB2 connects BRCA1 and BRCA2 in the G2/M checkpoint response’. Oncogene. doi:10.1038/s41388-018-0535-2.
[12] Xu, H., Di Antonio, M., McKinney, S., Mathew, V., Ho, B., O’Neil, N. J., Santos, N. Dos, Silvester, J., Wei, V., Garcia, J., Kabeer, F., Lai, D., Soriano, P., Banáth, J., Chiu, D. S., Yap, D., Le, D. D., Ye, F. B., Zhang, A., Thu, K., Soong, J., Lin, S. C., Tsai, A. H. C., Osako, T., Algara, T., Saunders, D. N., Wong, J., Xian, J., Bally, M. B., Brenton, J. D., Brown, G. W., Shah, S. P., Cescon, D., Mak, T. W., Caldas, C., Stirling, P. C., Hieter, P., Balasubramanian, S. and Aparicio, S. (2017) ‘CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours’, Nature Communications, 8(205). doi: 10.1038/ncomms14432.
[13] Zimmer, J., Tacconi, E. M. C., Folio, C., Badie, S., Porru, M., Klare, K., Tumiati, M., Markkanen, E., Halder, S., Ryan, A., Jackson, S. P., Ramadan, K., Kuznetsov, S. G., Biroccio, A., Sale, J. E. and Tarsounas, M. (2016) ‘Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds’, Molecular Cell. The Authors, 61(3), pp. 449–460. doi: 10.1016/j.molcel.2015.12.004.
[14] Rudkin, T. M. and Foulkes, W. D. (2005) ‘BRCA2: Breaks, mistakes and failed separations’, Trends in Molecular Medicine, 11(4), pp. 145–148. doi: 10.1016/j.molmed.2005.02.003.
[15] Mittica, G., Ghisoni, E., Giannone, G., Genta, S., Aglietta, M. and Valabrega,G. (2018) ‘PARP Inhibitors in Ovarian Cancer’, Recent Patents on Anti-Cancer Drug Discovery, 13(4), pp. 392–410. doi: 10.2174/1574892813666180305165256.
[16] Neff, R. T., Senter, L., Salani, R. (2018) ‘BRCA mutation in ovarian cancer: testing, implications and treatment considerations’, Therapeutic Advances in Vaccines, 9(6), pp. 259–261. doi: 10.1177/https.
[17] Venkitaraman, A. R. (2002) ‘Cancer susceptibility and the functions of BRCA1 and BRCA2’, Cell, 108(2), pp. 171–182. doi: 10.1016/S0092-8674(02)00615-3.
Enfermedades mitocondriales
escrito por Grupo6 | 10 enero, 2023
Por Marta García Arauzo y Miriam González Ferrero, Biología sanitaria UAH
Acaba de comenzar febrero, el mes de concienciación de las enfermedades raras. Aunque las enfermedades raras son aquellas con baja prevalencia; se estima que alrededor de un 7% de la población mundial se ve afectada por alguna, lo que resulta ser un gran número de personas. De hecho, cada 30 minutos nace un niño que para los 10 años habrá desarrollado una enfermedad mitocondrial. Las enfermedades mitocondriales son un grupo de trastornos asociados a fallos en el sistema de fosforilación oxidativa. Parte de los componentes de este sistema se codifican en el DNA mitocondrial, y mutaciones en él producirán patologías con síntomas bien definidos.
Cada 30 minutos nace un niño que para los 10 años habrá desarrollado una enfermedad mitocondrial.
Si quieres profundizar sobre las causas y conocer las enfermedades mitocondriales más importantes, ¡sigue leyendo!
Las mitocondrias son orgánulos citoplasmáticos con la función principal de generar energía en forma de ATP, mediante el proceso de fosforilación oxidativa en condiciones aerobias. Esto se realiza gracias a los complejos enzimáticos I-V de la cadena de transporte de electrones y a dos portadores de electrones (coenzima Q y citocromo c).
Figura 1. Cadena respiratoria de la membrana interna mitocondrial.
Otra función de las mitocondrias es contribuir a la apoptosis o muerte celular programada. Además, presentan funciones específicas de tipo celular como son el metabolismo del colesterol, la oxidación β de los ácidos grasos, la biosíntesis de esteroides sexuales y del hemo, y la termogénesis, entre otras.
Estos orgánulos presentan su propio sistema genético con toda la maquinaria necesaria para su expresión, es decir, replica, transcribe y traduce la información genética que contiene. Por ello se piensa que las mitocondrias tienen un origen endosimbiótico, donde una bacteria fue fagocitada por una célula eucariota ancestral.
El ácido desoxirribonucleico mitocondrial (mtDNA) es una molécula bicatenaria, circular, con un peso molecular de 11 000 000 daltons aproximadamente, sin intrones, se replica a partir de un solo origen y es muy pequeño. El genoma mitocondrial del ser humano se encuentra completamente secuenciado, está compuesto por 16 569 pares de bases que corresponden a un pequeño número de genes. Éstos se encuentran distribuidos entre la cadena pesada H (5 168 726 daltons) y la cadena ligera L (5 060 609 daltons).
