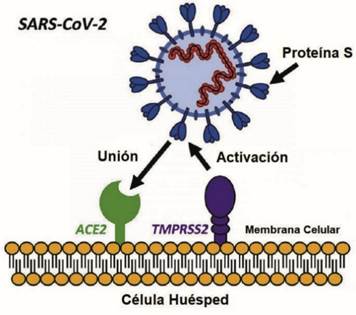
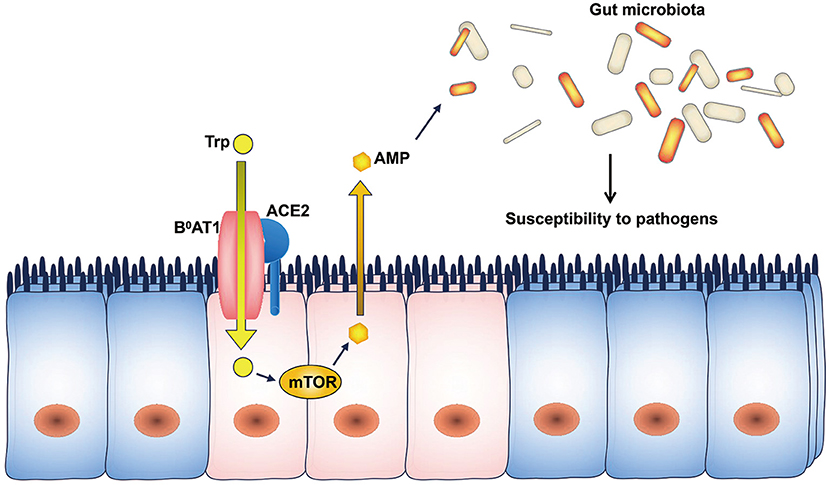
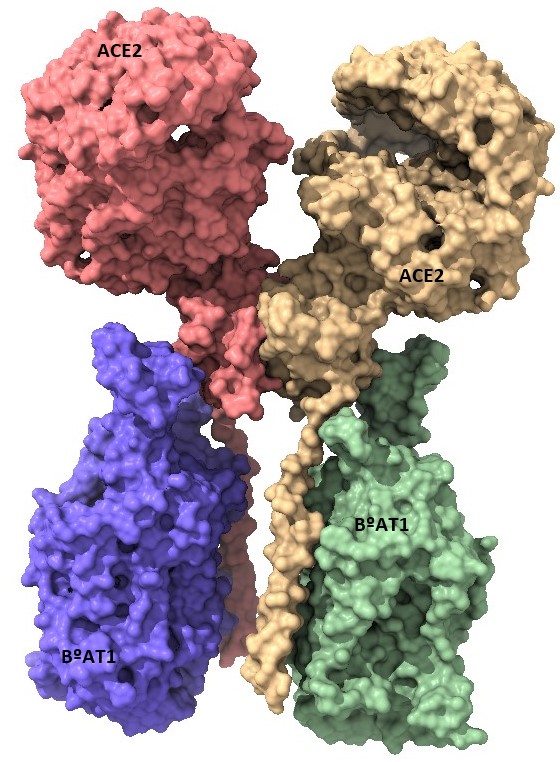
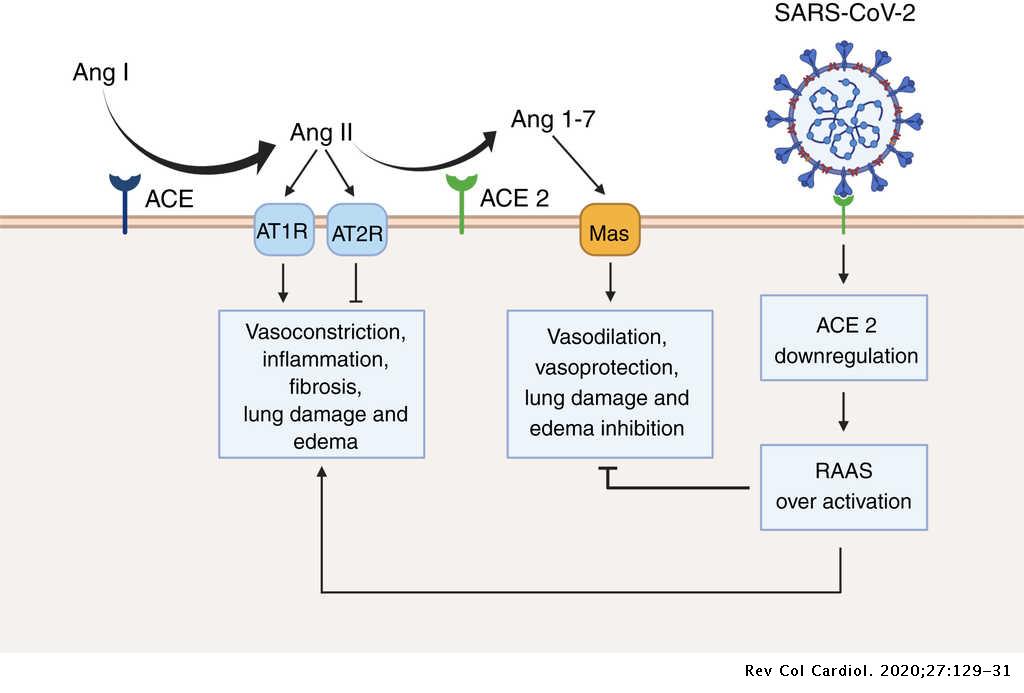
Por Rafael López Rodríguez y Elena Mozos Ruiz. Biología sanitaria, UAH
INTRODUCCIÓN
El ántrax es una enfermedad bacteriana causada por la Bacillus anthracis que afecta principalmente a los herbívoros, siendo humanos y carnívoros huéspedes accidentales. La bacteria produce tres proteínas, el antígeno protector (PA), el factor letal (LF) y el factor de edema (EF) Aunque rara vez aparece en humanos, sí que se dan casos de ántrax en algunos países no industrializados y últimamente se ha convertido en un peligro ya que puede ser utilizado como arma biológica.
ESTRUCTURA
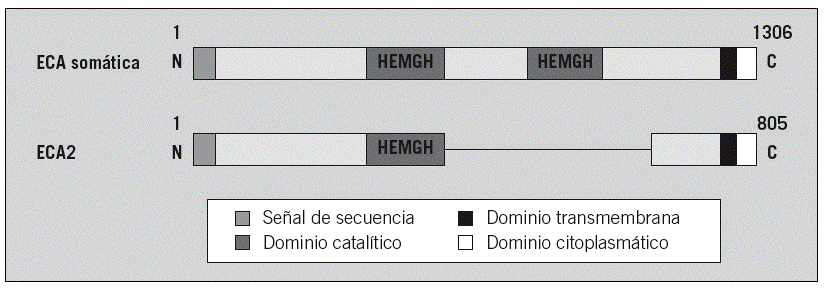
La enfermedad del ántrax es causada por la Bacillus anthracis,una bacteria gram positiva, aerobia y con cápsula. Dicha bacteria tiene la capacidad de formar esporas muy resistentes cuando hay una falta de nutrientes, las cuales pueden sobrevivir durante décadas. Una vez expuesta de nuevo a un ambiente rico en nutrientes, como la sangre de un animal, recuperan su forma activa
La enfermedad realmente será causada por las toxinas producidas por la Bacillus anthracis, así como algunas características de su cápsula
Cápsula:
La cápsula del B.anthracis está constituida por un polímero de poli-D-glutamato. No es tóxica, pero sí protege frente al sistema inmunitario del hospedador, ya que confiere resistencia contra la fagocitosis
Toxinas:
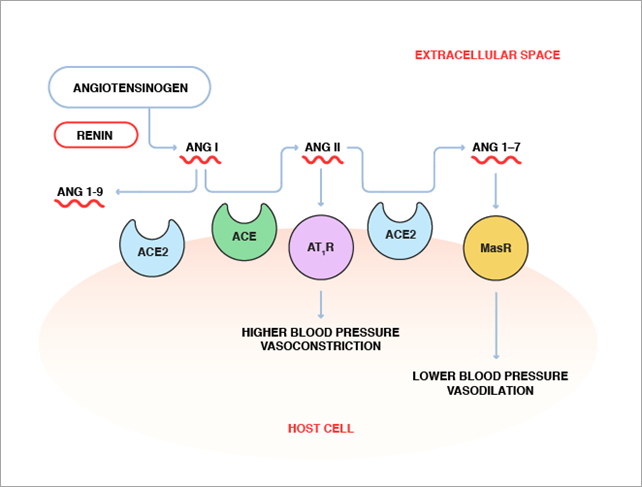
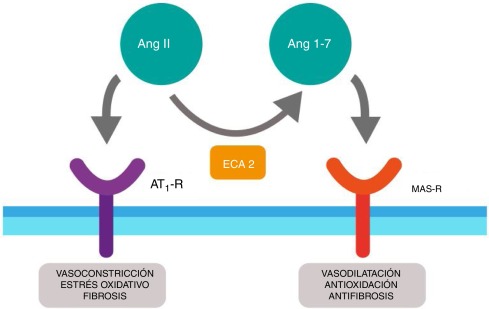
Individualmente ninguna de estas tres proteínas es tóxica, su toxicidad se debe a la expresión y secreción de dos toxinas. Una mezcla de antígeno protector y factor edema forma la toxina edemática (EdTx), que provoca edema. Una mezcla de antígeno protector y factor letal forma la toxina letal (LeTx), que causa la muerte
Esta toxina está formada por 3 componentes: el factor edema, factor letal y el antígeno protector
El factor edema (EF)
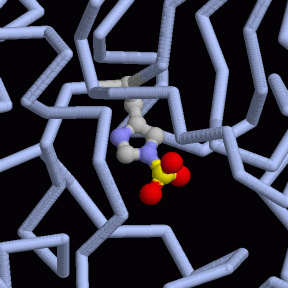
Es una adenilil ciclasa que utiliza el calcio como cofactor. El factor edema es activado cuando entra en la célula y se vincula a la calmodulina, una proteína muy común en nuestras células. Entonces, transforma el ATP a AMP cíclico, consumiendo ATP en el proceso. El AMPc interviene en la recepción de señales por parte de la célula, actuando como segundo mensajero dentro de ella. El EF provoca un gran aumento de AMPc intracelular y destruye el balance mantenido por las hormonas. Esto causa la aparición de un edema.
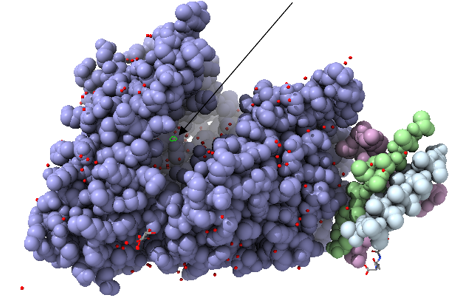
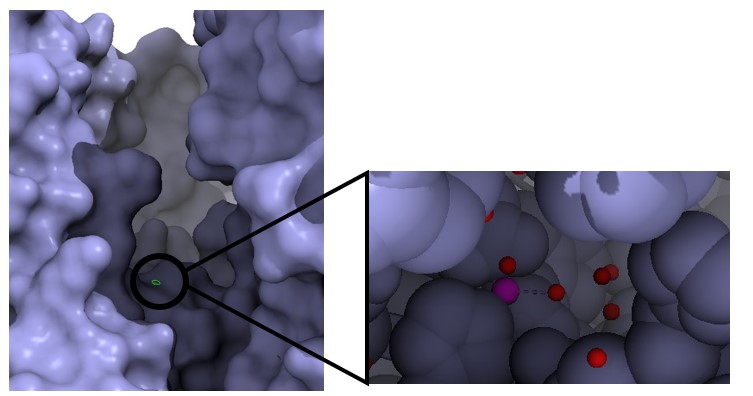
Estructura del factor edema de la toxina del ántrax. En naranja observamos la adenilil ciclasa. En rosa la calmodulina. De color blanco tenemos desoxiadenosina trifosfato, y por último en color verde, el calcio que actúa como cofactor. Imagen creada con ChimeraX a partir de PDB 1K90.
El factor letal (LF)
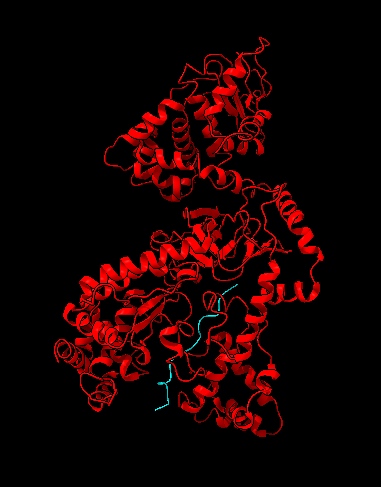
Es una proteasa (rompe enlaces peptídicos) que utiliza el zinc como cofactor y que es altamente específica. Esta realiza un corte en la región amino-terminal de una familia de proteínas quinasas activadas por mitógeno (MAPKK). De esta forma, el LF inactiva diferentes mecanismos de señalización celular
Esta proteína tiene 4 dominios: el dominio I participa en la unión a antígeno protector y el dominio II interviene en el reconocimiento y unión al sustrato. El dominio III está incluido dentro del dominio II, aunque su estructura consiste en una duplicación del dominio II. Y el dominio IV contiene el centro catalítico. Esta estructura revela una proteína que ha evolucionado por un proceso de duplicación, mutación y fusión de genes, dando lugar a una enzima con una actividad muy específica
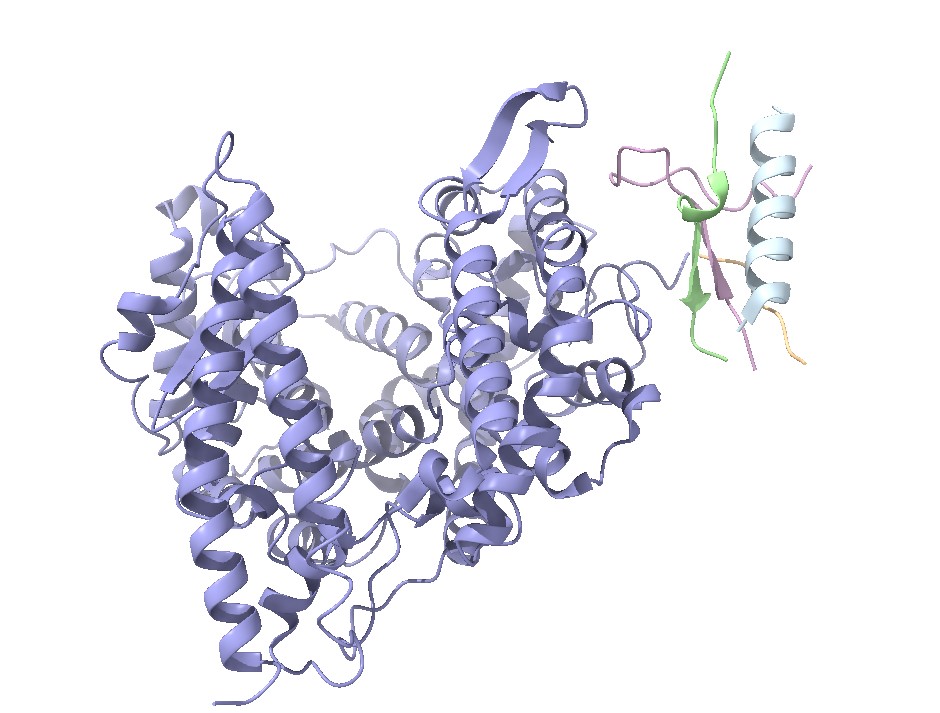
Estructura del factor letal de la toxina del ántrax. En rojo está señalado el factor letal y de color azul la MAPKK. Imagen creada con ChimeraX a partir de PDB 1JKY
El antígeno protector (PA)
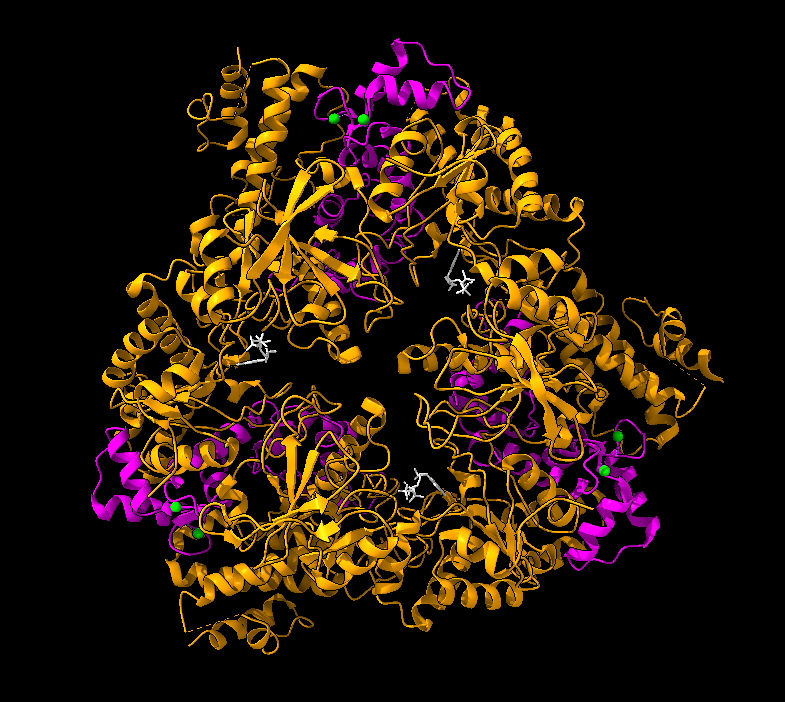
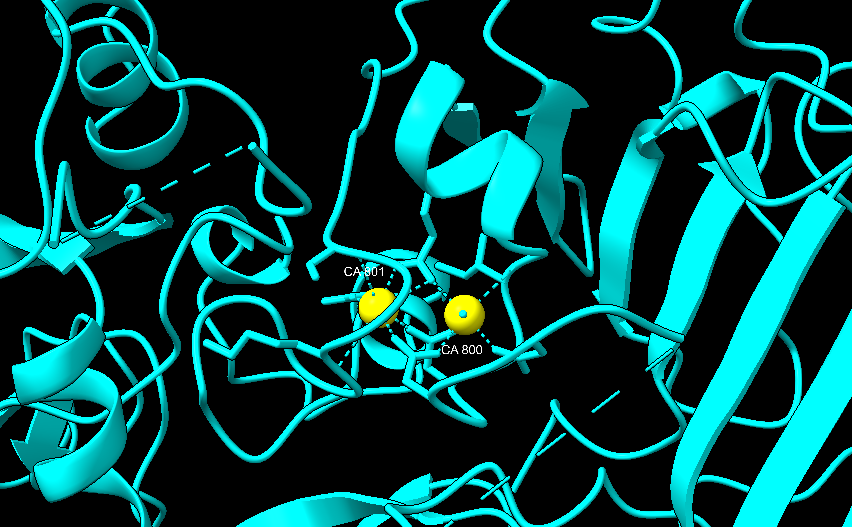
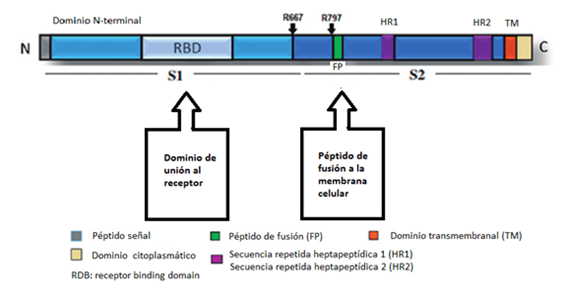
Es una proteína compuesta principalmente por láminas beta antiparalelas y que está formada por 4 dominios. El dominio I amino-terminal contiene la región de corte para la activación de proteasas, así como 2 iones de calcio. El dominio II, por otro lado, está implicado en la inserción a la membrana. El dominio III es pequeño y se desconoce su función. Por último, el dominio IV carboxilo-terminal, es el que participa en la unión al receptor. El antígeno protector forma un heptámero tras la eliminación de un fragmento amino-terminal del dominio I, esta estructura se inserta en la membrana y permite pasar al citosol a los factores tóxicos del ántrax.