Figura 3. DNA mitocondrial humano y sus mutaciones asociadas más comunes. [3]
La iniciación de la transcripción puede darse en ambas cadenas y se producirá un RNA precursor policistrónico, que será procesado para originar 13 mRNA individuales y 24 productos individuales de tRNA y rRNA.
El 93% de este ácido desoxirribonucleico comprende región codificante; el resto sería de DNA no codificante, que se piensa que actúa controlando la iniciación de la replicación y de la transcripción.
El DNA mitocondrial tiene información para 37 genes que codifican: 13 proteínas que forman parte de subunidades de 4 de los 5 complejos multienzimáticos del sistema de fosforilación oxidativa (sistema Oxphos), 22 ácidos ribonucleicos de transferencia (tRNAs) capaces de leer todo el código genético y 2 ácidos ribonucleicos ribosomales (rRNAs) componentes de los ribosomas específicos mitocondriales. Estos tRNAs y rRNAs se encargan de traducir las 13 proteínas codificadas por el mtDNA.
Figura 4. Subunidades de la cadena respiratoria codificadas por el mtDNA. [3]
El resto de polipéptidos que componen estos complejos, así como el complejo II completo, están codificados en el DNA nuclear. Por tanto, la biogénesis del sistema Oxphos requiere una regulación y expresión coordinada entre los genes del mtDNA y el DNA del núcleo celular.
Las subunidades que están codificadas en el DNA nuclear se sintetizan en el citosol, y deben ser transportadas a la mitocondria. Para ello, es imprescindible la proteína de choque térmico chaperonina, que despliega estas subunidades proteicas para que puedan atravesar la doble membrana mitocondrial en forma de largas cadenas. En la matriz mitocondrial, estas subunidades se vuelven a plegar para poder ensamblarse con los demás componentes que forman la cadena de transporte electrónico.
La genética del ácido desoxirribonucleico mitocondrial es muy peculiar, y presenta 5 aspectos fundamentales: poliplasmia, segregación mitótica, efecto umbral, herencia materna y una alta velocidad de mutación.
POLIPLASMIA
Cada mitocondria contiene entre 2 y 10 moléculas de mtDNA, y cada célula contiene múltiples mitocondrias. Por lo que hay un alto número de copias de mtDNA en cada célula (1 000 – 10 000), dependiendo del tejido. Al conjunto de mitocondrias de una célula se le denomina condrioma.
Normalmente, todas las mitocondrias poseen el mismo genoma, lo que se conoce como homoplasmia. Sin embargo, puede producirse una mutación en el mtDNA que afecte solo a algunas mitocondrias, así en una misma célula se encuentran mitocondrias con mtDNA mutado y mitocondrias con mtDNA normal; a esto se le denomina heteroplasmia.
También pueden existir células que en todas sus mitocondrias contengan mtDNA mutado, y la célula se encuentra en homoplasmia pero con mtDNA alterado.
SEGREGACIÓN MITÓTICA
El fenotipo de una línea celular puede variar durante la división celular, ya que las mitocondrias se distribuyen al azar entre las células hijas. Por tanto, si en una célula coexisten mtDNA normal y mtDNA mutado (heteroplasmia), tras varias divisiones celulares, se distribuyen distintas proporciones de genoma mitocondrial mutado y nativo en las células hijas. Así, se originarán tres genotipos diferentes: homoplasmia de mtDNA normal, heteroplasmia y homoplasmia de mtDNA mutado.
El fenotipo de una célula heteroplásmica depende del porcentaje de mtDNA mutado que presente. Si el número de moléculas de mtDNA alterado es relativamente bajo, se producirá una complementación con las moléculas de mtDNA normal y no habrá manifestación del defecto genético.
Cuando el mtDNA mutado sobrepasa un umbral determinado, sí se manifestará un fenotipo patógeno. De este modo, la producción de ATP está por debajo de los mínimos necesarios para el funcionamiento normal de los tejidos, ya que las proteínas codificadas en el mtDNA serán defectuosas, y esto origina una enfermedad en la persona.
El número de moléculas de mtDNA, es decir, el umbral, será distinto en cada órgano y tejido. Esto dependerá de la cantidad de energía requerida para su funcionamiento. Por ello, los tejidos que con mayor frecuencia se ven afectados son la visión, el sistema nervioso central, el corazón, el músculo esquelético, los islotes pancreáticos, el riñón y el hígado.
HERENCIA MATERNA
El mtDNA se hereda con un patrón vertical, no mendeliano, exclusivamente por vía materna. La madre transmite su genoma mitocondrial a todos sus hijos, pero solamente las hijas lo transmitirán a todos los miembros en la siguiente generación, y así sucesivamente.
Figura 6. Herencia materna de las enfermedades asociadas al mtDNA. Las mujeres afectadas corresponden a los círculos coloreados y los varones afectados corresponden a los cuadrados coloreados. [16]
Esto es debido al gran número de moléculas de mtDNA que contienen los óvulos (entre 100 000 y 200 000 copias), en comparación con unos pocos cientos que contienen los espermatozoides. Además, las mitocondrias que pueden entrar en el óvulo fecundado se eliminan por un proceso complejo en el que participa el sistema proteasoma ubiquitina.
En conclusión, las enfermedades vinculadas a mutaciones en el mtDNA son heredadas de manera exclusiva por línea materna.
ALTA VELOCIDAD DE MUTACIÓN
A diferencia de la recombinación de pares homólogos que tiene lugar en el núcleo, las moléculas de mtDNA no sufren recombinación, por lo que las mutaciones representan la única posibilidad de diversificación genética del mtDNA.
La tasa de mutación espontánea del mtDNA es de unas 10 a 20 veces mayor que en el DNA nuclear. Esto puede deberse a que en la mitocondria se producen continuamente especies reactivas de oxígeno (ROS), como consecuencia de la oxidación final de los compuestos carbonados. Por lo que el mtDNA es particularmente susceptible al daño oxidativo debido a este íntimo contacto con los radicales libres. Éstos dañarán este DNA que no está protegido por histonas y, además, presenta muy pocos mecanismos de reparación.
Algunas de las variaciones de la secuencia serán polimorfismos silentes que no tienen el potencial de un efecto patógeno, mientras que otras variaciones podrán ser mutaciones patógenas.
Las mitocondrias disfuncionales pueden ser eliminadas por digestión lisosomal o, también, las células que posean mitocondrias disfuncionales pueden sufrir apoptosis.
Figura 7. Múltiples vías de lesión del mtDNA que producen pérdida de función celular, apoptosis y envejecimiento. [16]
En relación al envejecimiento, la acumulación de las mutaciones en el mtDNA en células somáticas puede contribuir a enfermedades relacionadas con la edad, como el síndrome metabólico y diabetes, cáncer, trastornos cardiovasculares y degenerativos del sistema nervioso central (Alzheimer y Parkinson). Esto es debido a que estas mutaciones contribuyen a la declinación de la fosforilación oxidativa, y así disminución de la capacidad respiratoria de los tejidos. Por tanto, las mitocondrias se deterioran a lo largo de la vida, como consecuencia de la acumulación de mutaciones.
A su vez, la acumulación de mutaciones en el mtDNA producirá un sistema Oxphos ineficaz, con la posibilidad de generación excesiva de ROS, lo que hace que se cree un “círculo vicioso” de daño acumulativo en el genoma mitocondrial.
No obstante, dichas mutaciones somáticas del mtDNA no son transmitidas a la descendencia, por lo que el efecto hereditario de la mutagénesis del mtDNA requiere considerar aparte los acontecimientos en la línea germinal femenina.
Otras utilidades del genoma mitocondrial
Las variaciones en la secuencia de mtDNA entre diferentes individuos han resultado muy útiles para obtener un mayor conocimiento en otras áreas no relacionadas directamente con la clínica, como pueden ser estudios antropológicos, etnológicos y forenses.
En medicina forense, la genética mitocondrial se aplica para identificar personas a partir de restos humanos con abundante mtDNA, como cabello, huesos o dientes.
En el ámbito antropológico, el análisis comparativo del mtDNA entre distintos grupos étnicos del mundo permite establecer la manera en la que se desarrollaron las migraciones de grupos humanos a lo largo del planeta.
ENFERMEDADES DEL DNA MITOCONDRIAL
El término enfermedad mitocondrial agrupa a todas las afecciones relacionadas con alteraciones en el metabolismo oxidativo mitocondrial. El denominador común de estas patologías es la deficiencia en la biosíntesis de ATP, moneda energética de nuestro organismo.
El origen de estas enfermedades reside en mutaciones del mtDNA (mutaciones puntuales, deleciones o duplicaciones), y en alteraciones de la señalización genómica entre el DNA nuclear y el mitocondrial. Si la disfunción mitocondrial ocurre en la cadena respiratoria, estaremos hablando de citopatías mitocondriales.
Pinchando aquí puedes ver las enfermedades producidas por las mutaciones en los distintos genes relacionados con la cadena mitocondrial. Facilitado por la AEPMI (asociación de enfermos de patología mitocondrial).
Los déficits enzimáticos de la cadena respiratoria pueden ser únicos (cuando afectan a un solo complejo de la cadena respiratoria), o múltiples (afectan a varios). Las mutaciones del mtDNA suelen ser causadas por déficits múltiples, afectando a varios tejidos y causando la heterogeneidad de cuadros clínicos. De hecho; el mal funcionamiento de tres o más sistemas es característica principal del diagnóstico de las enfermedades mitocondriales.