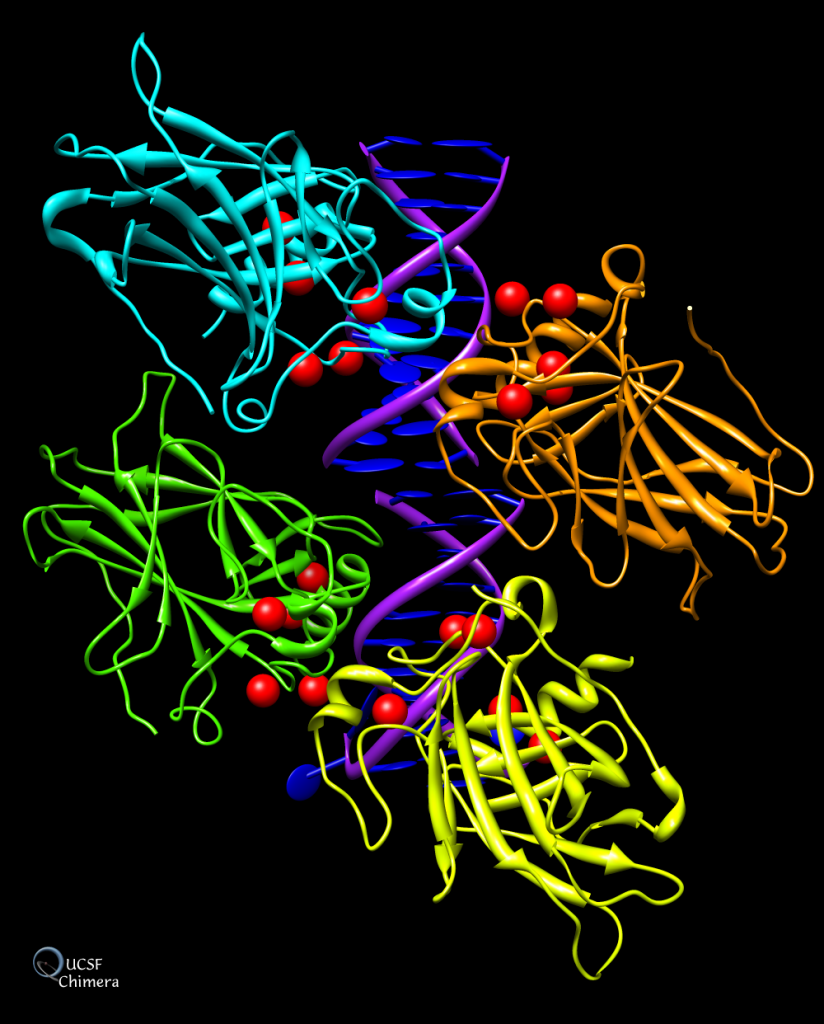
En azul estructura del antígeno protector de la toxina del ántrax. Imagen creada a partir de PDB 1ACC.En amarillo los iones de calcio del dominio I del antígeno protector de la toxina del ántrax. Imagen creada con ChimeraX a partir de PDB 1ACC.Estructura del heptámero formado por la unión 7 unidades del antígeno protector. Imagen creada con ChimeraX a partir del PDB 1TZO
MECANISMO DE ACCIÓN
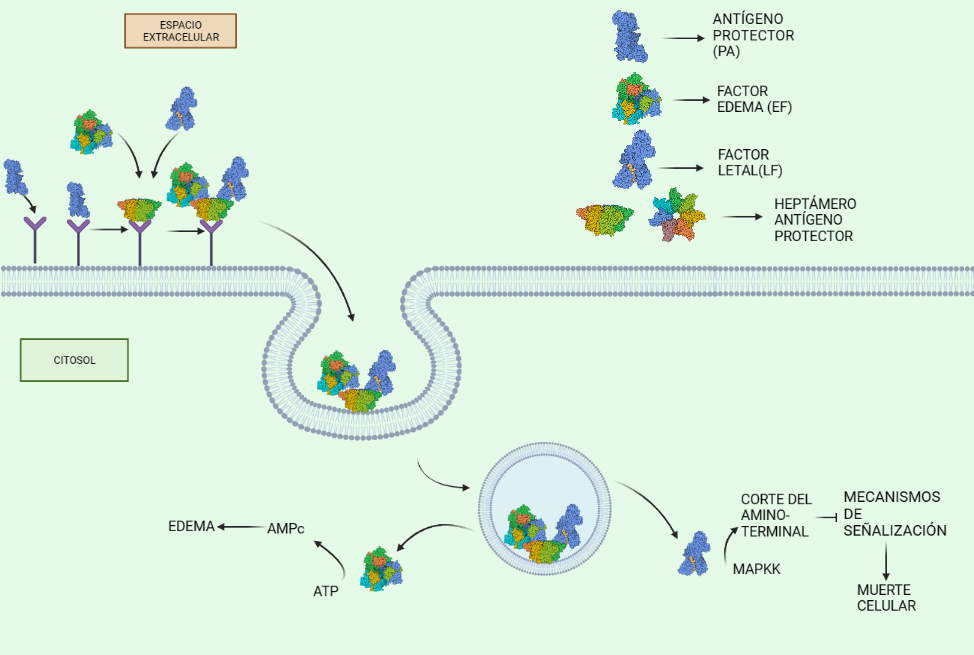
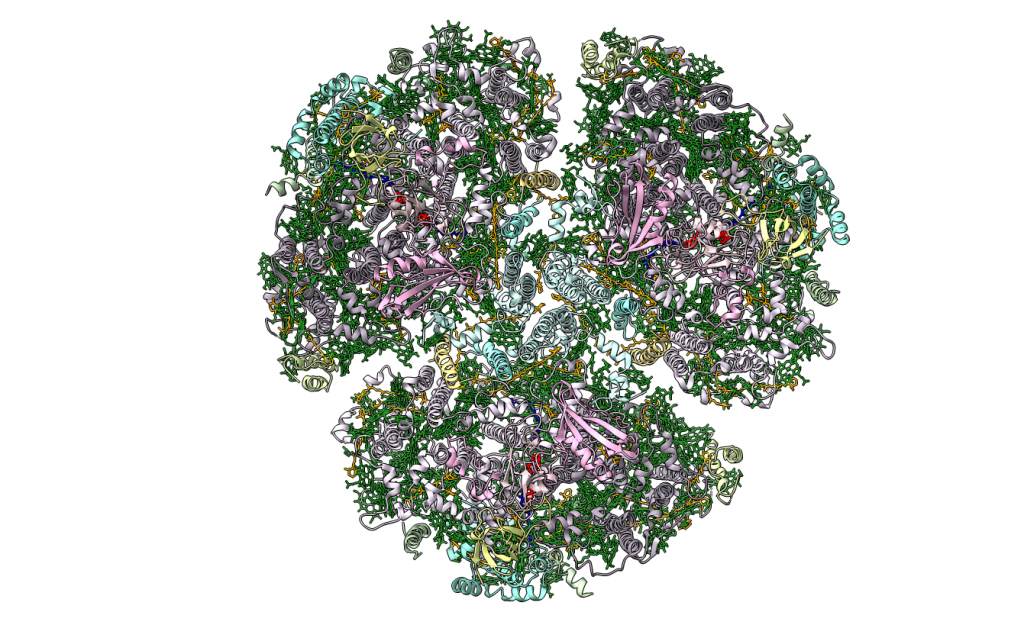
Ilustración del mecanismo de acción de los 3 componentes de la toxina del ántrax. Elaborado con Biorender.com.
El proceso es el siguiente. El antígeno protector es el mecanismo de unión a la célula. La bacteria lo secreta como una sola cadena, que llega hasta la membrana plasmática de una célula. Entonces, se une por lo menos a dos receptores de superficie celular distintos, y es cortado por una proteasa.
A continuación, se une a otras 6 copias de la proteína, formando un heptámero, que dará lugar a un poro. Y a este se unen EF y LF, ya que al separarse el fragmento amino-terminal, queda expuesto en PA un lugar de unión al cual se unen competitivamente EF y LF con alta afinidad. El complejo proteico formado ingresa a la célula por endocitosis mediada por receptor.
PA comienza a formar canales que producen la traslocación de EF y LF dentro del citosol, donde ejercen sus efectos tóxicos.
PATOGÉNESIS
Las personas se infectan con ántrax cuando las esporas ingresan a su organismo. Cuando las estas entran, pueden activarse, distribuyéndose por el individuo y produciendo toxinas. Esto puede ocurrir cuando una persona las respira, bebe agua o ingiere alimentos que están contaminados con ellas. Dichas esporas también pueden penetrar en el organismo a través de heridas o rasguños en la piel.
En el ser humano, el ántrax puede manifestarse de 3 formas distintas:
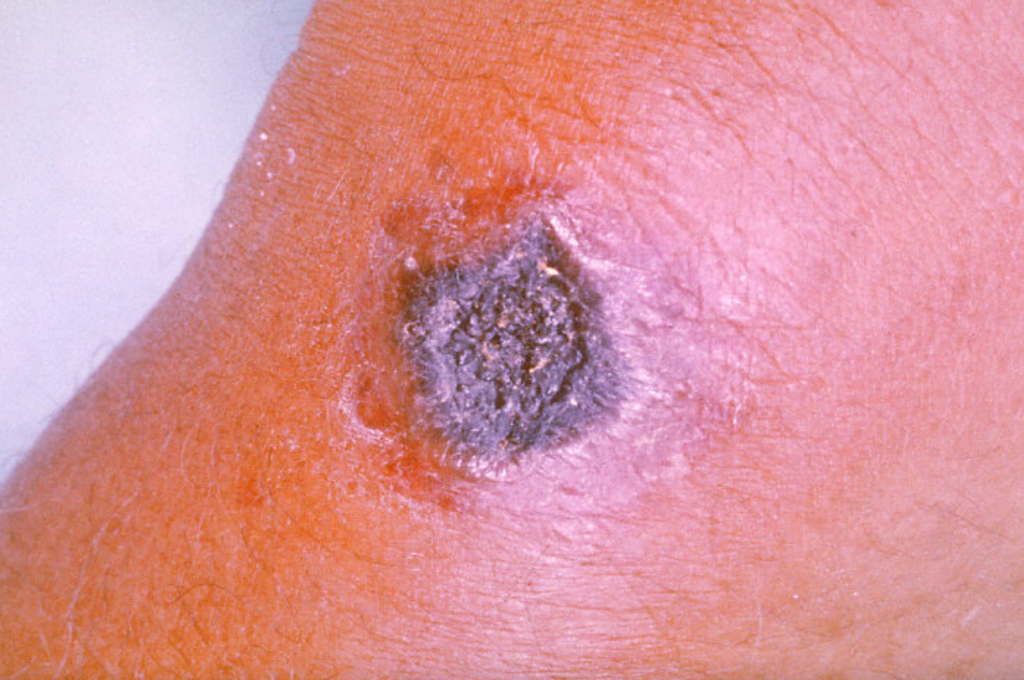
ÁNTRAX CUTÁNEO:
Es la forma más común de infección por ántrax y se considera también la forma menos peligrosa. En él, las esporas penetran la piel a través de una herida o rasguño. Sin embargo, también puede ocurrir al entrar en contacto con animales infectados o productos de origen animal contaminados.
La infección se desarrolla generalmente entre 1 y 7 días después de la exposición. Los síntomas incluyen la aparición de pequeñas ampollas o la aparición de un forúnculo cutáneo (úlcera) con un centro negro. Sin tratamiento, hasta el 20% de las personas con ántrax cutáneo corren riesgo de muerte. No obstante, con el tratamiento adecuado, prácticamente todos los pacientes con ántrax cutáneo sobreviven.
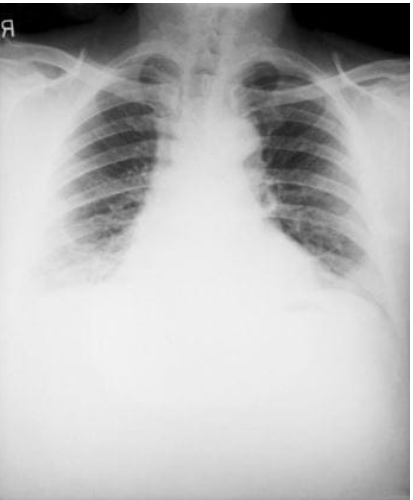
Cuando una persona inhala esporas de ántrax, puede desarrollar ántrax pulmonar. Este se inicia principalmente en los ganglios linfáticos en el pecho antes de distribuirse por el resto del cuerpo, lo que finalmente causa graves problemas respiratorios y posiblemente, la muerte
Sin tratamiento, sólo alrededor del 15% de los pacientes sobreviven. Sin embargo, con un tratamiento adecuado, aproximadamente el 55% de los pacientes sobreviven
Se da por el consumo de alimentos contaminados, mal cocinados o tratados. Las esporas de ántrax pueden afectar a la garganta y esófago, el estómago y los intestinos.
Sin tratamiento, más de la mitad de los pacientes con ántrax gastrointestinal mueren, pero la tasa de mortalidad se reduce hasta el 40% recibiendo tratamiento.
ÁNTRAX POR INYECCIÓN
Recientemente, se ha identificado otro tipo de ántrax en consumidores de heroína en el norte de Europa: el ántrax por inyección. Los síntomas pueden ser similares a los del ántrax cutáneo, pero el ántrax por inyección puede distribuirse por el organismo con mayor rapidez y es más difícil de reconocer y tratar que el ántrax cutáneo
ÁNTRAX COMO ARMA TERRORISTA
El ántrax es una toxina que tiene un gran potencial para ser utilizada como arma bioterrorista o en una guerra biológica. Esto es debido a que sus esporas poseen una gran resistencia a la temperatura, presión, pH y radiación ionizante además de poder sobrevivir un gran número de años en el agua y en la superficie.
El ántrax es utilizado como arma principalmente mediante el uso de aerosoles, provocando infección por ántrax pulmonar. Este tipo de la enfermedad es el más letal, al principio se manifiesta de forma parecida a una gripe, por lo que es difícil de diagnosticar, para cuando se reconoce la enfermedad suele ser demasiado tarde, pues normalmente la infección ya es sistémica, se produce un choque séptico y posteriormente la muerte.
Es considerada como el arma biológica más letal por la facilidad para producir y preservar sus esporas, a diferencia de otros agentes biológicos, que junto con su capacidad para contagiar y la resistencia de sus esporas previamente mencionada, la convierten en una amenaza terrorífica para toda la población.
En 2001 se produjo en Estados Unidos un ataque bioterrorista, esporas del ántrax fueron distribuidas mediante el servicio postal de forma intencionada, 22 personas fueron contagiadas produciendo 5 muertes. Este suceso cambió la forma en la que el sistema respondería ante una amenaza así, aumentando el dinero destinado a mejorar la capacidad de afrontar un ataque de este tipo
Petosa, C. et.al (1998) Protective antigen anthrax toxin doi: 10.2210/pdb1ACC/pdb
Pannifer, A.D. et.al (2001) Lethal factor anthrax toxin doi: 10.2210/pdb1JKY/pdb
Spencer, R. C. (2003). Bacillus anthracis. Journal of clinical pathology, 56(3), 182-187.
Guzmán Terán, C., Calderón Rangel, A., & Soto Gómez, E. (2017). ÁNTRAX: enfermedad aún vigente. Revista Avances En Salud, 1(2), 2017. https://doi.org/10.21897/25394622.1224
Pavan, María E., et.al. (2011). Bacillus anthracis: una mirada molecular a un patógeno célebre. Revista Argentina de Microbiología, 43(4),294-310.[fecha de Consulta 16 de Enero de 2022]. ISSN: 0325-7541. https://www.redalyc.org/articulo.oa?id=213021188010
Hanna, P., Meselson, M., Guillemin, J., & Dixon, T. (1999). Anthrax: review article. Massachussets Medical Society, 341, 815-826.
Spencer, R. C. (2003). Bacillus anthracis. Journal of clinical pathology, 56(3), 182-187.
ISOCITRATO DESHIDROGENASA, LA NUEVA ESPERANZA FRENTE AL CÁNCER
escrito por almala_1A | 20 febrero, 2023
Redactado por Alba Cerrato Benítez, Laura Blanco García y María Ayala Molina. GRADO EN BIOLOGÍA SANITARIA, UAH
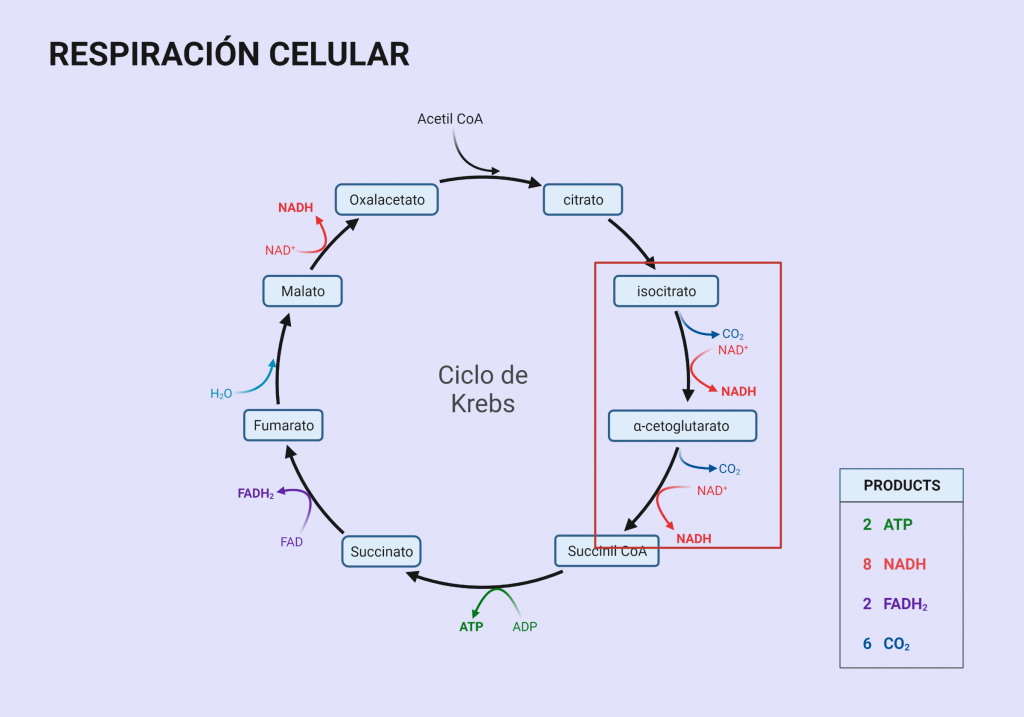
La isocitrato deshidrogenasa (IDH) es una enzima muy relacionada con el metabolismo de los carbohidratos. Esta enzima participa en la tercera reacción del ciclo de Krebs, catalizando el proceso de transformación del isocitrato en 2-oxoglutarato. Es la primera reacción de oxidación-reducción del ciclo en la que obtenemos poder reductor en forma de NADH. Esto hace que sea una importante proteína cuyas funciones explicaremos en el desarrollo de este trabajo.
La razón por la que escogimos esta enzima es por su importante papel en las dianas terapéuticas y en el desarrollo del cáncer. Recientes investigaciones han demostrado que la presencia de mutaciones en dos genes, IDH1 e IDH2 , se dan en los gliomas más comunes. Además, aunque no está muy claro si estas mutaciones están relacionadas con la esperanza de vida del paciente, existen estudios que apuntan a que las personas con estas alteraciones sobreviven durante más tiempo.
El estudio de este oncometabolito nos puede proporcionar información acerca del metabolismo de las células cancerosas y nos puede ayudar a descubrir nuevas líneas de investigación para el desarrollo de terapias contra el cáncer.
ESTRUCTURA
Actualmente, existen muchas isocitrato deshidrogenasas secuenciadas, sin embargo, el número de ellas con una estructura tridimensional resuelta está reducido. Concretando, solo encontramos las de los organismos Escherichia coli y Bacillus subtilis. Estas enzimas (IDH) catalizan la descarboxilación oxidativa del D-isocitrato, produciendo 2-oxoglutarato y CO2. Para ello es necesario NAD+ o NADP+ como coenzima y se produce NADH o NADPH, respectivamente.
Según su dependencia por la coenzima, podemos distinguir tres tipos fundamentales de IDH:
IDH NAD+ – dependientes: necesitan como coenzima al NAD+ para catalizar la reacción. Aparece fundamentalmente en las mitocondrias de organismos del Dominio Eucarya. También hay organismos del Dominio Bacteria.
IDH deshidrogenasas NADP+ -dependientes: requieren como coenzima al NADP+ para catalizar la reacción. Las encontramos en organismos de los tres Dominios: Eucarya, Bacteria y Archaea.
IDH con especificidad dual: utilizan tanto el NAD+ como el NADP+ como coenzima. Este tipo es el único que aparece en arqueas termófilos y en algunas bacterias.
En los seres humanos, existen tres isoformas de esta enzima:
IDH3 dependiente de NAD+, cataliza uno de los pasos del ciclo de Krebs en la mitocondria. Es necesaria para la respiración celular.
IDH1 y IDH2 son dependientes de NADP+, y por lo tanto aportan poder reductor a la célula en forma de NADPH. Se localizan en el citosol, y también en la mitocondria y el peroxisoma.
En el desarrollo de este trabajo nos centraremos en la estructura de la IDH1.
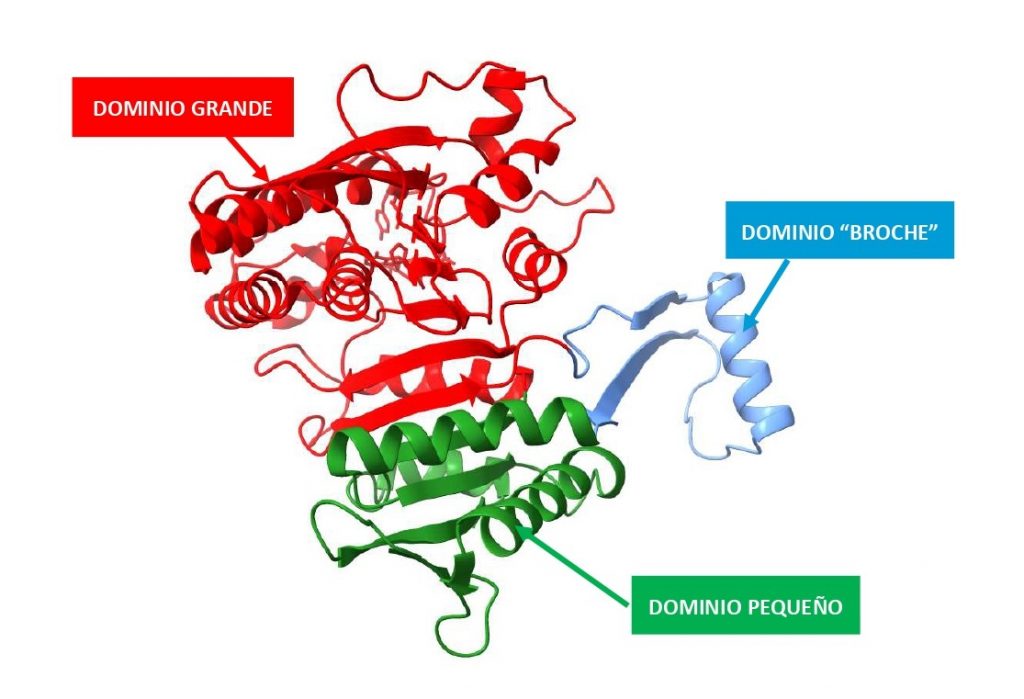
La IDH1 es una proteína α/β, es decir, la estructura de esta proteína está constituida por la alternancia de láminas beta y hélices alfa, cuyo núcleo es hidrófobo. Puede formar un dímero de dos subunidades iguales o un tetrámero formado por dos dímeros, este último parece ser la forma funcional de la proteína.
Podemos distinguir en cada subunidad 3 dominios: dominio grande, dominio pequeño y dominio “broche”. La dimerización y tetramerización que hemos mencionado anteriormente está mediada por interacciones a nivel de los dominios “broche” y de los dominios pequeños.
Zonas hidrofóbicas (azul) e hidrofílicas (amarillo) de la IDH 1. A partir de PDB 9ICD. Creado con ChimeraX.
Encontramos una lámina β que une los dominios grande y pequeño, y que además a ambos lados presenta dos surcos de naturaleza hidrofílica. En el surco formado por los dominios grande y pequeño de una subunidad y por el dominio pequeño de la subunidad adyacente encontramos el centro activo. Existen 2 sitios activos por cada dímero proteico.
Dominios de la enzima IDH 1. A partir de PDB 9ICD. Creado con ChimeraX
PAPEL BIOLÓGICO
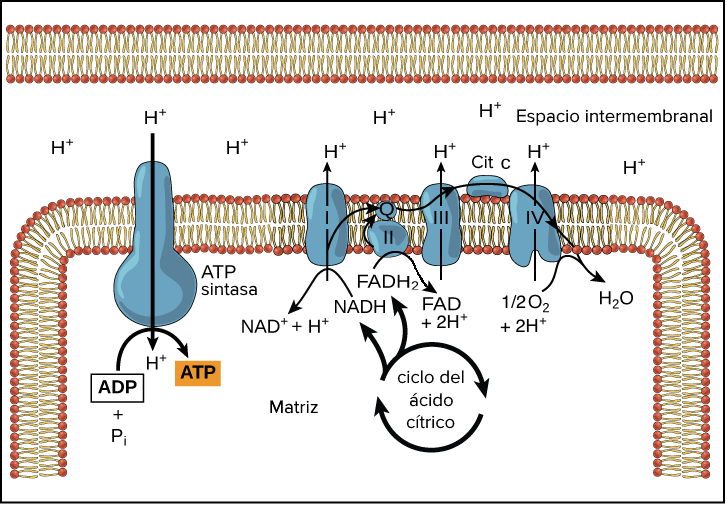
Las isocitrato deshidrogenasas son una familia de enzimas que catalizan la reacción de descarboxilación oxidativa de isocitrato en α-cetoglutarato (o 2-oxoglutarato) en el ciclo de Krebs. La regulación de la actividad enzimática de la isocitrato deshidrogenasa es muy importante para llevar a cabo las funciones biológicas. Estas enzimas tienen dos cofactores distintos: NAD+ y NADP+. En el caso de la IDH1, isoenzima en la que nos hemos centrado, el cofactor es el NADP+ (que utiliza como aceptor de electrones) y se encuentra principalmente en el citoplasma. Por lo tanto, otra de sus funciones será reducir NADP+ a NADPH. En el proceso de esta reacción se libera uno de los átomos de carbono del reactivo en forma de dióxido de carbono (CO2) y dos átomos de hidrógeno, uno de los cuales se transfiere al transportador (NADP+), que lo utilizará para impulsar la rotación de la ATP sintetasa. La reacción que la enzima lleva a cabo en el ciclo de Krebs cuenta con un intermedio de reacción que es el oxalosuccinato.
Esquema del Ciclo de Krebs, con la descarboxilación oxidativa del isocitrato en α-cetoglutarato señalada. Adaptado de “Krebs Cycle”, por BioRender.com (2023). Obtenido de https://app.biorender.com/biorender-templates
Además del papel catabólico que tiene la enzima en el ciclo de Krebs, se ha demostrado que tienen una función importante en la defensa de la célula contra el daño oxidativo a través de la generación de NADPH. Asimismo, la IDH citosólica contiene una secuencia de direccionamiento peroxisomal de tipo 1 que se encarga de dirigir diferentes proteínas hacia los peroxisomas. También se han encontrado isocitrato deshidrogenasas citosólicas (IDH1) en peroxisomas de levaduras y de células hepáticas humanas y de rata y se ha podido demostrar que son necesarias para la β-oxidación de ácidos grasos insaturados. Su función en la β-oxidación es provisionar con NADPH a los peroxisomas.
En algunas células bacterianas solo existe un tipo de isocitrato deshidrogenasa (IDH) dependiente de NADP+, cuyas funciones y estructuras también han sido estudiadas. En el caso de estos organismos, la enzima actúa en un punto muy importante entre el ciclo de Krebs y el ciclo de glioxilato. Mediante la regulación de su actividad, se puede regular la cantidad de sustrato de las dos reacciones. La fosforilación de un aminoácido de Serina (Ser) que se sitúa en el sitio de unión al que se une el ion isocitrato-metal, la actividad de la IDH de las bacterias es regulada. Sin embargo, el mecanismo regulador que controla la IDH1 en mamíferos no está claro. Aunque las enzimas en mamíferos y en bacterias se parecen, tan solo comparten el 20% de similitud en su secuencia. Sin embargo, la secuencia de la IDH en mamíferos tiene una Serina en la posición equivalente a la que poseen las bacterias en su secuencia. Esto sugiere que las IDH1 de mamíferos también puedan tener un sitio de unión que pueda ser fosforilado.
MECANISMO DE ACCIÓN
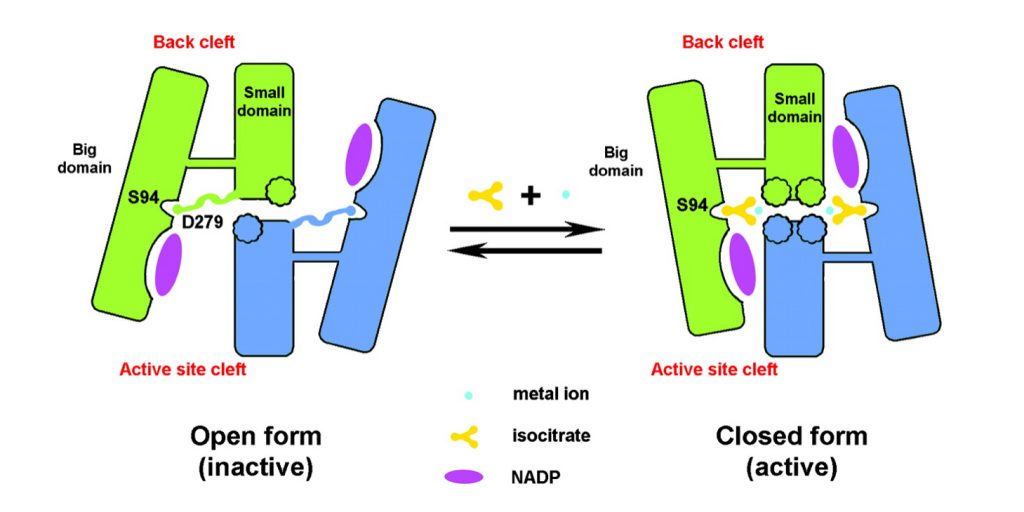
Las diferentes conformaciones que adopta el complejo enzimático provocan cambios en la estructura del centro activo. Podemos observar:
Conformación abierta (inactiva): Si no hay isocitrato, el surco del centro activo está abierto y el surco posterior cerrado. La interacción entre el Asp279 del dominio pequeño con la Ser94 del dominio grande estabiliza la estructura y, además, en la zona donde se encuentra el Asp279 encontramos una estructura extendida. Esa interacción impide la unión del isocitrato.
Conformación cerrada (activa): Si la concentración del isocitrato y del catión divalente llega a un cierto nivel, se une al centro activo. Rompiendo la interacción (puentes de hidrógeno) entre el Asp279 y Ser94, produciéndose la expulsión del Asp279 y la interacción con el Ca2+. La estructura extendida que se encontraba en la zona donde se encontraba, adopta una estructura en hélice α. Esto permite la entrada del isocitrato. Se cierra el centro activo y se abre el surco posterior.
Conformaciones inactiva y activa de la enzima IDH 1. Obtenida de http://proteinasestructurafuncion.usal.es/moleculas/IsocitratoDeshidrogenasas/index.html
Este cambio conformacional que se produce entre la forma activa e inactiva se debe a un movimiento de bisagra de las láminas β que se encuentra entre el dominio «broche» y el dominio pequeño.
IMPORTANCIA BIOMÉDICA: UNA ESPERANZA PARA EL CÁNCER
Los gliomas son los tumores primarios más comunes del sistema nervioso central, por lo que afectan a las funciones del corazón y la médula espinal. Según la Organización Mundial de la Salud, los gliomas se clasifican de I a IV según su grado de malignidad. Recientemente, se ha descubierto una posible relación de la aparición de los mismos con las mutaciones en el exón 4 de los genes que codifican las isocitratos deshidrogenasas 1 y 2. En el caso de la isocitrato deshidrogenasa 1, con los gliomas de grado II-III o la leucemia mieloide aguda entre otros.
El gen IDH1 se encuentra localizado en 2q33.3 y su ARNm contiene 10 exones para codificar la isocitrato deshidrogenasa 1 (414 aminoácidos). En relación con las mutaciones, nos interesa especialmente el residuo 132 de la proteína, correspondiente a una arginina altamente conservada en todas las isoformas de la enzima, que va a sufrir cambios a distintos aminoácidos.
Cambio de aminoácido
N (%)
R132H
92,7
R132C
3,6
R132S
1,8
R132G
0,9
R132L
0,5
R132V
0,5
Tabla 1: Mutaciones en IDH1. En la primera columna, se indican los cambios de aminoácidos; y en la segunda, el porcentaje de aparición de cada mutación con respecto al número total de mutaciones descritas. Marcada en rosa se encuentra la mutación más común. (Balss, 2008).
Como hemos observado, la mutación más frecuente es la que cambia una arginina por una histidina (R132H). Pero, ¿Qué consecuencias lleva asociadas dicha mutación? ¿Tanta importancia tiene el cambio de un único aminoácido?
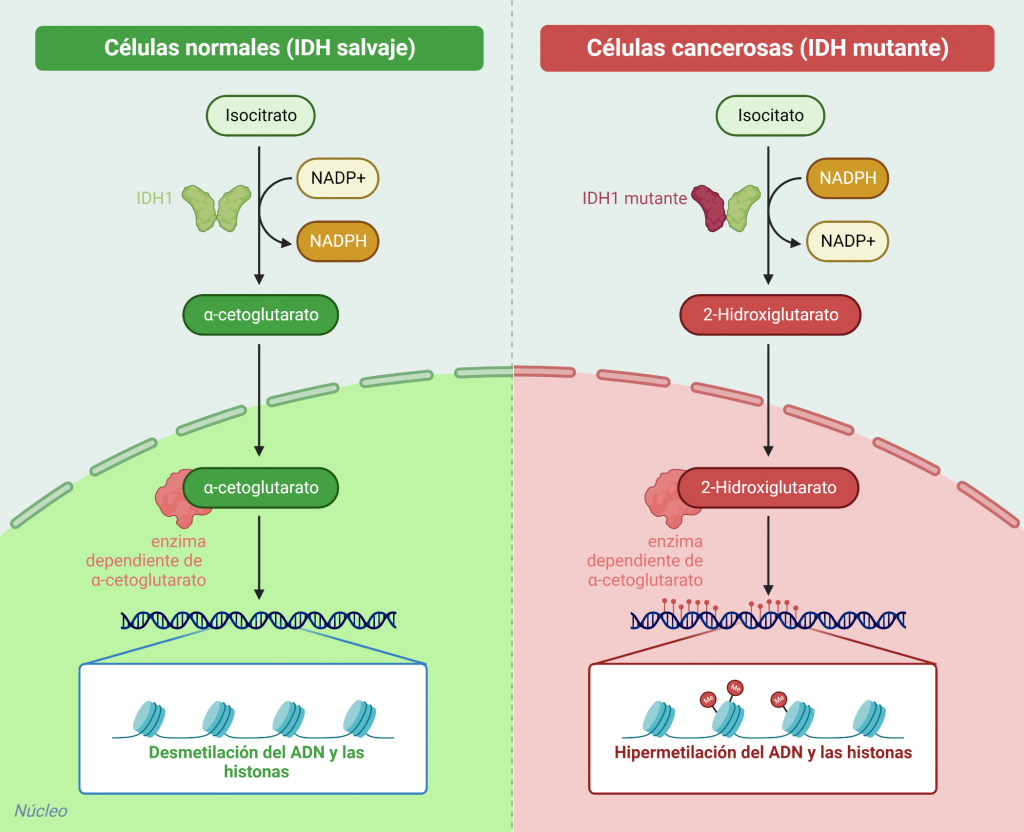
Como se ha mencionado con anterioridad, la isocitrato deshidrogenasa salvaje(no mutante), está involucrada en la descarboxilación oxidativa del isocitrato a 2-oxoglutarato y dióxido de carbono con la reducción de NADP+ a NADPH. El NADPH obtenido se utiliza para la producción de glutatión reducido, un tripéptido encargado de neutralizar los radicales libres, toxinas y oxidantes que se generan en las células, favoreciendo la supervivencia celular y la anti-apoptosis.
Sin embargo, lo que ocurre al cambiar un aminoácido (arginina) del sitio activo de la enzima por otro (histidina) es que la enzima pierde su capacidad de catalizar la conversión de isocitrato a 2-oxoglutarato. En su lugar, la enzima adquiere la capacidad de catalizar la reducción dependiente del NADPH del 2-oxoglutarato a 2-hidroxiglutarato. Una de las consecuencias de la producción excesiva del oncometabolito, 2-hidroxiglutarato, es la formación e incremento de la malignidad de gliomas.
Esquema comparativo de la catálisis de la isocitrato deshidrogenasa 1 salvaje y mutante. Adaptado de «IDH Mutant in Cancer Cells”, por BioRender.com (2023). Obtenido de https://app.biorender.com/biorender-templates
Además, el 2-hidroxiglutarato, al ser tan parecido en estructura al 2-oxoglutarato, funciona como un inhibidor de las enzimas dependientes de 2-oxoglutarato. Estas enzimas tienen un papel muy importante en el mantenimiento del equilibrio de las histonas y la metilación del ADN por lo que, un exceso de 2-hidroxiglutarato podría causar lahipermetilación de histonas y ADN y el bloqueo de la diferenciación celular.
Diversos estudios indican que la probabilidad de que un paciente que presenta dicha mutación se recupere, es mayor que la de un paciente que no la presenta en el caso de gliomas de bajo grado. Para el resto de gliomas, la situación varía: en algunas ocasiones resulta en un valor de pronóstico desfavorable y, en otras, aún no se ha encontrado relación. Además, ha resultado ser una herramienta útil para diferenciar los gliomas de otras lesiones benignas y diferenciar entre gliomas primarios y secundarios.
Estos descubrimientos han abierto un nuevo campo de investigación e innovación farmacológica: la búsqueda de inhibidores de las IDH mutantes.