Desde que en la década de 1960 se observaron por primera vez acúmulos musculares de mitocondrias en individuos con intolerancia al ejercicio, posteriormente se han ido relacionando las disfunciones mitocondriales con numerosas enfermedades: trastornos neurodegenerativos, cardíacos, enfermedades neurometabólicas, cáncer, obesidad, etc. Al limitarse el nivel de ATP, suelen verse más afectados los tejidos con alta necesidad energética, como cerebro, músculo, hígado, corazón, riñones y SNC. Entre las manifestaciones clínicas más frecuentes se encuentran la sordera, ceguera, diabetes, ataxia, debilidad muscular, disfunciones hepáticas y pancreáticas y fallos en el corazón, riñones e hígado.
Esta gran variedad de manifestaciones clínicas supone un reto para el diagnóstico de enfermedad mitocondrial. Cuando en un paciente se presentan uno o más de estos síntomas, la sospecha clínica debe ser ratificada por pruebas metabólicas específicas de disfunción mitocondrial, y finalmente, tras realizar pruebas de confirmación morfológicas, histoenzimáticas, bioquímicas o genéticas, sus resultados nos pueden conducir al diagnóstico de disfunción mitocondrial. En la siguiente imagen puedes consultar el protocolo a seguir desde la sospecha médica.
Figura 8. Tomada del artículo de referencia [7].
Existen síndromes clínicos específicos, bien definidos, de los que vamos a introducir los más representativos.
CPEO: Oftalmoplejía externa progresiva crónica
El signo principal de este síndrome es la blefaroptosis, también llamada ptosis palpebral o párpado caído, junto con paresia (debilidad) simétrica de los músculos extraoculares. También suele acompañarse de miopatía proximal de las extremidades, intolerancia al ejercicio y debilidad muscular.
Figura 9. Paciente CPEO en posición de mirada hacia arriba. Es visible la ptosis palpebral. Imagen tomada del artículo de referencia [10].
El CPEO está causado por deleciones grandes y normalmente espontáneas del mtDNA. Se ha observado que muchas de las deleciones encontradas están delimitadas por repeticiones directas de 3 a 13 nucleótidos. Esto podría significar que la presencia de estas repeticiones conlleva a errores en la replicación, que serían los responsables de la deleción.
Algunos casos también ocurren por mutaciones puntuales, como la del gen del tRNA LeuUUR en A3243 (la A proviene de la base adenina y el número representa la posición del nucleótido 3,243). En un ínfimo porcentaje de individuos se ve afectada la región del DNA mitocondrial con genes que codifican para rRNA.
Este síndrome aparece antes de los primeros 20 años de vida.
Síndrome de Kearns-Sayre
Es una enfermedad variante de la CPEO más grave y peculiar que también aparece antes de los 20 años de edad. Además de oftalmoplejía progresiva crónica y retinitis pigmentaria atípica se suma uno de los siguientes problemas: bloqueo cardíaco, síndrome cerebeloso (alteración funcional del cerebelo) e hiperproteinorraquia, que consiste en una cantidad de proteína superior a 100 mg/L en LCR. Al conjunto de estos tres problemas multisistémicos se denomina la tríada clásica.
La siguiente imagen es útil para explicar la retinopatía pigmentaria:
Figura 10. En la zona superior se presenta una imagen captada por una persona con visión no afectada y el mismo paisaje visto por una persona con retinosis pigmentaria. Debajo vemos dos cortes transversales del ojo, correspondientes a cada uno de los casos. En el caso del ojo afectado aparecen manchas más oscuras en todo el globo ocular, identificadas como agregados de pigmentos de la retina. Imagen y pie de foto tomados del artículo de referencia [11].
MERRF o síndrome de epilepsia mioclónica con fibras rojo-rasgadas
Las manifestaciones clínicas de este síndrome son epilepsia mioclónica o mioclonías, convulsiones generalizadas y miopatía con presencia de fibras rojo-rasgadas.
Con estos síntomas, realizar un electroencefalograma es muy interesante para el diagnóstico, pues registra las mioclonías y puede determinar su origen.
Las mioclonías son movimientos musculares involuntarios originados por una actividad anormal de la corteza sensomotora. Las fibras rojas forman unidades motoras tónicas, de contracción lenta. Son resistentes a la fatiga y de ellas dependerá el correcto mantenimiento del equilibrio y la postura y los movimientos de poca intensidad. Por eso, la disfuncionalidad de las fibras rojas conlleva errores en este tipo de ejercicios.
Figura 11. Cortes histológicos de músculo deltoides izquierdo teñidos con técnica tricrómica de Gomori, donde se observa citoplasma y fibras musculares teñidas de rojo y colágeno color azulado, así como la presencia de atrofia de fibras musculares y de las típicas fibras rasgadas propias de la enfermedad mitocondrial. Imagen y pie de foto tomados del artículo de referencia [12].
En algunos pacientes aparecen síntomas clínicos adicionales como demencia, sordera neurosensorial, neuropatía sensitiva, atrofia óptica, fallo respiratorio y cardiomiopatía.
Es un síndrome de curso progresivo que aparece tanto en la infancia como en edad adulta. Como es de herencia materna, podemos estudiarlo mediante pedigríes.
MERRF se asocia principalmente a mutaciones puntuales del gen del tRNA Lys en la posición A8344G (transición de adenina por guanina en la posición 8,344) o, de forma minoritaria, en T8356C. La mutación A8344G altera específicamente el bucle TᴪC del tRNA Lys, lo cual reduce la síntesis proteica celular. Todas las mutaciones son en forma heteroplásmica. Los jóvenes con un 95% de heteroplasmia pueden desarrollar este síndrome, mientras que los adultos de entre 60-70 años o más podrán hacerlo con solo un 60% del DNA mutado. La razón de que haya personas que nacen con fenotipo normal y desarrollan MERRF en la edad adulta puede ser que, con la edad, la capacidad oxidativa de la mitocondria se reduce, promoviendo la aparición de la enfermedad.
Figura 12. En este gráfico se observa cómo la gravedad de las manifestaciones clínicas de MERRF es directamente proporcional a la capacidad oxidativa de la mitocondria. También puedes ver en el pedigrí la herencia materna del síndrome. Extraído del artículo de referencia [8].
MELAS: Síndrome de encefalomiopatía mitocondrial con acidosis láctica y episodios de accidentes cerebro-vasculares
Con solo leer el nombre, ya nos hacemos una idea de los síntomas de la enfremedad. Básicamente, el acrónimo MELAS nos explica que este síndrome consiste en encefalomiopatía mitocondrial, acidosis láctica e ictus cerebrales semejantes a apoplejías. Estos ictus son reversibles y pueden ser visualizados a través de resonancia magnética o PET. Los recién nacidos con este síndrome son sanos, sin embargo, durante los primeros años de vida dejan de crecer súbitamente y se empiezan a presentar los síntomas comentados. Estas manifestaciones suelen acompañarse de convulsiones generalizadas, dolor de cabeza, sordera, demencia y, a veces, de fibras rojo-rasgadas.
Figura 13. La fila superior muestra la imagen por resonancia magnética de un paciente de MELAS, con alta actividad en corteza temporal y horizontal y en sustancia blanca. La fila inferior muestra la presencia de fibras rojo-rasgadas en diafragma (izquierda) y corazón (derecha) teñidos con tinción de Gomori. Imagen tomada del artículo de referencia [14].
MELAS, al igual que MERRF, se hereda por vía materna. Esta enfermedad ha sido asociada fundamentalmente con mutaciones en el gen del tRNA LeuUUR del mtDNA. Alrededor del 80% de los casos son causados por una transición en A3243G, pero se han encontrado otras con menor incidencia, como A3271G, también en el tRNA LeuUUR y alguna en genes codificantes de proteínas, todas en forma heteroplásmica.
Si lo recuerdas, la transición A3243G también la hemos visto en CPEO (oftalmología progresiva externa crónica), y además también aparecen sordera, miopatía o diabetes; es decir, esta mutación tiene efectos fenotípicos muy variables.
Al ser dañado el tRNA, la síntesis de proteínas mitocondriales se verá comprometida.
LHON: Neuropatía óptica de Leber
Se caracteriza por la pérdida de la visión bilateral, indolora, aguda o subaguda, debido a la atrofia del nervio óptico. Aparece en la segunda o tercera década de la vida de hombres y mujeres, más frecuentemente en los primeros. A parte de la afectación de la visión, también puede aparecer neuropatía retrobulbar y periférica, síndrome piramidal y cerebeloso, el fondo de ojo con edema, tortuosidad de los vasos retinianos e incluso trastornos en la conducción cardiaca.
Figura 14. Estas dos fotografías tomadas de la fuente [9] de la bibliografía, muestran el fondo de ojo al inicio de la enfermedad (izquierda) y diez meses más tarde (derecha). En este periodo se puede apreciar cómo el nervio óptico se ha ido atrofiando.
El síndrome de Lohn fue la primera enfermedad humana de herencia materna que fue asociada con una mutación puntual del mtDNA. Estas mutaciones se pueden encontrar de forma homo y heteroplásmica y se han descrito en gran número. Esto es relevante pues las manifestaciones clínicas dependen de la mutación existente. Las tres mutaciones primarias o patogénicas verdaderas están en genes que codifican para proteínas constituyentes del complejo I de la cadena respiratoria. Son G3460A, T14484C y G11778A, esta última asociada al 50% de los casos y responsable de la forma más severa de la enfermedad.
MILS: Síndrome de Leigh de herencia materna
Consiste en encefalomielopatía necrosante subaguda; es decir, lesiones necróticas cerebrales en tálamo, ganglios basales, tronco cerebral y médula espinal. La esperanza de vida es muy limitada y suele aparecer durante el primer año de vida.