REFERENCIAS / BIBLIOGRAFÍA
Balss, J., Meyer, J., Mueller, W., Korshunov, A., Hartmann, C. y von Deimling, A. (2008). Análisis de la mutación del codón 132 de IDH1 en tumores cerebrales. Acta neuropathologica , 116 , 597-602.
Dang, L., White, D. W., Gross, S., Bennett, B. D., Bittinger, M. A., Driggers, E. M., Fantin, V. R., Jang, H. G., Jin, S., Keenan, M. C., Marks, K. M., Prins, R. M., Ward, P. S., Yen, K. E., Liau, L. M., Rabinowitz, J. D., Cantley, L. C., Thompson, C. B., Heiden, M. G. V., & Su, S. M. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature, 462(7274), 739-44. https://doi.org/10.1038/nature08617
Etxaniz Ulazia, O. (2017). Investigación de las mutaciones de los genes de IDH1 y 2 en los gliomas de bajo grado [Tesis doctoral]. Universidad Autónoma de Barcelona.
García Martínez, A. (2016). Estudio de biomarcadores en gliomas y su utilidad clínica [Tesis doctoral]. Universidad de Alicante.
PDB101: Molecule of the Month: Isocitrate Dehydrogenase. (s. f.). RCSB: PDB-101. https://pdb101.rcsb.org/motm/129
Liu, Z., Yao, Y., Kogiso, M., Zheng, B., Deng, L., Qiu, J. J., Dong, S., Lv, H., Gallo, J. M., Li, X. N., & Song, Y. (2014). Inhibition of Cancer-Associated Mutant Isocitrate Dehydrogenases: Synthesis, Structure–Activity Relationship, and Selective Antitumor Activity. Journal of Medicinal Chemistry, 57(20), 8307-8318. https://doi.org/10.1021/jm500660f
¿Qué es el Glutatión Reducido y para qué sirve? (s. f.). https://www.tarracofarma.com/que-es-el-glutation-reducido-y-para-que-sirve
Reitman, Z. J., & Yan, H. (2010). Isocitrate Dehydrogenase 1 and 2 Mutations in Cancer: Alterations at a Crossroads of Cellular Metabolism. JNCI Journal of the National Cancer Institute, 102(13), 932-941. https://doi.org/10.1093/jnci/djq187
Ricaurte O, Neita K, Valero D, Ortega-Rojas J, Arboleda-Bustos CE, Zubieta C, Penagos J, Arboleda G. Estudio de mutaciones en los genes IDH1 e IDH2 en una muestra de gliomas de población colombiana. biomedica [Internet]. 1 de mayo de 2018 [citado 1 de febrero de 2023];38(Sup1):86-92. Disponible en: https://revistabiomedica.org/index.php/biomedica/article/view/3708
Robert, T. (s. f.-c). Dpto. Bioquímica y Biología Molecular. http://proteinasestructurafuncion.usal.es/moleculas/IsocitratoDeshidrogenasas/index.html
Sumihito Nobusawa, Takuya Watanabe, Paul Kleihues, Hiroko Ohgaki; Mutaciones de IDH1 como firma molecular y factor predictivo de glioblastomas secundarios. Clin Cancer Res 1 de octubre de 2009; 15 (19): 6002–6007. https://doi.org/10.1158/1078-0432.CCR-09-0715
UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. Protein Sci. 2021 Jan;30(1):70-82.
Vista de Estudio de mutaciones en los genes IDH1 e IDH2 en una muestra de gliomas de población colombiana. (s. f.-b). https://revistabiomedica.org/index.php/biomedica/article/view/3708/3915
Xu, X., Zhao, J., Xu, Z., Peng, B., Huang, Q., Arnold, E. y Ding, J. (2004). Las estructuras de la isocitrato deshidrogenasa dependiente de NADP citosólica humana revelan un nuevo mecanismo de autorregulación de la actividad. Revista de química biológica , 279 (32), 33946-33957.
Zachary J. Reitman, Hai Yan, Isocitrate Dehydrogenase 1 and 2 Mutations in Cancer: Alterations at a Crossroads of Cellular Metabolism, JNCI: Revista del Instituto Nacional del Cáncer , volumen 102, número 13, 7 de julio de 2010, páginas 932–941, https ://doi.org/10.1093/jnci/djq187
APP: la ladrona de recuerdos
escrito por entropia_01 | 20 febrero, 2023
Elaborado por Candela Martínez, María Orenes y Almudena López
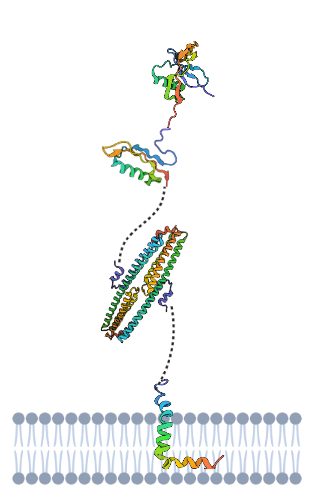
Estructura de los dominios extracelulares y transmembrana de la proteína APP. Diseñada por María Orenes partir de PDB y BioRender
INTRODUCCIÓN
La APP, proteína precursora del péptido beta-amiloide (Amyloid-beta Precursor Protein por sus siglas en inglés), es una glucoproteína transmembrana conservada evolutivamente desde Caenorhabditis elegans hasta los humanos.
En humanos, el gen está localizado en el cromosoma 21 (curiosamente, el cromosoma del síndrome de Down, hecho muy importante, como veremos más adelante) y formada por 770 aminoácidos repartidos en diferentes dominios proteicos. Se encuentra en diversos tejidos destacando el nervioso, pues se piensa que participa en la neurogénesis y maduración del cerebro, plasticidad, formación y reparación sináptica, además del transporte del hierro.
Es procesada por diferentes proteasas, las: α, β y γ. Los fallos en los cortes de sus dominios son los causantes de la formación de placas amiloides, que principalmente consisten en el cúmulo de beta-amiloide en el tejido cerebral, causando degeneración neuronal o más comúnmente llamado, enfermedad de Alzheimer (Cho et al., 2022).
ESTRUCTURA
Tenemos que la APP es una proteína dividida en tres dominios, unidos por conectores flexibles formados por cadenas de aminoácidos sin estructura secundaria. Un dominio extracelular, otro transmembrana (el más importante en la formación de la enfermedad) y otro dominio hacia el medio intracelular. Estos dos últimos dominios forman un fragmento denominado C99 cuya fragmentación es decisiva en la generación de placas amiloides.
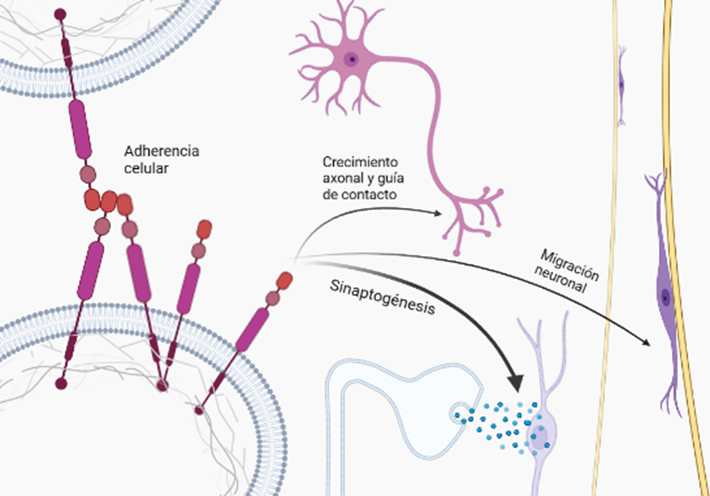
Cuando la proteína está intacta, actúa como proteína receptora enviando señales a otras proteínas del interior celular, además, es capaz de unirse a estructuras del exterior de la célula como la heparina y laminina, permitiendo la adhesión celular.
Por otro lado, la división en fragmentos de la misma también hace que sea funcional; división que se lleva a cabo por unas proteínas proteasas denominadas secretasas, que cortan a ambos lados del péptido transmembrana. Por un lado los dominios externos, de mayor tamaño, son liberados fuera de la célula ayudando a controlar el crecimiento de las neuronas, y por el otro, el dominio del interior celular se libera a este, ayudando a la síntesis de proteínas en el núcleo.
Y por último, el dominio transmembrana, el cual es el causante de la demencia, pues cuando se suelta de la membrana se introduce en la célula pudiendo cambiar su conformación a beta-lámina, y por acumulación de la misma se generan las placas amiloides. (Burley et al., 2017)
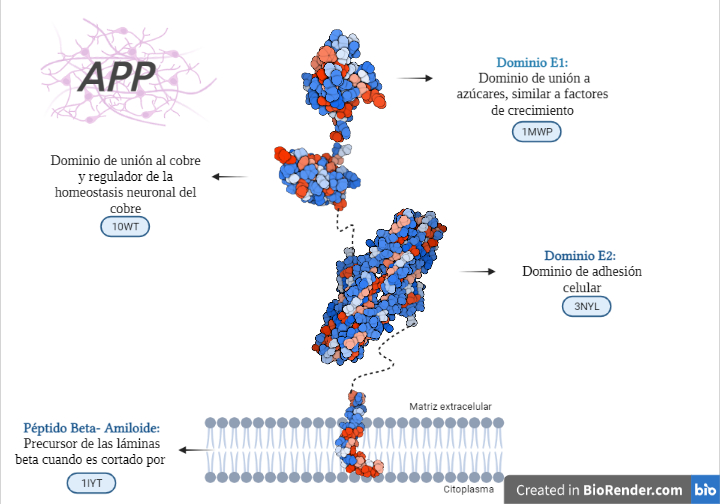
Dominio más externo: PROTEÍNA DE UNIÓN A AZÚCAR:
Es también llamado dominio N-terminal de la APP o estructura cristalina del dominio similar al factor de crecimiento N-terminal de la proteína precursora de Alzheimer.
Esta es la estructura cristalina del dominio de unión a heparina. Entre sus funciones se encuentra la estimulación del crecimiento de neuritas.
Se trata de una estructura muy cargada con capacidad de interaccionar con glucosaminoglicanos, mientras que su parte hidrófoba se estima que tiene función de dimerización o unión de ligandos.
Dada la semejanzas estructurales entre la proteína y los factores de crecimiento que contienen cisteína se cree que este dominio N-terminal es capaz de llevar a cabo una función similar a uno de estos.
Dominio intermedio: DOMINIO DE UNIÓN AL COBRE- REGULADOR DE LA HOMEOSTASIS NEURONAL DEL COBRE:
El cobre es uno de los principales generadores de radicales libres en nuestro cerebro, también conocido como estrés oxidativo.
Nuestras células han desarrollado sistemas para transportar estos metales, causantes de las especies oxidantes.
En el caso de esta proteína, se encarga de regular la homeostasis del cobre neuronal, pues la unión de la APP con el cobre disminuye la formación de beta amiloide.
Esto se ha observado en ratones que sobreexpresan esta proteína, y que poseen menor cantidad de cobre en el cerebro.
Dominio más cercano a membrana: DOMINIO DE ADHESIÓN CELULAR:
Se trata de un dímero antiparalelo del dominio E2 de la proteína.
Hay estudios que han demostrado que la APP puede formar dímeros, siendo este dominio uno de los contribuyentes a la formación de esto.
La estructura antiparalela se produce tras la dimerización de las dos subestructuras de E2 mediante la unión del N-terminal de un monómero con el C-terminal del segundo monómero.
Se sugiere la idea de que la dimerización de esta viene inducida por la unión de heparina. (Lee S et al., 2011)
Dominio transmembrana: PÉPTIDO BETA AMILOIDE. SIMILAR A UN DOMINIO DE FUSIÓN DE VIRUS:
En términos generales, los principales causantes de la enfermedad del Alzheimer son los péptidos beta amiloides (Aβ o Abeta) que proceden de la escisión proteolítica de la APP.
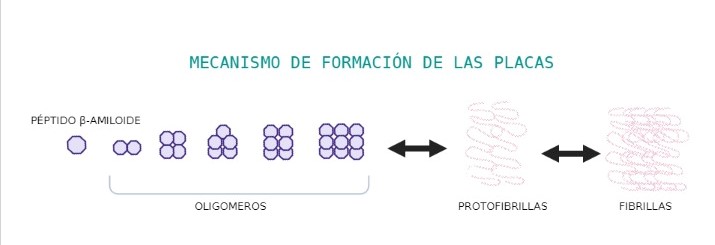
Se ha visto que in vitro, Abeta puede sufrir cambios conformacionales, pasando de estructura soluble a láminas beta fibrilares, las cuales se agregan, siendo neurotóxicas.
Algunos estudios han visto que se trata de una estructura formada por dos tramos helicoidales, unidos por un puente flexible, pero de los cuales no se conoce con precisión la longitud y posición.
La similitud que presenta esta estructura junto con la secuencia del resto C-terminal con el dominio de unión de hemaglutinina de influenza muestra la posibilidad de su capacidad neurotóxica. (Crescenzi, Orlando et al., 2022)
Estructura de los diferentes dominios de la APP con diferenciación entre partes polares (azules) y apolares (rojas). Creada por María Orenes a partir de ChimeraX y BioRender.com
FUNCIÓN BIOLÓGICA
Las funciones biológicas de la APP no han sido identificadas del todo desde el descubrimiento de esta proteína, pero sigue siendo objeto de numerosas investigaciones. Aunque su rol en la enfermedad del Alzheimer es la más conocida, cumple muchas otras funciones relevantes además de la vía patogénica tales como: la neurogénesis (nacimiento de nuevas neuronas), la maduración del cerebro, el crecimiento de las neuronas, la plasticidad, y la formación y reparación sináptica, de ahí que sea tan abundante en el cerebro en relación con otras zonas del cuerpo. También la interacción célula a célula, la adhesión célula-sustrato, apoptosis, el metabolismo de calcio o la transducción de señales a través de la membrana. Todas ellas continúan aún bajo estudio. (Zhang et al., 2011).
Una de las isoformas de la APP, como es la APP659, está involucrada en la maduración del cerebro durante la embriogénesis. Además, se requiere en la formación de la sinapsis neuromuscular; ya que se localiza con receptores de la acetilcolina. La abundancia de la APP durante la sinaptogénesis sugiere un papel funcional en la formación de la red neuronal y la participación en el crecimiento y restauración neuronal después de traumas cerebrales, ya que ayuda a la organización de las fibras neuronales. También interacciona con numerosas proteínas regulando el tráfico celular y el procesamiento de señales.
Imagen de la APP con su dominio intracelular interactuando con elementos del citoesqueleto y algunas de sus funciones en el cerebro. Creado por Candela Martínez con BioRender.
Otra función neurotrófica atribuida a la APP es la propiedad ferroxidasa y de tráfico de hierro, por lo que puede que alguno de sus fragmentos se encuentren regulados por este metal, el cual actúa además en el metabolismo de la APP al regula la expresión de su ARNm. De igual manera actúa el cobre sobre la APP, sugiriendo que la APP participe en la homeostasis del cobre y sea junto con el hierro una posible diana terapéutica en el Alzheimer. (Nalivaeva NN & Turner AJ, 2013)
A continuación hablaremos con más profundidad sobre algunas de sus funciones:
Adhesión celular
Una de las funciones mencionada previamente es la de adhesión celular e interacción célula-célula, necesarias para diferentes etapas del neurodesarrollo, como la construcción de la red neuronal y madurez funcional de numerosas etapas complejas de migración de neuronas a la apropiada capa de la placa cortical y la acción conjunta de las CAM (moléculas de adhesión celular) y APP. Algunas CAM como las integrinas, se localizan en los conos de crecimiento y las dendritas neuronales, y la interacción con la APP proporciona la tracción necesaria para la generación de las espinas dendríticas y el cono de crecimiento (GC) en el axón, que detectará el entorno para la correcta formación de la sinapsis. (Sosa et al., 2017).
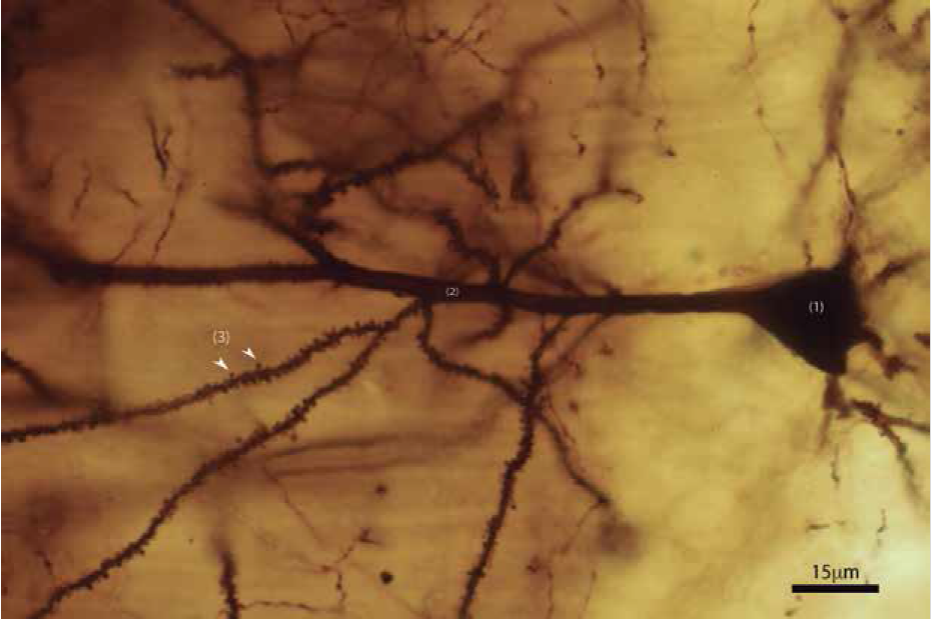
Espinas dendríticas. Imagen de Valencia Segura, R. K., Colín Barenque, L., & Fortoul van der Goes, T. I. (2018). Las espinas dendríticas, su función y algunas alteraciones. Revista de la Facultad de Medicina (México), 61(1), 46-55.
Cono de crecimiento. Imagen de https://www.researchgate.net/publication/318827764/figure/fig2/AS:522578831261696@1501603910083/Figura-2-4a-cono-de-crecimiento-en-el-extremo-de-un-axon-en-proceso-de-elongacion-b.png
Esquema de la APP, (a) donde la región extracelular es capaz de unirse a diferentes componentes de la matriz extracelular y (b) interactuar con otras moléculas de adhesión celular. Imagen de Sosa et. al, The physiological role of the amyloid precursor protein as an adhesion molecule in the developing nervous system.
Esto implica un importante equilibrio y coordinación entre adhesión y movilidad, procesos mutuamente dependientes, ya que en caso de romperse esa relación con un exceso o insuficiencia de adhesión, se puede provocar una alteración en la organización normal de la red neuronal. En los casos de síndrome de Down (DS), donde el cromosoma 21 se encuentra duplicado y donde se localiza la APP sobreexpresada, da lugar a un desequilibrio que puede culminar en neurodegeneraciones similares al Alzheimer. Hay evidencias de que la APP presenta características importantes en sus dominios similares a las moléculas de adhesión celular (CAM), como la capacidad de generar una interacción con proteínas de andamio asociadas al citoesqueleto en el dominio citoplasmático, y de formar dímeros en el dominio extracelular. Esta capacidad de dimerización viene determinada por la capacidad del dominio extracelular de la APP (E2) de formar estructuras cristalinas junto con otros miembros de la familia APP, fundamental para la interacción transcelular. (Soba et al., 2005)
Por lo que la acción conjunta y mecánica de vinculación con elementos de la ECM (membrana extracelular) y la señalización, genera que la APP participe en la remodelación de las adherencias celulares así como en la organización dinámica del citoesqueleto subcortical, al coordinarse con lamelipodios y filopodios contenidos en este.