Se puede acompañar de alteraciones respiratorias y de la deglución (por estar afectado el tronco cerebral), además de parálisis oculomotoras, atrofia óptica, leucodistrofia, movimientos involuntarios o hiperproteinorraquia (disminuyendo así la conducción nerviosa). Además, las habilidades psicomotoras adquiridas tienden a empeorar.
El síndrome de Leigh está causado por la transversión de T por G en el gen de la subunidad 6 de la ATPasa, T8993G.
Figura 15. Necrosis del encéfalo. Imagen tomada del artículo de referencia [13].
Hasta aquí hemos comentado las enfermedades mitocondriales más representativas, pero cabe destacar que, en numerosas ocasiones, no es posible catalogar dentro de los síndromes específicos a todos los pacientes diagnosticados con enfermedad mitocondrial. Esto se debe fundamentalmente a la enorme diversidad en las manifestaciones clínicas que cada paciente desarrolla. Además, el fenotipo clínico de un paciente mito evoluciona a lo largo del tiempo, desarrollando otros síntomas y signos diferentes a los que presentaba cuando se realizó el diagnóstico.
En cuanto al tratamiento, no existen medidas curativas que reviertan la disfunción mitocondrial, sino que se seguirá un tratamiento farmacológico y nutricional destinado a enlentecer el proceso de degeneración natural. También es importante la terapia psicológica y de mantenimiento para lograr la mayor calidad de vida de los pacientes.
Como hemos visto, casi la totalidad de estas enfermedades se manifiestan en las primeras dos décadas de vida. Por esto, es importante que, especialmente los niños, se enfrenten de manera positiva y esperanzadora al diagnóstico recibido. Una herramienta útil para ellos puede ser el siguiente cuento redactado por la Obra Social del Hospital Sant Joan de Déu, que explica la enfermedad de una forma amena y lúdica.
Es muy importante que todos los enfermos de estas patologías se sientan acompañados y comprendidos. Para ello, resulta esencial la concienciación poblacional de estas enfermedades, de cara no solo a conseguir financiación para seguir investigando, sino también para acercar el conocimiento de las mismas a todas las personas, aunque no les afecte directamente. Existen varias asociaciones que ayudan a conseguir esto, como la Asociación de Enfermos de Patologías Mitocondriales (AEPMI).
Además, la página web de una fundación australiana contiene numerosos testimonios de personas afectadas por enfermedades mitocondriales, lo cual es otra forma muy útil de que un mayor número de personas conozcan estas patologías.
También se realizan numerosas campañas como carreras solidarias #RunForMito, recogida de tapones o dispositivos tecnológicos en desuso, venta de productos solidarios, o campañas de iluminación como #LightUpForMito, que se realizó con razón de la Semana mundial de la concienciación de enfermedades mitocondriales, celebrada en septiembre, y a la que se sumaron numerosas asociaciones y ayuntamientos, consiguiendo promover la sensibilización muchas personas y dando voz a todas las familias afectadas.
Bibliografía
Solano, A., Playán, A., López-Pérez, M. J. and Montoya, J. (2001) ‘Enfermedades genéticas del ADN mitocondrial humano’, Salud Pública de México, 43(2), pp. 151–161. doi: 10.1590/s0036-36342001000200010.
Dra. Aglais Arredondo Falagán, I Lic. Gleymis Venet Cadet,I Dra. Dra. Olivia Román Guerra I y MsC. Eglis Yanet Ramírez Delgado. (2012) Bases moleculares de las enfermedades mitocondriales, Medisan, 16(5), pp. 795–805.
Barrera-Ramírez, C. F., Barragán-Campos, H. M., Sánchez-Guerrero, J., García-Ramos, G., Vega-Boada, F. and Estañol, B. (1999) ‘El otro genoma: El concepto clínico de las citopatías mitocondriales o enfermedades de la fosforilación oxidativa’, Revista de Investigación Clínica, 51(2), pp. 121–134.
Boveris, A., Costa, L. and Junqueira, V. (2000) ‘Envejecimiento mitocondrial. La teoría del envejecimiento mitocondrial por la acción continua de los radicales libres del oxígeno y del nitrógeno’, Ciencia e Investigación, pp. 13–23. Available at: http://revistasinvestigacion.unmsm.edu.pe/index.php/farma/article/view/4393/4457.
Jesus, E. P., Carmen, G. L., Oscar, B. B. and Manuel, C. G. (2014) ‘Protocolos Diagnóstico Terapeúticos de la AEP: Neurología Pediátrica’, Revista de Neurología, 58(6), p. 288. doi: 10.33588/rn.5806.2013478.
Feillet, F., Schmitt, E., Gherardi, R. and Bonnemains, C. (2014) ‘Enfermedades mitocondriales’, EMC-Pediatría, 49(2), pp. 1–12. doi: 10.1016/s1245-1789(14)67271-1.
Campos, Y., Pineda, M., García Silva MT., Montoya J., Antoni L. Andreu (2014) ‘Enfermedades mitocondriales’, Revista de Neurología, 58(06), p. 288. doi: 10.33588/rn.5806.2013478.