Proteínas adaptadoras citoplasmáticas
Para determinar las funciones de la APP es importante saber con qué proteínas interactúa. Se ha encontrado que la familia de proteínas FE65 de los conos de crecimiento y sinapsis interactúan con ella a través de su C-terminal citoplasmático, por eso estas proteínas adaptadoras han sugerido roles para la APP en la dinámica del cono de crecimiento, la migración neuronal y la señalización intracelular. Además, FE65 asociada con el dominio intracelular (AICD) de la APP puede afectar a la transcripción de genes, ya que APP AICD-FE65 se libera de la membrana y se dirige al núcleo donde puede alterar la transcripción de genes. (Hoe et al., 2008). Aunque todavía quedan muchas dudas sobre los efectos en el tráfico y procesamiento de este tipo de proteínas adaptadoras en la APP, existen estudios que analizan este tipo de cuestiones (Borg et al. 1996; Parisiadou y Efthimiopoulos 2007).
Unión de ligandos
Los procesos funcionales de la APP sugieren que funcione como un receptor transmembrana de determinados ligandos que afectan a su procesamiento. Exámenes recientes han demostrado que el principal factor genético de riesgo de la enfermedad del Alzheimer (EA) de inicio tardío es la apoliproteína E (ApoE) debido a las interacciones funcionales y estructurales con la APP, ya que estas asociaciones pueden afectar a la producción de la proteína beta amiloide (Aβ). En ausencia de la ApoE, la producción de Aβ, la secreción de APP y la estabilidad de los fragmentos de APP C-terminal se ven afectados. (Pietrzik et al., 2002)
La ApoE se encuentra mediada por una familia de receptores de lipoproteínas de baja densidad (LDLR) que se encuentran a su vez divididas en dos subgrupos. Actúan en la transducción de señales y la neurotransmisión, además, todos se encuentran dentro de las placas seniles y se han asociado genéticamente con la Enfermedad de Alzheimer.
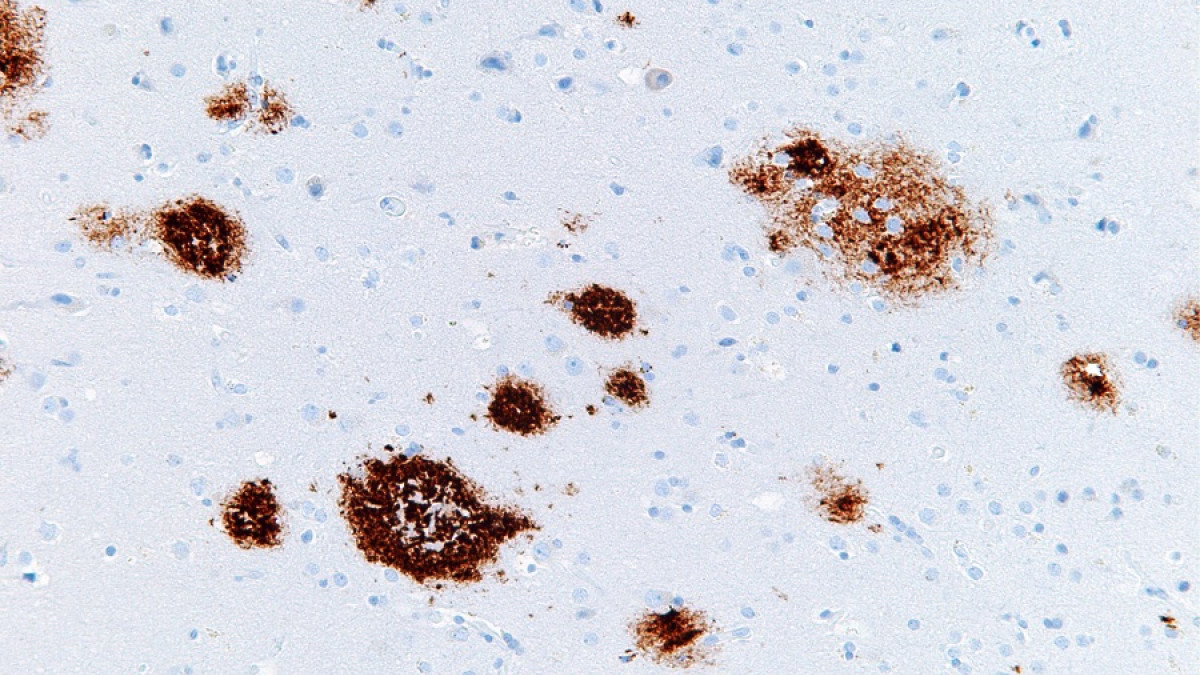
Placas seniles. Imagen de Curiosoando.com (Actualizado el 16 noviembre, 2018). «¿Qué son las placas neuríticas o placas seniles?». Disponible en https://curiosoando.com/que-son-las-placas-neuriticas-o-placas-seniles
La APP y los receptores de la apoE comparten una serie de rasgos estructurales y funcionales. Ambas son proteínas transmembrana de tipo I y presentan grandes dominios extracelulares y pequeños dominios citoplasmáticos, que en el caso de los receptores de la ApoE pueden ser numerosos mientras que en la APP observamos sólo uno para un número de proteínas adaptadoras (Stolt y Bock 2006). Además, comparten un patrón de proteólisis y de escisión superficial constitutiva e inducible que generan formas solubles que nos hace pensar cada vez más que la APP puede actuar como receptor. Una vez estudiemos y comprendamos cómo funciona la ApoE podremos quizás saber si la APP lo hace de manera similar.
Apoptosis
Aunque no se sabe con claridad los mecanismos de muerte celular en la enfermedad del Alzheimer (EA), se asocia con la pérdida general de neuronas y mutaciones en el gen de la APP que conducen a un aumento de producción de la Aβ. La APP-BP1, identificada como una proteína de unión a la APP, es la subunidad reguladora de la enzima E1 activadora de una proteína similar a la ubiquitina NEDD8 que participa en una vía para la ubiquitinación. (Chen et al., 2003), y juega un papel importante en la progresión del ciclo celular y la supervivencia.
APP-BP1 es un gen cuyas proteínas codificadas al interactuar con la APP impulsa la transición de la fase S a la M en células neuronales en división y causa apoptosis en neuronas por la vía de conjugación de NEDD8 (neddilación). La sobreexpresión de la APP-BP1 en neuronas primarias que conducen a la apoptosis puede ser bloqueada con la inhibición de la neddilación y la coexpresión de un péptido que compite con la APP-BP1 por unirse a la APP. Se observa una sobreexpresión de la APP-BP1 en el hipocampo cerebral de las personas con EA, ya que participa como hemos mencionado antes, en la activación de NEDD8. (Chen et al., 2000)
MECANISMOS DE ACCIÓN
Formas tempranas de la EA:
Se han descubierto tres genes causantes de formas tempranas de la Enfermedad del Alzheimer: el gen que codifica para la proteína precursora del péptido β-amiloide (APP), el gen de la presenilina 1 (PSEN1) y el de la presenilina 2 (PSEN2).
Gen codificante para APP:
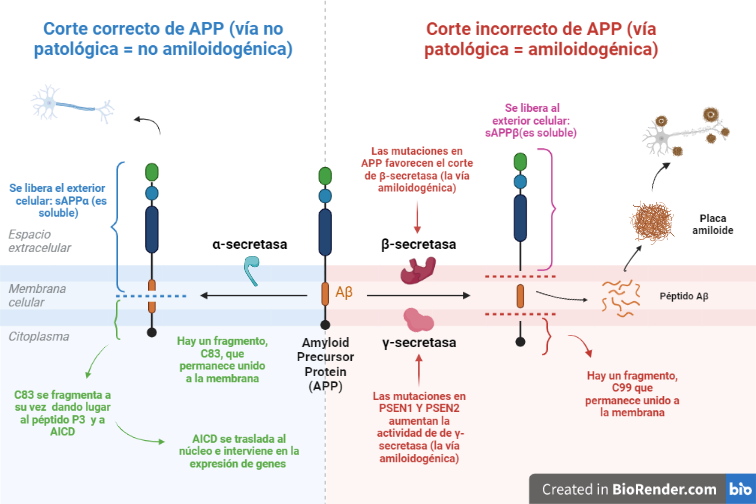
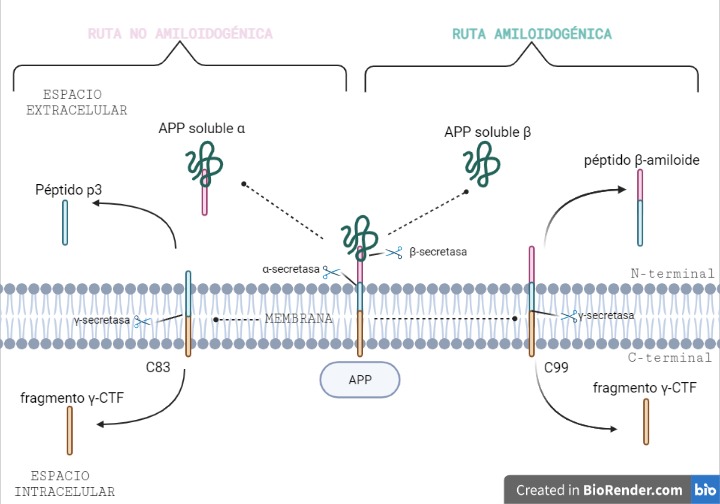
Vía no amiloidogénica:
Por esta vía tiene lugar el corte de alfa-secretasa (entre los aminoácidos 687 y 688). Este da lugar a dos fragmentos: uno de ellos es sAPPα, el cual es soluble y se expulsa al exterior celular; mientras que el otro permanece en la membrana, y se conoce como C83.
Y es este último fragmento el que posteriormente será procesado por la γ-secretasa, cortando por los aminoácidos 712, 714 o 715, que corresponden con los residuos 40, 42 o 43 del péptido Aβ.
Esta vía es conocida como no amiloidogénica, es decir, no patológica, ya que como producto final no se obtiene un agregado peptídico.
Vía amiloidogénica:
Por otra parte, existe otra vía donde intervienen la beta-secretasa y la γ-secretasa.
El corte de β-secretasa se produce entre los aminoácidos 671 y 672, liberando al exterior celular el fragmento, sAPPβ, el cual también es soluble.
Al igual que en la vía anterior, en la membrana permanece otro fragmento, llamado C99, es procesado también por la γ-secretasa, cortando por los mismos aminoácidos 712, 714 o 715, dando lugar a tres posibles péptidos. Estos péptidos se conocen como: Aβ40, Aβ42 o Aβ43, y pueden formar agregados que forman las fibras insolubles de los depósitos de amiloide.
Vías amiloidogénica y no amiloidogénica de la APP. Creada por Almudena López a partir de BioRender.com
Genes de las presenilinas: PSEN1 y PSEN2
En 1995 se clonó el gen responsable del 50% de los casos de la EA. Este se llamó inicialmente S182 pero poco después pasó a llamarse presenilina 1 (PSEN1). Hoy en día, existen 177 mutaciones en PSEN1, las cuales causan EA a edades muy tempranas.
Posteriormente, se localizó en el cromosoma 1 una secuencia genética muy similar a PSEN1. Este gen, conocido como presenilina 2 (PSEN2), es responsable de una proporción muy pequeña de los casos de EA autosómicos dominantes. Además, actualmente se sabe que las presenilinas son un cofactor del complejo multiproteico de la γ-secretasa.
Formas tardías de EA:
Alrededor del 95% de los casos de EA aparecen a edades avanzadas.
Por el momento, el único gen que se ha relacionado con la EA tardía es el que codifica para la apolipoproteína E (APOE). El gen APOE, se localiza en el cromosoma 19 y codifica para tres isoformas : ε2, ε3 y ε4, las cuales se diferencian entre sí por la presencia de los aminoácidos cisteína o arginina en las posiciones 112 y 158 de la APOE.
De forma que si las frecuencias de cada una de las isoformas son comparadas entre pacientes, atendiendo a criterios de edad, se percibe un incremento del alelo ε4 en pacientes con EA tardía respecto a controles sanos de edades avanzadas. Esto quiere decir, que un paciente que contenga una copia del alelo ε4 tiene un riesgo de contraer la enfermedad mayor, y este será aún mayor si posee dos copias del alelo. (Setó-Salvia et al., 2010)
RELACIÓN CON LAS DROGAS
El consumo en exceso de drogas por el abuso de opiáceos es un problema extendido por todo el mundo. Sin embargo, a pesar de no conocerse gran información sobre la neuropatología que causa este consumo, estudios han concluido que la neuroinflamación que se genera podría desembocar en una neurodegeneración prematura de los cerebros de los consumidores.
Tras investigaciones de los cerebros de jóvenes consumidores , se aplicó un análisis inmunohistoquímico detallado del hipocampo, el tronco encefálico y los ganglios basales para tau hiperfosforilada, β-amiloide, proteína precursora de β-amiloide (βAPP) y ubiquitina, el cual denotó un exceso de ovillos neurofibrilares (NFTs); los cuales son causantes de la degeneración neuronal y disfunción sináptica.
A diferencia de los no consumidores, en quienes los NFTs solamente se localizaron en la corteza entorrinal; en aquellos que sí consumían drogas también se localizaron en el subículo, la neocorteza temporal, el núcleo basal de Meynert y el locus coeruleus.
Además, los consumidores de drogas dieron positivo en βAPP en mayor proporción que en los no consumidores, aunque prácticamente no se localizaron placas de beta-amiloide. (Ramage et al., 2005)
IMPLICACIÓN DE LA PROTEÍNA TAU
Papel de la proteína TAU en la Enfermedad del Alzheimer. Creada por Almudena López a partir de BioRender.com (Vallés-saiz, L., 2022)
DAÑO CEREBRAL
La encefalopatía traumática crónica (ETC) es una enfermedad neurodegenerativa que se asocia con lesiones cerebrales traumáticas leves repetitivas. De forma patológica se caracteriza por la acumulación anormal de la proteína TAU.
Para determinar el alcance de la extensión del depósito del péptido β amiloide (Aβ), se realizaron estudios en atletas y militares veteranos con diagnóstico de ETC. Gracias a estos se descubrió que más del 52% de los pacientes diagnosticados con CTE portaban depósitos del péptido Aβ. Además, esta acumulación de este péptido tenía lugar a una velocidad mayor que en aquellos que no diagnosticados con CTE. E incluso, un subgrupo cumplió con los criterios de diagnóstico tanto para CTE como para EA.
En conclusión, estos resultados proponen que existe una alteración y aceleración en el depósito de Aβ en un conjunto de individuos con CTE si lo comparamos con el envejecimiento normal, y que Aβ mantiene una fuerte relación con la progresión patológica y clínica de CTE, independientemente de la edad. (Stein et al., 2015)
TRATAMIENTOS
Antioxidantes:
Se ha demostrado que el estrés oxidativo, es decir, la acumulación de productos derivados de la cadena de transporte de electrones, tales como: el radical hidroxilo, superóxido o el peróxido de hidrógeno (localizados en la mitocondria) son responsables de estrés oxidativo. Este es a su vez precedente de las lesiones que se generan en la Enfermedad de Alzheimer. Además, los radicales libres podrían ser los causantes de cambios en pares de bases del ADN, o incluso de daños en las cadenas de ADN. Es por esto que uno de los tratamientos se basa en combatir este estrés oxidativo para lograr paliar el deterioro cognitivo de la enfermedad.
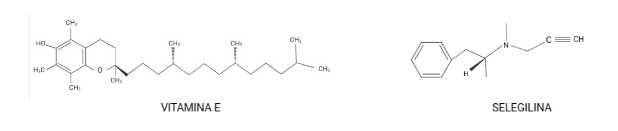
Durante un trabajo realizado por el Alzheimer’s Disease Cooperative Study, en el que se usaron los antioxidantes: α-tocoferol (mayormente conocido como vitamina E) y selegilina, se señaló un posible efecto positivo de la vitamina E en la EA.
Una de las principales conclusiones fue la prolongación del tiempo en el que ocurrían procesos, como por ejemplo la pérdida de la capacidad para realizar algunas actividades de la vida cotidiana (basándose en la escala de puntuación clínica de la demencia) era más largo en aquellos que tomaban vitamina E que en comparación con quienes tomaban selegilina, ambos fármacos o placebo. Sin embargo, el uso de vitamina E o selegilina no causó alteraciones en el deterioro cognitivo. Estos fármacos se seleccionaron porque la vitamina E es un “scavenger” (destructor) de radicales libres; y por su parte la selegilina inhibe la desaminación oxidativa. (Shah et al., 2008)
Moléculas nombradas anteriormente. Creación propia a partir de BioRender.com
Agentes nootrópicos:
Los agentes nootrópicos son derivados cíclicos del ácido γ-aminobutírico (GABA), este es un aminoácido no proteico que se localiza en altas concentraciones en el SNC de los mamíferos, cuya su función principal es actuar como un neurotransmisor inhibidor. Algunos de estos ejemplos pueden ser : piracetam, pramiracetam, aniracetam u oxiracetam. En concreto el primero de ellos, ha manifestado beneficios a corto y largo plazo, principalmente en los relacionado a las funciones de la atención y la memoria. Asimismo, estudios en animales suscitan que estimulan la síntesis y liberación de AcH y que desarrollan una función sobre los receptores de glutamato AMPA. (Santos-Espinosa et al., 2018)
El glutamato es el principal neurotransmisor excitatorio en las neuronas corticales e hipocampales.
La glutamina sintetasa, se oxida en los cerebros de las personas con EA, lo que conduce a un exceso de glutamato. La activación excesiva de los receptores NMDA por el glutamato, aumenta la vulnerabilidad de las neuronas del SNC; lo que concluye en degeneración neuronal.
Un ejemplo de estos fármacos es la memantina, esta se emplea para el bloqueo de los canales NMDA controlados por glutamato, inhibiendo así su activación patológica, pero manteniendo la activación fisiológica.
Inhibidores de la acetilcolinesterasa:
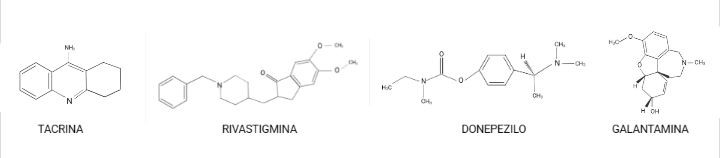
Como por ejemplo: tacrina, donepezilo, rivastigmina y galantamina son los principales tratamientos contra el Alzheimer (leve a moderada). Aunque poseen distintas propiedades farmacológicas, todos ellos inhiben la acción de la AChE.
Estos evitan la degradación del neurotransmisor (acetilcolina); de forma que favorecen un aumento de los niveles de ACh en la hendidura sináptica y facilitan los efectos de los neurotransmisores tanto nicotínico como muscarínico, lo que en ambos casos contribuye a mejorar la cognición. Sin embargo, en estudios realizados a largo plazo, se ha observado un deterioro progresivo de los pacientes, lo que nos indica que estos fármacos nos proporcionan una mejora de los síntomas de la enfermedad solamente durante un periodo de tiempo, y que, por tanto, no actúan sobre los mecanismos patogénicos de la enfermedad. (Golimstok et al., 2006)
Esquemas de las moléculas nombradas anteriormente. Creación propia a partir de BioRender.com
ALZHEIMER EN LA ACTUALIDAD
La enfermedad causada por el mal corte de esta proteína es una de las principales causas de demencia en nuestro país, así como en el mundo. Según datos del SEN (“Sociedad Española de Neurociencia”), hoy en día en España unas 800.000 personas sufren la enfermedad, incrementando cada año aproximadamente en 40.000 nuevos casos. No obstante, se estima que entre el 30 y el 40% de los casos totales están aún sin diagnosticar.
En el año 2015, alrededor de 47 millones de personas sufrían demencia en el mundo, y conforme a las proyecciones de población, si las cifras actuales se mantienen, en 2050 la cifra podría llegar a 130 millones de personas. En el caso de España, entre un 3 y un 4% de la población de entre 75 y 79 años sufren Alzheimer, aumentando a 34% en mayores de 85. En mayores de 65, la SEN estima que alrededor de un 15 % de la población sufre deterioro cognitivo leve, que puede deberse a la enfermedad en el 50% de los años aproximadamente.
A continuación, se exponen citas del Dr. Juan Fortea, Coordinador del grupo de Estudio de Conducta y Demencias de la Sociedad Española de Neurología (SEN):
“La enfermedad de Alzheimer es la primera causa de demencia neurodegenerativa en el mundo y supone un problema sanitario de primer orden. Además, debido a que es una enfermedad cuya prevalencia aumenta exponencialmente a partir de los 65 años, ante el progresivo envejecimiento de la población española, urge el desarrollo de políticas sanitarias destinadas a garantizar el adecuado diagnóstico y acceso a los tratamientos presentes y futuros en nuestro país, así como la puesta en marcha de registros nacionales que permitan precisar la verdadera prevalencia e incidencia del Alzheimer”.
Dr. Juan Fortea, Coordinador del grupo de Estudio de Conducta y Demencias de la Sociedad Española de Neurología (SEN).
“Se estima que la mitad de los casos de la enfermedad de Alzheimer se puede atribuir a nueve factores de riesgo potencialmente modificables: diabetes mellitus, hipertensión arterial en edad media de vida, obesidad e edad media de vida, tabaquismo, inactividad física, depresión, actividad cognitiva o bajo nivel educativo, la hipoacusia y el aislamiento social, por lo que una reducción de entre un 10 y un 15% en dichos factores de riesgo podrían potencialmente prevenir entre 1 y 3 millones de casos de Alzheimer en el mundo”.
Dr. Juan Fortea, Coordinador del grupo de Estudio de Conducta y Demencias de la Sociedad Española de Neurología (SEN).
Referencias
Burley, S. K., Berman, H. M., Kleywegt, G. J., Markley, J. L., Nakamura, H., & Velankar, S. (2017). Protein Data Bank (PDB): the single global macromolecular structure archive. Protein Crystallography, 627-641.
Chen, Y., Liu, W., McPhie, D. L., Hassinger, L., & Neve, R. L. (2003). APP-BP1 mediates APP-induced apoptosis and DNA synthesis and is increased in Alzheimer’s disease brain. The Journal of cell biology, 163(1), 27-33.
Chen, Y., McPhie, D. L., Hirschberg, J., & Neve, R. L. (2000). The amyloid precursor protein-binding protein APP-BP1 drives the cell cycle through the SM checkpoint and causes apoptosis in neurons. Journal of Biological Chemistry, 275(12), 8929-8935
Cho, Y., Bae, H. G., Okun, E., Arumugam, T. V., & Jo, D. G. (2022). Physiology and pharmacology of amyloid precursor protein. Pharmacology & Therapeutics, 108122..
Crescenzi, Orlando et al. “Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain.” European journal of biochemistry vol. 269,22 (2002): 5642-8. doi:10.1046/j.1432-1033.2002.03271.x
Golimstok, A. (2006). Tratamiento de la enfermedad de Alzheimer. Rev. Hosp. Ital. B. Aires Vol, 26(4), 132.
Hoe, H. S., & William Rebeck, G. (2008). Functional interactions of APP with the apoE receptor family. Journal of neurochemistry, 106(6), 2263-2271.
Lee S, Xue Y, Hu J, Wang Y, Liu X, Demeler B, Ha Y. The E2 domains of APP and APLP1 share a conserved mode of dimerization. Biochemistry. 2011 Jun 21;50(24):5453-64. doi: 10.1021/bi101846x. Epub 2011 May 26. PMID: 21574595; PMCID: PMC3120129.
Nalivaeva NN, Turner AJ. The amyloid precursor protein: a biochemical enigma in brain development, function and disease. FEBS Lett. 2013 Jun 27;587(13):2046-54. doi: 10.1016/j.febslet.2013.05.010. Epub 2013 May 16. PMID: 23684647
Pietrzik, C. U., Busse, T., Merriam, D. E., Weggen, S., & Koo, E. H. (2002). The cytoplasmic domain of the LDL receptor-related protein regulates multiple steps in APP processing. The EMBO journal, 21(21), 5691-5700.
Ramage, S. N., Anthony, I. C., Carnie, F. W., Busuttil, A., Robertson, R., & Bell, J. E. (2005). Hyperphosphorylated tau and amyloid precursor protein deposition is increased in the brains of young drug abusers. Neuropathology and applied neurobiology, 31(4), 439-448.
Santos-Espinosa, A., Manzanarez-Quin, C. G., Reyes-Díaz, R., Hernández-Mendoza, A., Vallejo-Cordoba, B., & González-Córdova, A. F. (2018). Ácido γ-aminobutírico (GABA) producido por bacterias àcido lácticas en alimentos fermentados. Interciencia, 43(3), 175-181.
Setó-Salvia, N., & Clarimón, J. (2010). Genética en la enfermedad de Alzheimer. Revista de Neurología, 50(6), 360-364.
Shah, R. S., Lee, H. G., Xiongwei, Z., Perry, G., Smith, M. A., & Castellani, R. J. (2008). Current approaches in the treatment of Alzheimer’s disease. Biomedicine & Pharmacotherapy, 62(4), 199-207.
Soba, P., Eggert, S., Wagner, K., Zentgraf, H., Siehl, K., Kreger, S., … & Beyreuther, K. (2005). Homo‐and heterodimerization of APP family members promotes intercellular adhesion. The EMBO journal, 24(20), 3624-3634.
Sosa, L. J., Cáceres, A., Dupraz, S., Oksdath, M., Quiroga, S., & Lorenzo, A. (2017). The physiological role of the amyloid precursor protein as an adhesion molecule in the developing nervous system. Journal of neurochemistry, 143(1), 11-29.
Stein, T. D., Montenigro, P. H., Alvarez, V. E., Xia, W., Crary, J. F., Tripodis, Y., … & McKee, A. C. (2015). Beta-amyloid deposition in chronic traumatic encephalopathy. Acta neuropathologica, 130(1), 21-34.
Vallés-Saiz, L. (2022). Nuevas funciones para la proteína tau en el Sistema Nervioso Central y tejidos periféricos.
TRIPSINA: LA PRIMERA «ENZIMA»
escrito por Carmen_1B | 20 febrero, 2023
Por Cristina Díaz Fraile, Yolanda García del Castillo López y Carmen Fernández Montiel. 1ºB de Biología Sanitaria
Para que sea más fácil relacionar los términos, y por lo tanto, reducir la energía necesaria para entender bien el tema, vamos a introducir primero lo que es una enzima.
Las enzimas son moléculas orgánicas proteicas que se encargan de catalizar reacciones. Estas reaccionan con el sustrato, formando un complejo enzima-sustrato que ocasionará que mediante un “juego” entrópico y entálpico se reduzca la energía necesaria de activación. Hay muchos tipos de enzimas que se encargan de diferentes reacciones (aunque una misma puede servir para varios sustratos debido a que no son tan específicas). La tripsina pertenece a un grupo de enzimas llamadas peptidasas o proteasas.
Esta enzima fue descubierta por el fisiólogo alemán Wilhelm Kühne mientras realizaba sus estudios sobre los mecanismos digestivos en 1876. De hecho, fue él quien introdujo la palabra enzima al investigar sobre la propia tripsina.
LA TRIPSINA: UNA ENZIMA PROTEASA
Para entender su función y mecanismo precisa conocer que se trata de una enzima proteasa, es decir, una enzima que es capaz de catalizar la ruptura de las cadenas polipeptídicas en unidades proteicas más pequeñas, como lo son los péptidos o los aminoácidos, el monómero estructural de las proteínas. Esta enzima se encuentra en humanos y otros animales como los rumiantes. Es producida por el páncreas, pero se secreta al duodeno ya que es una enzima digestiva esencial.
Figura 1: Funcionamiento de la tripsina (creado a partir de BioRender)
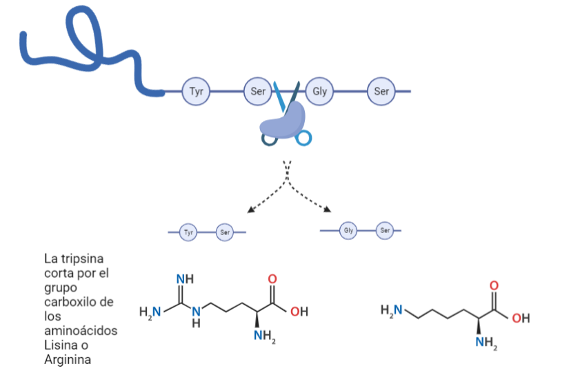
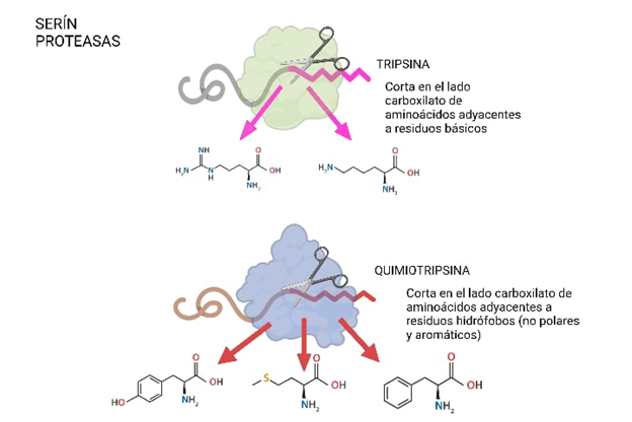
En concreto, la tripsina resulta ser una serín proteasa debido a la presencia del aminoácido serina en su centro activo, el cual será esencial para la catálisis. Las serín proteasas son capaces de reconocer la secuencia primaria de la cadena polipeptídica y cortarla por el lado carboxilo de aminoácidos específicos dependiendo de cuál sea la enzima concreta que actúe: por ejemplo, la tripsina corta en el lado carboxilato de aminoácidos adyacentes a residuos básicos como lo son la lisina y la arginina, mientras que la quimiotripsina lo hace junto a residuos no polares y aromáticos, como la tirosina, metionina o fenilalanina.
Figura 2: Funcionamiento serín proteasas (creado a partir de BioRender)
OPTIMIZACIÓN
Esta enzima tiene un rango de pH óptimo entre el 7 y el 8, aunque más concretamente es de 7,7. Asimismo, actúa con una temperatura óptima de 37 grados Celsius. De hecho, a unos 25º la actividad de la tripsina es 1.87 menor que a la temperatura óptima. Por lo tanto, es una reacción favorable en cuanto a la termodinámica, pero como tiene una energía de activación muy alta, cinemáticamente hablando es desfavorable.
Un efector en la actividad de la proteasa está relacionada con el calcio que estabiliza la estructura terciaria, ya que la tripsina tiene un lugar de unión al calcio. Este hace que no se formen agregados de tripsina, favoreciendo la actividad de esta.
MECANISMO: LA TRIADA CATALÍTICA
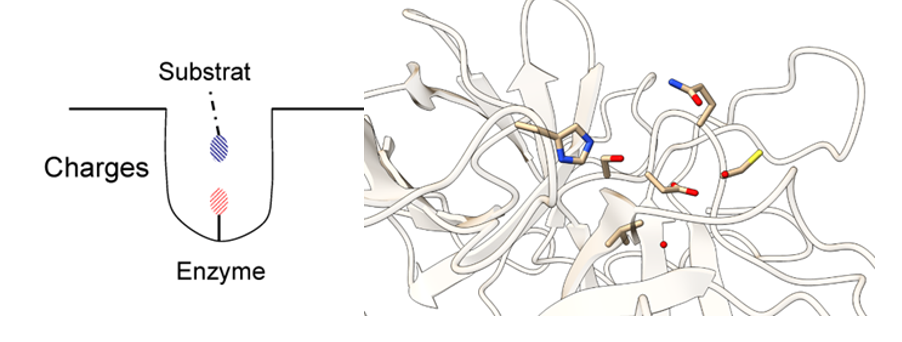
Para realizar su función, la tripsina y las enzimas hidrolasas en general poseen una triada catalítica en su centro activo, que son residuos de tres aminoácidos que funcionan en conjunto en la grieta catalítica del sitio activo para poder llevar a cabo la catálisis.
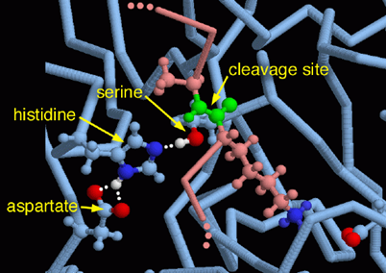
En el caso de la tripsina, esta triada está constituida por serina, histidina y aspartato. Además, posee un “bolsillo” (Substrate-binding pocket) cerca del centro activo formado por un carboxilo de la cadena lateral de un ácido aspártico. Este bolsillo tiene una fuerte relación con lo ya mencionado anteriormente, puesto que si la tripsina tiene afinidad por cortes de aminoácidos básicos (carga positiva), el hecho de que el residuo del bolsillo sea ácido hace que ambas cargas (la positiva de la base y la negativa del ácido) interaccionen electrostáticamente.
Figura 3: Centro activo de la tripsina. A la izquierda se aprecia el Substrate-binding pocket, imagen sacada de Sjøli, Stian. (2011). Kinetic and docking studies of inhibitors targeting the catalytic zinc in MA clan enzymes. La figura de la derecha muestra la triada catalítica de la tripsina vista en Chimera X a partir de PDB-101 entry: 2PTC
La conservación de esta triada permite la clonación de la secuencia consenso donde los oligonucleótidos degenerados se utilizan en una cadena de reacciones del material genético de la polimerasa para poder reconocer tripsinas. De hecho, la tripsina se ha utilizado para observar la especificidad de sustrato entre las serinas proteasas.
Una vez explicado los componentes esenciales para la catálisis vamos a centrarnos en las interacciones que tienen lugar en la triada para generar un entorno favorable que promueva la ruptura de los péptidos.
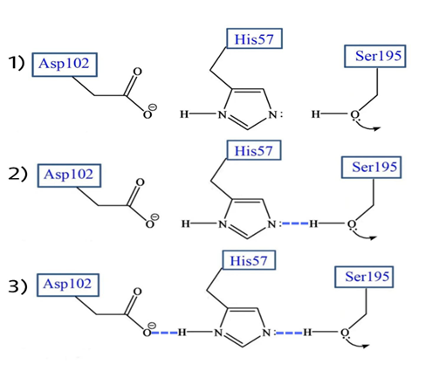
La serina195 es un aminoácido muy reactivo, no esencialmente por su naturaleza sino por el entorno que le rodea que le confiere la alta reactividad. En primer lugar, el grupo hidroxilo (OH) de la serina establece un puente de hidrógeno con el anillo imidazol de la histidina. El aspartato, que se encuentra en el lado contrario del anillo de imidazol, establece una unión entre su carga negativa y el hidrógeno del anillo.
Figura 4: Modo de actuación de la triada catalítica por pasos. (De elaboración propia)
La histidina por tanto polariza el grupo OH de la serina y acepta su protón, dejando al Oxígeno con una carga negativa que incrementa la capacidad nucleofílica de la serina. El nitrógeno de la Histidina queda cargado positivamente y será el aspartato que con su carga negativa estabilice dicha carga para hacer de la histidina un mejor aceptor de protones.
Figura 5: Centro activo de la tripsina. Imagen sacada de PDB-101 doi:10.2210/rcsb_pdb/mom_2003_10
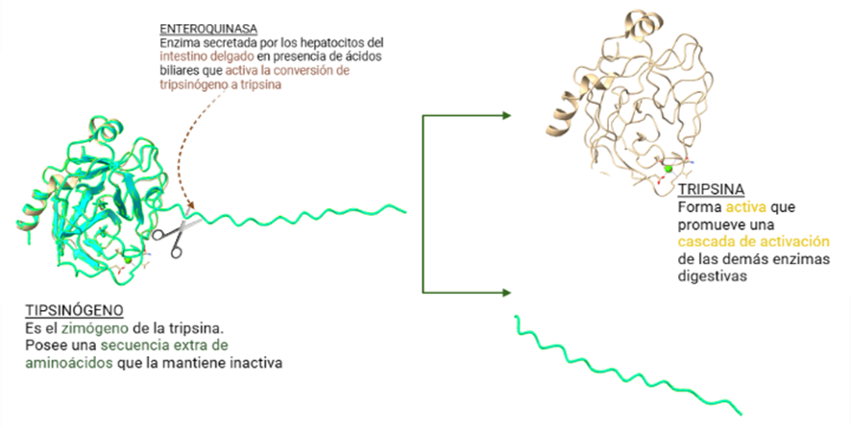
SISTEMA DE REGULACIÓN: EL TRIPSINÓGENO
Sin embargo, todo el mecanismo que envuelve a las enzimas digestivas, entre las que se encuentra la tripsina, no está siempre en funcionamiento, sino que existen unas formas inactivas llamadas zimógenos que necesitan ser activadas. Las tripsinas se secretan en forma de tripsinógeno ya que es una forma de protección para intentar prevenir una actividad prematura del páncreas, pudiendo desencadenar una reacción inflamatoria que provoca pancreatitis. El tripsinógeno es formado en los aparatos de Golgi, empaquetados en forma de zimógenos granulados, y secretados por exocitosis.
Figura 6: Modo de actuación del tripsinógeno. (Figura realizada con BioRender)
Por otra parte, en estos gránulos también podemos encontrar inhibidores de tripsina secretoras pancreáticas (PSTI) para la inactivación de algunas tripsinas activas. Además, el tripsinógeno puede ser también activado por catepsinas (que pueden intervenir en la pancreatitis aguda), proteasas lisosomales que se activan a pH bajo. Sin embargo, estas no se encuentran en los mismos compartimentos de la célula. El mecanismo catalítico es un ataque nucleofílico.
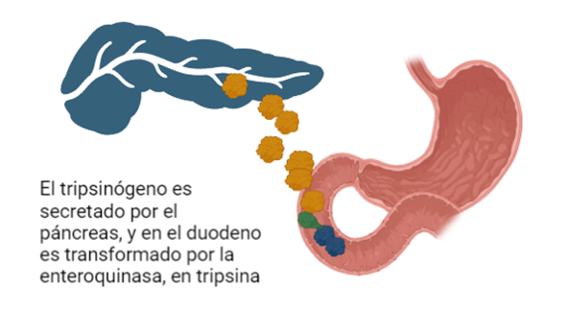
En el paso desde la forma inactiva hacia la activa actúa otra serín proteasa, la enteroquinasa (también conocida como enteropeptidasa), cuya localización se encuentra en el duodeno y que es secretada por los enterocitos una vez la comida ingerida sale del estómago. La enteropeptidasa convierte al tripsinógeno en su forma activa la tripsina, provocando la activación en cascada de las enzimas digestivas pancreáticas.