Montoya, J. (2000) ‘Diseases of the mitochondrial DNA’, Revista de Neurología, 31(4), pp. 324–333. doi: 10.33588/rn.3104.2000236.
Calderón-Garcidueñas, A. L., Perez-Loria, O., Alberto-Sagastegui, J. and Farias- Garcia, R. (2000) ‘Oftalmoplejia progresiva externa secundaria a miopatía mitocondrial. Presentación de un caso y revisión de la literatura’, Gaceta Médica de México, 136(3), pp. 267–271.
Delgado Pelayo Sari (2007) Retinosis Pigmentaria. Preguntas y Respuestas, Revista Médica MD.
Miranda Nava, G. and Ortega Ponce Fabiola, E. E. (2010) ‘Epilepsia mioclónica con fibras rojas rasgadas’, Revista Mexicana de Neurociencia, 11(3), pp. 243–245.
Gutiérrez, A., Saldaña Martínez, A., García Ramírez, R., Rayo Mares, D., Carreras, M., López Pérez, M. J., Montiel Sosa, J. F. (2009). Síndrome de Leigh causado por la mutación G14459A del ADN mitocondrial en una familia mexicana. Revista de neurología.(Ed. impr.), pp. 248-250.
Conforto, A. B., Yamamoto, F. I., Oba-Shinjo, S. M., Pinto, J. G. C., Hoshino, M., Scaff, M. and Marie, S. K. N. (2007) ‘Screening for melas mutations in young patients with stroke of undetermined origin’, Arquivos de Neuro-Psiquiatria, 65(2 B), pp. 371–376. doi: 10.1590/S0004-282X2007000300001.
Donaire de la Torre, C. (2015). Enfermedades mitocondriales: una forma aplicada de enseñar genética. Trabajo Fin de Máster. Universidad de Jaén.
El suicidio del actor Robin Williams en 2014 conmocionó al mundo del cine y a todos los que nos gustaba como actor y disfrutamos muchas de sus películas. A mi me gustó especialmente en El Club de los Poetas Muertos, El Indomable Will Hunting y Despertares, donde dió vida al neurólogo, gran amante de la Química y divulgador de la Ciencia que fué Oliver Sacks.
Robin Williams, encarnando a Oliver Sacks, hablando sobre la Tabla Periódica a Robert de Niro en Despertares
Algún tiempo después, su familia reveló que Robin Williams padecía un tipo de demencia, asociada a la enfermedad de Parkinson, llamada demencia con cuerpos de Lewy. Según cuentan, esta enfermedad degenerativa pudo estar detras del suicidio de Williams. Es muy complejo hablar sobre la demencia con cuerpos de Lewy en una entrada de blog, por lo que solo voy a dar unas pinceladas, centrándome en un aspecto fundamental: la importancia de la estructura de las proteínas.
Fijate en ésta imagen:
La gran mancha redonda oscura es un cuerpo de Lewy incluido en una célula de una parte del cerebro de un paciente con demencia con cuerpos de Lewy. Estos cuerpos son masas de proteínas agregadas. Estas masas de proteínas forman en primer lugar unas estructuras que se denominan agresomas (de agresión). La formación de éstos cuerpos es una consecuencia de las propiedades de las proteínas y aquí entramos en el meollo de la cuestión: las proteínas, para realizar su función tienen que plegarse dando lugar a unas estructuras tridimensionales características. Estas estructuras tridimensionales son las que dan sentido y funcionamiento a las proteínas. Lo malo es que muchas proteínas tienden, de modo natural, a plegarse «mal», siguiendo su tendencia natural. Al fin y al cabo una proteína es un tipo de poliamida, como el nylon de las fibras de tejidos sintéticos. Y, como esas fibras, las proteínas tienden a formar fibras insolubles y resistentes. Cuando una proteína en la célula se pliega mal, la célula la marca como defectuosa, para eliminarla antes de que pueda provocar daños. Este marcaje se hace con una pequeña proteína llamada ubiquitina. Las proteínas marcadas se dirigen hacia un sistema de eliminación llamado proteasoma.
Una vista simplificada del sistema de eliminación de proteínas defectuosas. Las proteínas mal dobladas pueden formar agregados insolubles, que se almacenan en el IPOD, o bien se marcan (ubiquitina) y se ponen en «cuarentena» para su revisión o eliminación en el JUNQ. Si las proteinas marcadas se agregan, se «tiran al punto limpio», llamado agresoma. Cuando las células tienen defectos para procesar el «punto limpio» agresoma, éste crece y se convierte en un cuerpo de Lewy, produciéndose la enfermedad. Imagen tomada de M Takalo, A Salminen, H Soininen, M Hiltunen, A Haapasalo, Am J Neurodegener Dis 2013;2(1):1-14.