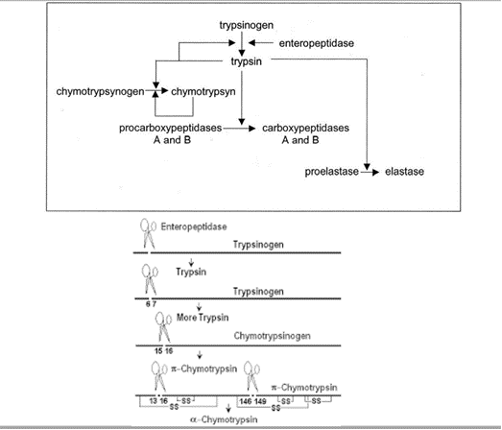
Figura 7: Proceso por el cual el tripsinógeno pasa a tripsina gracias a la actuación de la enteroquinasa. (Creando con BioRender y Chimera X a partir de PDB-101 entry:2ptc y 1tgs)Figura 8: Cascada de activación de enzimas digestivas. Imagen creada por el Dr. César Ángel Menor Salvan
INHIBIDORES
Los inhibidores son unas proteínas, (y un tipo de inhibidor serina) que reducen la actividad de la tripsina, controlando las reacciones. Son inhibidores irreversibles y suicidas. Usados principalmente como “controladores de fallos” para evitar que el tripsinógeno se convierta en tripsina antes de su secreción. Sin embargo, también pueden ser considerados negativos en el caso en el que intervenga en la digestión cuando está funcionando correctamente.
Estos inhibidores los podemos encontrar en muchos tipos de alimentos, como patatas, judías, cacahuetes, maíz dulce, cereales y soja. Asimismo, también se han encontrado en la leche materna humana que ayuda siendo un protector del intestino para los bebés. Ha sido comprobado que el calor puede reducir la actividad de los inhibidores, sobre todo en la soja y productos del estilo. Por ejemplo, hirviendo la soja durante 14 minutos, inactivas el 80 % de los inhibidores y durante 30, el 90%.
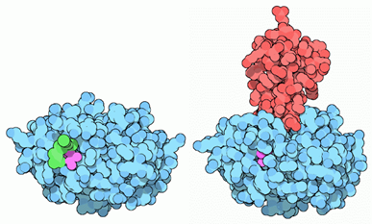
Figura 9: Tripsinógeno (izquierda), tripsina con inhibidor proteico (derecha, rojo). Este inhibidor es generado por el páncreas e inactiva los posibles restos de tripsina en el intestino una vez ha acabado su función. Imagen sacada de PDB-101 doi:10.2210/rcsb_pdb/mom_2003_10
Como se ha mencionado previamente, la tripsina tiene inhibidores de su actividad (PSTI). En cambio, es posible que la actividad de la tripsina exceda la capacidad de los inhibidores, provocando que se desencadene una cascada de reacciones en la que se activan proteasas que puedan provocar daños en las células ya que activan el receptor 2 (PAR-2), haciendo que se sinteticen citoquinas y la regulación de la exocitosis a través de un feedback negativo.
Mediante estudios, han descubierto la importancia de la formación del complejo con la tripsina mediante unos experimentos, donde observaron que los inhibidores modificados reaccionaban mucho más lento que los “normales”.
Algunos usos de estos inhibidores son:
Como ya se ha comentado, la tripsina se usa en los cultivos y en la preparación de muestras, pues los inhibidores se encargan de que la tripsina no destroce más de lo que es necesario, las células y proteínas que se van a observar.
Los inhibidores vegetales se usan para repeler insectos, ya que provoca que el intestino de estos no funcione correctamente.
UTILIDAD EN BIOMEDICINA
En los inicios de la proteómica se usaba para separar proteínas tras un proceso de electroforesis en gel, para la secuenciación de péptidos y la identificación por espectrometría de masas. Que se pueda hacer este proceso se debe a que las tripsinas no les afecta el inhibidor fenilalanilclorometilcetona (TPCK). Esto se tiene en cuenta por la importancia de la especificidad del corte.
Actualmente la tripsina se usa en tecnologías de los alimentos, sigue utilizándose en el análisis proteómico, en la preparación de cultivos (la tripsinización, que es un proceso en el que se usa la tripsina tanto para la proteólisis en la que se separan las células adherentes del recipiente, como para pasarlas a otros), separación de tejidos en células constituyentes. Es un modulador de la capacidad alergénica de la soja y de la producción de péptidos antihipertensivos, junto a ser un identificador de enfermedades como la pancreatitis, la fibrosis quística, el cáncer… Estas enfermedades originan malabsorción, es decir, cuando hay una disminución de la producción de tripsinas, y como consecuencia, la incapacidad para digerir y absorber los nutrientes correctamente.
Por otra parte, científicos han descubierto que la tripsina puede estar asociada al Alzheimer y con las proteínas de larga duración, ya que estas al tener más “vida”, es más probable que surja una isomerización de la proteína, originando que sea más difícil de digerir, pues las enzimas digestivas (como la tripsina) tienen una tolerancia relativa a los isómeros, provocando problemas que pueden derivar al Alzheimer.
La tripsina a su vez está relacionada con la coagulación de la sangre. Por eso, ayuda en la eliminación de tejido muerto para que cicatricen mejor las heridas (desbridamiento), esto puede deberse a que degrada las proteínas del tejido. También ha habido muchos estudios que parecen indicar que la tripsina puede ser utilizada en lesiones traumáticas para reducir edema.
APLICACIONES DE LA TRIPSINA
Las enzimas son imprescindibles en una gran cantidad de industrias como la agrícola, alimentaria, farmacéutica, biomédica… Esto conlleva a que surja la necesidad de disponer de dichas enzimas. Es por esto por lo que la tripsina es utilizada para una enorme variedad de investigaciones relacionadas con este tipo de industrias.
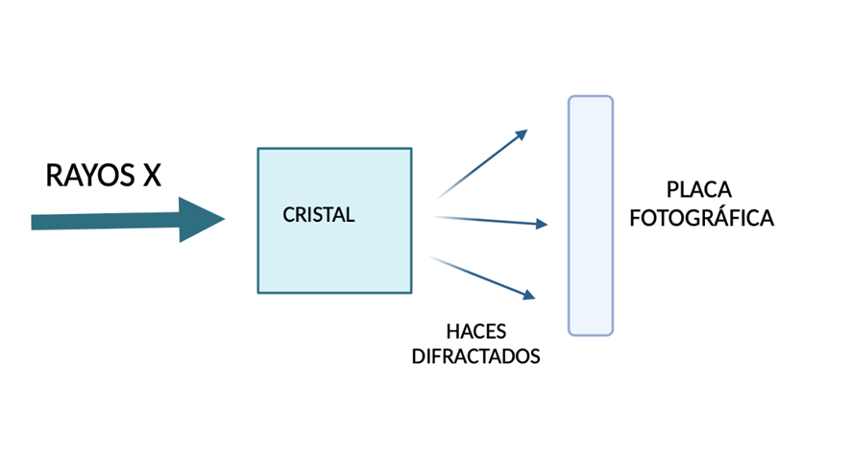
Actualmente, se está estudiando la aplicación de la tripsina en varios ámbitos entre los que destaca la aplicación biomédica en la difracción de Rayos x en cuanto a complejos proteína-inhibidor. Esta enzima tiene un gran interés gracias a las similitudes que presenta con otras enzimas de la misma familia y que la haría útil para la sustitución de enzimas como el Factor X (factor de coagulación). Gracias a esto, también se ha podido estudiar su unión a un inhibidor que proviene de las semillas de girasol.
Figura 10: Difracción de rayos X. (De elaboración propia)
Asimismo, se han investigado algunas aplicaciones que puede presentar en la agricultura, participando como inhibidor en la soya y el garbando, siendo mucho más efectiva en la soya ya que ha sido capaz de modificar y aumentar la presencia de láminas beta plegadas, bucles, alfa hélices y cadenas no ordenadas además de reducir el número de estructuras agregadas de esta misma llevando a la modificación de su perfil electroforético.
Campo de girasoles. (De elaboración propia)
Por otro lado, la tripsina ha sido estudio de la tesis de Elena Sofia Escobar Barrera de la Universidad nacional abierta y a distancia. Dicha tesis esta principalmente basada en la creación de tratamientos para destruir o impedir la aparición del herpesvirus-bovino tipo I en programas de transferencia de embriones. Dicho virus es capaz de unirse a los espermatozoides causando patologías a los futuros embriones. Existen varias formas de prevención, siendo la tripsina una solución enzimática que actúa como desinfectante. Aunque presentaba ciertos inconvenientes como el desarrollo de otras patologías, para evitar que pudiese causar otro tipo de infecciones en los embriones se llevó a cabo el desarrollo de una tripsina recombinante (obtenida del maíz).
Y con esto vamos terminando para que no sea muy difícil de digerir toda esta información.
REFERENCIAS
-Asociación Americana de Química Clínica. Malabsorción. Actualizado el 11 de noviembre de 2019. -Aviles Gaxiola, S. Efecto de agentes reductores y/o procesamiento térmico sobre la actividad de inhibido de tripsina y solubilidad de concentrados proteicos de soya o garbanzo. -Cohen, Maja; Davydov, Olga; Fluhr, Robert (2019-02-05). «Plant serpin protease inhibitors: specificity and duality of function». Journal of Experimental Botany. Society for Experimental Biology (OUP). 70 (7): 2077–2085. doi:10.1093/jxb/ery460. ISSN 0022-0957. doi:10.2210/rcsb_pdb/mom_2003_10 -Escobar Barrera, E. S. Eficiencia del uso de la Tripsina como tratamiento para combatir el Herspes virus Bovino-1 en programas de transferencia de embriones. -Hirota, M., Ohmuraya, M., & Baba, H. (2006). The role of trypsin, trypsin inhibitor, and trypsin receptor in the onset and aggravation of pancreatitis. Journal of gastroenterology, 41(9), 832-836. -Hirota, M., Ohmuraya, M., & Baba, H. (2006). The role of trypsin, trypsin inhibitor, and trypsin receptor in the onset and aggravation of pancreatitis. Journal of gastroenterology, 41(9), 832-836. -Huang, H. L.; Hsing, H. W.; Lai, T. C.; Chen, Y. W.; Lee, T. R.; Chan, H. T.; Lyu, P. C.; Wu, C. L.; Lu, Y. C.; Lin, S. T.; Lin, C. W.; Lai, C. H.; Chang, H. T.; Chou, H. C.; Chan, H. L. (2010). «Trypsin-induced proteome alteration during cell subculture in mammalian cells». Journal of Biomedical Science. 17 (1): 36. doi:10.1186/1423-0127-17-36. PMC 2873939. PMID 20459778. -Ito C, Yamaguchi K, Shibutani Y, et al. Acciones antiinflamatorias de proteasas, bromelina, tripsina y su preparación mixta. Nihon Yakurigaku Zasshi. 1979; 75 (3): 227-237. -John A. Williams, in Encyclopedia of Gastroenterology, 2004.Kadam, S.S., and Smithard, R.R. (1987). «Effects of heat treatments on trypsin inhibitor and hemagglutinating activities in winged bean». Plants Foods for Human Nutrition. 37 (2): 151–159. doi:10.1007/BF01092051. S2CID 84193551. -Kaur, J., & Singh, P. K. (2020). Trypsin detection strategies: A review. Critical Reviews in Analytical Chemistry, 1-19. -Kühne, Wilhelm (1877). «Über das Verhalten verschiedener organisirter und sog. ungeformter Fermente» [On the behavior of various organized and so-called unformed ferments (Sobre el funcionamiento de varios fermentos organizados, considerados aún no formados)]. Verhandlungen des naturhistorisch-medicinischen Vereins zu Heidelberg. Neue Folge [nueva serie] (en alemán) (Heidelberg) 1: 194-198. -Liu, KeShun (2012-12-06). Soybeans: Chemistry, Technology, and Utilization. Springer. ISBN 978-1-4615-1763-4. -Ma W, Tang C, Lai L. Especificidad de tripsina y quimotripsina: correlación dinámica controlada por el movimiento del bucle como determinante. Biophys J. 2005; 89 (2): 1183-93. doi: 10.1529 / biophysj.104.057158 -Mathews, C. K.; Van Holde, K.E et Ahern, K.G (2003). Bioquímica (3 edición) -Miller PC, Bailey SP, Barnes ME, Derr SJ, Hall EE. Los efectos de la suplementación con proteasa en la función del músculo esquelético y DOMS después de correr cuesta abajo. J Sports Sci. 2004; 22 (4): 365-72. doi: 10.1080 / 02640410310001641584October 2003, David Goodsell -Olsen, Jesper V.; Ong, Shao-En; Mann, Matthias (2004-06). «Trypsin Cleaves Exclusively C-terminal to Arginine and Lysine Residues». Molecular & Cellular Proteomics (en inglés) 3 (6): 608-614. doi:10.1074/mcp.T400003-MCP200. Consultado el 6 de septiembre de 2021. -Ozawa, K., & Laskowski Jr, M. (1966). The reactive site of trypsin inhibitors. Journal of Biological Chemistry, 241(17), 3955-3961.Patricia JJ, Dhamoon AS. Fisiología, digestión. [Actualizado el 7 de julio de 2019]. En: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 enero. -Rick, W. (1974). Trypsin. In Methods of enzymatic analysis (pp. 1013-1024). Academic Press. -Santiago García, R. (2000). Inhibidores de serinproteasas: estudios estructurales de complejos tripsina-inhibidor. -Shah D, Mital K. El papel de la tripsina: quimotripsina en la reparación de tejidos. Avances en terapia. 2018; 35 (1): 31-42. doi: 10.1007 / s12325-017-0648-a -Shaw PC. El uso de una formulación de tripsina-quimotripsina en fracturas de la mano. Br J Clin Pract. 1969 1 de enero; 23 (1): 25-6. Silverman, Gary A.; Bird, Phillip I.; Carrell, Robin W.; Church, Frank C.; Coughlin, Paul B.; Gettins, Peter G.W.; Irving, James A; Lomas, David A.; Luke, Cliff J.; Moyer, Richard W.; Pemberton, Philip A.; Remold-O’Donnell, Eileen; Salvesen, Guy S.; Travis, James; Whisstock, James C. (2001). «The Serpins Are an Expanding Superfamily of Structurally Similar but Functionally Diverse Proteins». Journal of Biological Chemistry. American Society for Biochemistry and Molecular Biology (Elsevier). 276 (36): 33293–33296. doi:10.1074/jbc.r100016200. ISSN 0021-9258. PMID 11435447. S2CID 18684515. -Silzel, J. W., Ben-Nissan, G., Tang, J., Sharon, M., & Julian, R. R. (2022). Influence of Asp Isomerization on Trypsin and Trypsin-like Proteolysis. Analytical Chemistry, 94(44), 15288-15296. -Sistema de salud de Nemours Children. Estudio observacional prospectivo a largo plazo de la incidencia de la colonopatía fibrosante. -Smith, C., Van Megen, W., Twaalfhoven, L., & Hitchcock, C. (1980). The determination of trypsin inhibitor levels in foodstuffs. Journal of the Science of Food and Agriculture, 31(4), 341-350. -Trypsin (Huber, R., & Bode, W. (1978). Structural basis of the activation and action of trypsin. -Wirnt, R. (1965). Trypsin. In Methods of enzymatic analysis (pp. 807-818). Academic Press.
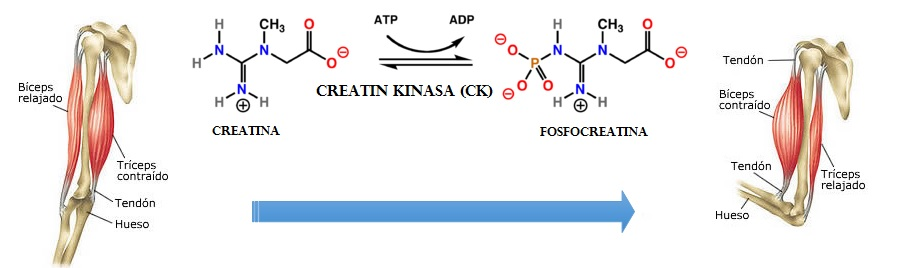
La fosfoglicerato mutasa, ¿de la glucólisis a la intolerancia al ejercicio físico?
escrito por elizabeth.cairos_1A | 20 febrero, 2023
Elizabeth Cairós Escobar. Biología Sanitaria, UAH.
INTRODUCCIÓN
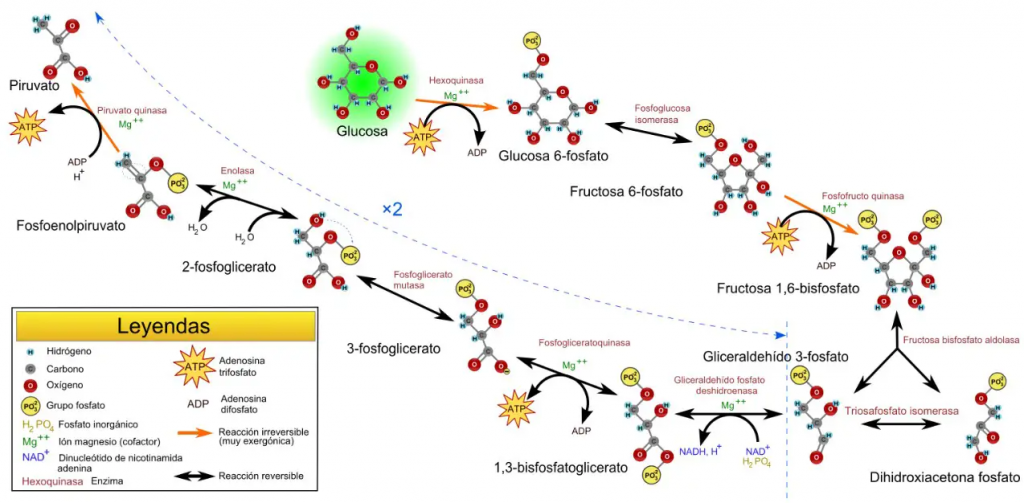
La fosfoglicerato mutasa (PGM) se trata de una enzima que interviene en el metabolismo de carbohidratos. Este comprende, dentro de los mecanismos de degradación (es decir, catabolismo), los procesos de la glucólisis y la glucogenólisis, que destacan como los principales, aunque la enzima metabólica que vamos a tratar hace presencia en el primero de los dos procesos mencionados. Por esta razón, ¿sabemos lo qué es la glucólisis?
La glucólisis se trata de un proceso metabólico, que cuenta con diez etapas, en el que se transforman moléculas de glucosa en piruvato. Forma parte del catabolismo central de la glucosa y, como resultado, se obtienen dos moléculas de piruvato por cada molécula de glucosa transformada.
Del mismo modo, este proceso genera, de manera anaerobia, dos moléculas de energía en forma ATP, a partir de dos de ADP, y dos moléculas de poder reductor en forma de NADH, a partir de dos de NAD+.
Concretamente, la PGM cataliza el octavo paso de este proceso.
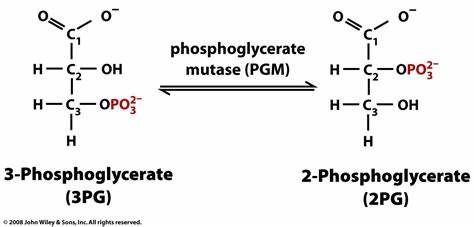
Como se indicó en el apartado introductorio, la fosfoglicerato mutasa es una enzima presente en el proceso metabólico de la glucólisis y es la encargada de la conversión de moléculas de 3-fosfoglicerato (3PG) en moléculas de 2-fosfoglicerato (2PG). Además, es importante señalar que esta transformación se lleva a cabo mediante un intermediario metabólico, que se trata del 2,3-bisfosfoglicerato.
Pero, ¿cuál es el verdadero funcionamiento de la PGM y cómo ocurre la reacción?
Si hablamos de manera amplia y general, la principal función de esta enzima es desplazar el fosfato hacia un lugar clave en el centro de la molécula de 3PG, con la finalidad de contribuir al proceso final de obtención de energía que, recordando, se da en forma de ATP. De igual forma, incidir en que esta translocación ocurre, concretamente, del tercer carbono de la molécula al segundo, y da como resultado la formación de 2-fosfoglicerato.