Cuando las proteínas marcadas como defectuosas se agregan, la célula las amontona en una especie de contenedor, o «punto limpio», que es el agresoma, para evitar que provoquen daños en la célula. La célula entonces elimina la basura de éste punto limpio en un proceso llamado autofagocitosis.
Si falla el proceso de limpieza del «punto limpio» por un defecto, la basura se acumula, forma una masa intratable y el agresoma se convierte en cuerpos de Lewy.
Como vengo diciendo, todo este proceso depende de cómo se doblen las proteínas y de la costumbre natural de las proteínas para formar agregados insolubles de fibras (como las poliamidas que se usan en tejidos). En el caso de la enfermedad que padecía Robin Williams, una de las proteínas responsables de la formación de los cuerpos de Lewy y de la demencia es la alfa-sinucleína. En condiciones normales, la alfa-sinucleína es una pequeña proteína esencial para el desarrollo del cerebro, ya que tiene una función fundamental en algunos procesos celulares de las neuronas. En condiciones patológicas, la alfa-sinucleína es una de las responsables de la neurodegeneración y la demencia asociada a la enfermedad de Parkinson y las demencias por cuerpos de Lewy. Y todo se debe a la física de la estructura de la proteína y su tendencia a plegarse mal. Voy a visualizarla para enseñartela, con ayuda del software Chimera y los datos del Protein Data Bank.
alfa-sinucleína normal:
Es una proteína sencilla, formada por una cadena en la que se ve, en rojo, una de las formas estructurales fundamentales que forman las proteínas: la alfa-hélice. Dos alfa-hélices se conectan por una cadena simple (un motivo muy básico en las proteíans que se llama hélice-giro-hélice) y queda otra larga cadena simple. En ésta forma, la alfa-sinucleína puede hacer su función fisiológica y es soluble (las alfa-hélices dan lugar a proteínas solubles).
Pero cuando la alfa-sinucleina se pliega mal, algo a lo que esta proteína tiene tendencia debido a su gran simplicidad, tiende a formar otro motivo estructural diferente: la lámina beta. La lámina beta forma estructuras agregadas e insolubles y está favorecida termodinámicamente en estas cadenas cortas de proteína. Las cadenas cortas y simples de proteínas tienden de modo natural a formar fibrillas insolubles. Es decir, el esfuerzo energético para formar la sinucleína funcional de arriba es mucho mayor y, si se la deja a su aire, el fallo en el plegamiento conduce a la:
alfa-sinucleína agregada:
Este es un agregado o fibrilla de alfa-sinucleína. En morado están las láminas beta, que se organizan paralelamente unas con otras, precipitando una masa de proteínas. Forma como «pelusillas» que se van acumulando, agregándose unas a otras, ya que las láminas beta son «pegajosas» y tienden a pegarse unas a otras. La célula marca estas «pelusillas» y las guarda en el agresoma para eliminarlas. Si la célula tiene un defecto para limpiar el agresoma, es cuando se produce la enfermedad.
Cuerpos de Lewy en células de pacientes. B: imagen por microscopía electrónica de un cuerpo de Lewy en el que se observan las masas de «pelusa» de las proteínas agregadas. Mirad la imagen de la estructura anterior e imaginad cómo esas masas se van acumulando hasta formar la «pelusa» de la imagen. La imagen está tomada de éste artículo de Lancet.
¿cómo provocan los cuerpos de Lewy los problemas neurodegenerativos y la demencia?. Pues parece ser que la masa de proteína precipitada interfiere con las funciones celulares, acumula iones de hierro que provocan un fuerte estrés oxidativo que termina matando la célula, dando lugar a la pérdida de las funciones de la zona afectada (motoras, cognitivas) y dando lugar a los síntomas típicos de estas enfermedades.
Al final la demencia por cuerpos de Lewy es una consecuencia natural de la propia Física de las proteínas: la termodinámica nos dice que las cadenas de proteínas, en especial las pequeñas, tienden a formar fibras de láminas beta (como la seda o el nylon). Las células invierten mucha energía y complejos mecanismos para plegar correctamente las proteínas y, cuando el plegamiento falla (por razones termodinámicas o por mutaciones en la cadena que alteran la estructura funcional) hay todo un sistema de control de calidad y eliminación. La enfermedad es el resultado de una cadena de fallos en todo este complejo sistema de construcción, control de calidad y limpieza de las proteínas. Y es el resultado de las propiedades naturales de las cadenas de proteínas, por eso es tan difícil de prevenir y tratar. Resulta extraño pensar que Robin Williams murió por culpa de una pequeña proteína que simplemente se plegó siguiendo su tendencia natural. Si ésto no te hace sentir frágil e indefenso, sometido a las leyes de la Naturaleza, no se que mas podría hacerlo.
Esto nos lleva a otra de las grandes preguntas de la Ciencia: si las cadenas de proteína tienden naturalmente a formar «pelusas» de fibrillas, ¿como se originó, al principio de la evolución, el sistema de estructuras que dió lugar a las proteínas funcionales?