Sin embargo, si se profundiza más en el funcionamiento de la PGM, el proceso de conversión del que se encarga se lleva a cabo mediante la isomerización irreversible del 3PG y la intervención de un cofactor que, en este caso, se trata del Mg+2. Asimismo, esta reacción de conversión ocurre en dos etapas.
Primeramente, la fosfoglicerato mutasa, que ha sido previamente fosforilada en un sobrante del aminoácido histidina, cede dicho fosfato al grupo hidroxilo (OH–) del carbono en la posición 2 del 3PG. De esta manera, se genera el 2,3-bisfosfoglicerato (el intermediario metabólico de la reacción).
Seguidamente, en el segundo paso de la conversión, la molécula de 2,3-bisfosfoglicerato le transfiere a la enzima su fosfato del carbono 3, obteniéndose como resultado tanto la liberación de la PGM fosforilada de nuevo como el producto final de la reacción que esta enzima cataliza (la molécula de 2-fosfoglicerato).
Por otro lado, y como información adicional, ya una vez la fosfoglicerato mutasa sea poseedora del fosfato adicional (el cedido por la molécula de 2,3-bisfosfoglicerato), esta queda activa por un breve periodo de tiempo, de apenas dos minutos. Durante este corto intervalo realiza su función en repetidas ocasiones antes de que pierda el fosfato y, por consecuente, tenga que ser suministrado de nuevo
ESTRUCTURA DE LA ENZIMA
La estructura de la PGM no es común a todos los organismos (depende de la especie a la que se esté haciendo referencia), ya que puede adoptar formas variables y diferir en el número de subunidades que la forman. Estas subunidades pueden ser del tipo M o B y cuentan con una localización concreta en los tejidos, aunque puede variar durante el desarrollo del individuo.
En el caso de los humanos, la enzima fosfoglicerato mutasa está formada por dos subunidades idénticas, pero se puede observar en las imágenes inferiores que en otros seres vivos esto no es así.
Estructura cuaternaria de fosfoglicerato mutasa de levadura. Imagen realizada con ChimeraX (código de la proteína: 3PGM).
Hidrofobicidad de aminoácidos de fosfoglicerato mutasa de levadura. Imagen realizada con ChimeraX (código de la proteína: 3PGM).
*La fosfoglicerato mutasa de levadura (PDB 3PGM), al igual que la de humanos, está compuesta por subunidades idénticas, aunque en el primero de los casos cuenta con 4 subunidades.
Aún así, la PGM en el filo de los mamíferos cuenta con 3 isoenzimas (enzimas que son diferentes en cuanto a su secuencia de aminoácidos, pero que catalizan la misma reacción), cuyo origen recae en las diferentes maneras de unión de las subunidades de la enzima.
Por otro lado, para su funcionamiento, esta enzima, en las células humanas, utiliza una molécula especial del aminoácido histidina que es el que, como se señaló anteriormente, coloca el fosfato, después de haber sido extraído, en el lugar específico de la enzima.
No obstante, la fosfoglicerato mutasa hace, en realidad, la acción contraria a la histidina, ya que incorpora un fosfato a la molécula de 3PG (para que se una al otro que ya posee dicha molécula) y luego se encarga de quitarlo.
Como es lógico, para añadir dicho fosfato la PGM primero tiene que hacerse con él, por lo que la 2,3-bisfosfoglicerato es la molécula intermedia encargada de suministrárselo.
Asimismo, la PGM posee un átomo de Mg+2 como cofactor.
IMPORTANCIA DE LA PGM EN CUANTO AL EJERCICIO FÍSICO
Hasta ahora se ha hecho referencia a la fosfoglicerato mutasa y a su implicación en el metabolismo de azúcares, más concretamente en la glucólisis, mas ¿cuál es su relación con la actividad física?
Partiremos de la base de que, si se dan déficits en enzimas relacionadas con el metabolismo lipídico y de azúcares a causa de la herencia de enfermedades genéticas autosomales recesivas, se pueden desarrollar enfermedades conocidas como miopatías metabólicas.
Las miopatías metabólicas conllevan la manifestación de frecuentes crisis de mialgias (conocidas popularmente como dolores musculares), calambres y rigidez en los músculos. Este último síntoma se traduce en una difícil contracción muscular, causada por la disminución energética tras la reducción del trifosfato de adenosina (TFA) o adenosín trifosfato (ATP), un nucleótido imprescindible en la obtención de energía en las células, mediante la fosforilación oxidativa en la mitocondria.
Los déficits enzimáticos que causan estas miopatías metabólicas pueden deberse a fallos en el metabolismo de carbohidratos (normalmente producidos por ejercicio físico en periodos menores a 10 minutos y de elevada intensidad) o por trastornos en el metabolismo de lípidos (actividad física por más de 10 minutos de tiempo y de baja intensidad). Con esto, pondremos el foco de atención en los déficits enzimáticos causados por el primer tipo de fallos, ya que, como se ha visto a lo largo de toda esta entrada, la fosfoglicerato mutasa participa en la catálisis metabólica de azúcares.
Así, se tiene que los déficits enzimáticos a los que hacemos referencia pueden ser causados por la deficiencia de miofosforilasa (glucogenosis tipo V), fosfofructocinasa muscular (glucogenosis tipo VII), beta enolasa (glucogenosis tipo XIII), fosfoglicerato quinasa (glucogenosis tipo IX), fosfoglicerato mutasa (glucogenosis X) y/o lactato deshidrogenasa (glucogenosis tipo XI).
Generalmente, las miopatías causadas por estas enzimas comienzan a manifestarse en el periodo de la adolescencia y, en todas ellas, las concentraciones de creatina cinasa (CK), una sustancia que libera el cuerpo cuando se somete a un estrés físico considerable, varían y se elevan.
Por otro lado, y centrándonos exclusivamente en la fosfoglicerato mutasa, la deficiencia de esta enzima causa la que se denomina Enfermedad de almacenamiento de glucógeno (o glucogenosis tipo X). Se trata de una enfermedad no demasiado común que causa calambres musculares debido al ejercicio, la mioglobinuria (excreción de mioglobina en la orina a causa de una mioglobinemia procedente de la destrucción de la musculatura estriada, según el Diccionario Médico de la Clínica Universidad de Navarra) y la presencia de agregados tubulares durante biopsias en el músculo.
Estudios de investigación han concluido que en esta patología aumentan considerablemente los niveles de CK (y es que, en episodios de mioglobinuria aguda, los valores de la CK pueden elevarse hasta 200 veces más de los parámetros normales) y, por el contrario, no lo hacen los niveles de lactato.
A día de hoy no existe tratamiento farmacológico para esta enfermedad, sino que simplemente se trata con una buena hidratación, la toma frecuente de glucosa y la alcalinización de la orina. No obstante, hay una serie de medidas recomendadas para prevenir las crisis de calambres musculares, como evitar el ejercicio físico que las produzca y el ayuno, ya que ambos factores predisponen a su aparición.
Finalmente, el pronóstico de esta enfermedad es, por norma general, bueno (hay casos excepcionales en los que se produce una insuficiencia renal aguda por el aumento de mioglobina en sangre a causa de una rabdomiólisis – rotura o descomposición del tejido muscular – grave), aunque se recomienda el estudio de los polimorfismos de esta enzima para seguir recabando información sobre ella y permitir el avance de futuras investigaciones.
Además de la implicación de la PGM en las miopatías metabólicas, también hay determinados estudios que han investigado las alteraciones de esta enzima y sus isoenzimas en tumores de distintos órganos.
A modo de breve resumen, estos estudios han determinado un descenso en la actividad de la fosfoglicerato mutasa en los tumores cerebrales y un incremento de la misma en los otros tipos de quistes.
[2] Escribano, M. Z., & Soria Aznar, M. (s/f). Trabajo Fin de Grado POLIMORFISMOS GENÉTICOS EN EL METABOLISMO GLUCOLÍTICO RELACIONADOS CON LA FATIGA CANDIDATOS A SER ESTUDIADOS EN UNA MUESTRA DE DEPORTISTAS DE ÉLITE. Unizar.es. Recuperado el 29 de enero de 2023, de https://zaguan.unizar.es/record/57619/files/TAZ-TFG-2016-1007.pdf
[3] INSERM US14 — TODOS LOS DERECHOS RESERVADOS. (s/f). Orphanet: Enfermedad de almacenamiento de gluc�geno por deficiencia de fosfoglicerato mutasa. Orpha.net. Recuperado el 29 de enero de 2023, de https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=ES&Expert=97234
[4] Mioglobinuria. (s/f). Cun.es. Recuperado el 29 de enero de 2023, de https://www.cun.es/diccionario-medico/terminos/mioglobinuria
[5] Molecule of the month: Glycolytic enzymes. (s/f). RCSB: PDB-101. Recuperado el 29 de enero de 2023, de https://pdb101.rcsb.org/motm/50
[6] Papazian, O., & Rivas Chacón, R. (2013). Miopatías metabólicas. Revista de neurologia, 57(S01), 65. https://doi.org/10.33588/rn.57s01.2013257
[7] Pich, N. D. (1997). Actividad e isoenzimas de la fosfoglicerato mutasa. Alteraciones tisulares en los tumores humanos. Alteraciones sericas en el infarto de miocardio. Universitat de Barcelona.
[8] Zerón, H. M., Morales, P. V., Salgado Alday, M. E., Mendieta, H., & Felipe, Z. (s/f). Mioglobinuria. Reporte de un caso y revisión de la literatura. Core.ac.uk. Recuperado el 29 de enero de 2023, de https://core.ac.uk/download/pdf/161648906.pdf
[9] (S/f). Azucareraprofesionales.es. Recuperado el 29 de enero de 2023, de https://www.azucareraprofesionales.es/wp-content/uploads/2018/04/Libro-Blanco-del-Azucar-Indice-Interactivo.pdf
ACE2: UN ALIADO DEL COVID
escrito por beatriz.sendra-1d | 20 febrero, 2023
Por Beatriz Sendra y Nerea Villegas, Biología Sanitaria UAH.
1. INTRODUCCIÓN
Figura 1. Proteína ACE2 con átomo de Zinc del centro activo rodeado de verde. Imagen creada con ChimeraX, con el código PDB: 1R42.
La ACE2 o enzima convertidora de angiotensina-2, es una carboxipeptidasa dependiente de zinc que se expresa en la membrana (Figura 1). Esta enzima tiene como principal función la participación en el sistema de renina-angiotensina, inhibiendo la angiotensina II por vía zimógeno que ha sido previamente activada por la ACE, provocando entre otras cosas, la bajada presión arterial. (24)
Se trata de una proteína que se encuentra integrada en la membrana plasmática (22) y se expresa en numerosos tipos celulares, entre los que cabe destacan los endotelios del corazón y vasos sanguíneos (sistema cardiovascular), las células epiteliales de los pulmones y la superficie de las células del riñón (9, 17, 27). Esta amplia distribución puede ser causa de la gran especificidad de sustrato que tiene la ACE2 y le permite participar en muchas rutas peptídicas (24).
Además de su función en el sistema de renina-angiotensina, la ACE2 funciona también como facilitador del transporte de aminoácidos y tiene un papel importante en la recepción del síndrome respiratorio agudo severo (SARS-CoV-2).
El descubrimiento de la ACE2 ha sido reciente, hace apenas 20 años y fue posterior al descubrimiento de la ACE, a pesar de la gran correlación que existe en la actividad de ambas en el sistema de renina-angiotensina (6, 24). La ACE2 fue el primer homólogo de la ACE (4, 22). (Figura 2)
Figura 2. Similitudes y diferencias entre dominios de ACE y ACE2. (20)
Este trabajo se centra en las funciones del ACE2 y en la relevancia que tiene en relación al COVID. Para ello, se ha estructurado en 5 grandes secciones. La Sección 2 está dedicada a la estructura y mecanismos de la catálisis de la ACE2. La Sección 3 se centra en las principales funciones de esta proteína. Seguidamente, en la Sección 4 se comentan las implicaciones biomédicas fundamentales. En la Sección 5 se resumen algunas de las conclusiones más importantes. Se termina el trabajo con la relación de referencias que se han consultado, y en las que esencialmente nos hemos basado, para el desarrollo del contenido.
2. ESTRUCTURA Y MECANISMOS
Figura 3. Proteína ACE2 con dominios de diferente colores. Imagen creada con ChimeraX, con el código PDB: 1R42.
Como ya se ha comentado en la introducción, la ACE2 es una proteína transmembrana que atraviesa una sola vez la membrana (unipaso), y que está compuesta por 805 residuos de aminoácidos. Tiene tres dominios principales: extracelular, transmembrana e intracelular. El dominio extracelular esta formados por dos dominios: el dominio metalopeptidasa y el dominio de colectrina. (Figura 3)
El dominio metalopeptidasa se localiza en el extremo N-terminal y está formada principalmente por estructuras de alfa-hélice, aunque también contiene algunas beta-láminas. Además, es el que más glicosilaciones sufre durante su postraducción. Es en este dominio donde se une el cofactor Zn, el cual participa en la catálisis de la proteína. Los residuos de unión a este cofactor forman la secuencia consenso de la proteínas y se corresponde con HEXXH siendo H histonas, E glutamina y X cualquier aminoácido con carga. Esta secuencia consenso es común en todas las metalopeptidasa de la familia M2. (23)
Por otro lado, encontramos el dominio colectrina más cercano al extremo C-terminal el cual está compuesto por beta-láminas. Es en este dominio donde se encuentra el sitio de unión del virus SARS-CoV-2. Además, seguido de él se encuentra el dominio transmembrana constituido mayoritariamente por estructura de alfa-hélice. Este dominio transmembrana es el que menos residuos abarca de todos pues únicamente los aminoácidos situados entre las posiciones 741-761 atraviesan la membrana. (2, 12, 14) (Figura4)
El último dominio, el dominio intracelular, se encuentra en el extremo C-terminal y contiene varios motivos lineales, como motivos de señal que interaccionan con proteínas con importante participación en vías de endocitosis y autofagia. (2)
Figura 4. Dominios de la ACE2. (2)
El centro activo de esta proteína se encuentra en el dominio extracelular metalopeptidasa y es en él donde se encuentra el sitio de unión del zinc. (Figura 5)
Figura 5. Centro activo de ACE2 y la unión de su cofactor de Zinc al centro activo. Imagen creada con ChimeraX, con el código PDB: 1R42.
Las principales modificaciones postraduccionales son las N-glicosilaciones en las asparaginas. Una de esas glicosilaciones, en concreto la de la asparagina 90, puede reducir la infección por SARS-CoV-2. Además, para mantener estable la estructura, se forman puentes disulfuro entre cisteínas. (14)
En cuanto a las isoformas, la ACE 2 tiene dos. Una de ellas no es funcional como carboxipeptidasa mientras que la otra no es funcional como receptor del coronavirus.
La ACE2 presenta una actividad catalítica que se caracteriza por tener un pH de trabajo óptimo de 6,5 y además ser potenciado por la presencia de aniones monovalentes, lo que la relaciona con la actividad de la ACE. Los aniones monovalentes que potencian la actividad de la ACE2 son el anión cloruro(Cl-)y fluoruro (F-), ambos aumentan la actividad de la ACE2 como unas 10 veces. En cambio, existen otros aniones monovalentes como el anión bromuro, que no afectan a la actividad de la proteína.
La actividad proteolítica ya mencionada consiste únicamente en la eliminación del extremo C-terminal (residuo), y es por ello que se dice que es una carboxipeptidasa. La actividad proteolítica de la hidrólisis de péptidos no tiene la misma eficiencia catalítica con todos los péptidos por igual. Uno de los sustratos con los que tiene más afinidad es con la angiotensina II, integrante del sistema de renina-angiotensina. (25)
3. FUNCIONES
La ACE2 realiza una gran variedad de funciones. Sin embargo, las más importantes se pueden resumir en tres : la participación en el sistema renina-angiotensina; la recepción del SARS-Cov2 y el transporte de aminoácidos.
En primer lugar, la ACE2 tiene como principal función su participación en el sistema renina-angiotensina. Se trata de un conjunto de reacciones llevadas a cabo en la sangre que tienen una relevancia muy importante en el equilibrio de la homeostasis y regulación de la presión arterial. (Figura 6)
Figura 6. Sistema de reacciones del sistema angiotensina-renina y sus efectos finales. (29)
El sustrato angiotensinógeno es un zimógeno liberado a la circulación del hígado que es activado y transformado por la enzima renina a la angiotensina I. La renina es secretada por unas células de los glomérulos de los riñones cuando se reduce el flujo sanguíneo renal. La angiotensina I es cortada por la ACE en la superficie endotelial de los vasos sanguíneos dando lugar a la angiotensina II. Este paso puede ser inhibido por el fármaco IECA debido a que es un inhibidor de la ACE. En condiciones normales, la angiotensina II, interacciona con el receptor AT-1, estimulando una serie de reacciones intercelulares de vías de señalización que desencadenan una serie de respuestas celulares entre las que destacan el aumento de la presión arterial, inflamación y fibrosis. Este paso puede ser inhibido por la ARA-II ya que esta molécula es un inhibidor de la angiotensina 2.
Sin embargo, los efectos que causa la angiotensina II necesitan ser regulados y es por eso que el sistema cuenta con otra vía que provoca efectos contrarios, que pueden ser inducidos cuando se bloquea el receptor AT-1 por la acción de la ARB. En esta vía interviene la ACE2, desactivando y degradando la angiotensina II en angiotensina (1-7), que va a ser reconocida por el receptor MAS, provocando una serie de reacciones intercelulares que desarrollaran un conjunto de respuestas celulares con efecto contrario a las producidas por la vía AT1, como la bajada de la presión arterial, antiinflamación y antifibrosis (Figura 7)
Figura 7. Esquema de transformación de la angiotensina 2 en angiotensina (1-7) a través de la ACE2 y sus efectos al unirse con su correspondiente receptor (13).
La ACE2 puede degradar también la Angiotensina I a angiotensina (1-9) (el cual a su vez es transformado en angiotensina (1-7) por la ACE), pero la eficiencia catalítica de esta degradación es muy inferior que la que tiene con la degradación de la angiotensina II. (2, 20)
En segundo lugar, funciona como receptor del SARS-CoV2 y también del SARS-CoV. Como cualquier otro virus, necesita de la maquinaria de la célula para poder reproducirse y por ello necesitan entrar a la célula. En el caso del SARS-CoV2, ingresa en la célula usando ACE2 como receptor. La ACE2, detecta un dominio de unión al receptor (RBD) que contiene al que se une a través de su dominio del extremo C-terminal (región del dominio extracelular), formándose el complejo RBD-ACE2 (28). Así es como interacciona la espícula del virus con la ACE2.
Figura 8. Dominios de la proteína S de la espícula de coronavirus. (1)
La proteína de la que proviene la espícula S del SARS-COV-2 consta de dos subunidades. En la primera (S1) se localiza el dominio RBD previamente mencionado que está relacionado con la unión al dominio receptor de la ACE2, y el reconocimiento de virus. La segunda subunidad (S2) está relacionada con la fusión de las membranas, la del virus con la de la célula diana. (Figura 8)
Figura 9. Esquema de entrada del SARC-Cov-2 a las células a través de ACE2 y TMPRSS2. (16)
La entrada del virus a la célula huésped se da gracias a la interacción de la proteína S de la espícula del virus y la ACE2 y está regulada por la proteína TMPRSS2. Esta actúa como una proteasa cuando el virus se une a la ACE2. Lo que hace es cortar en un par de puntos concretos de la S2 de la espícula, de forma que se produce la fusión de las membranas, promoviendo la entrada del virus a la célula. (13, 16) (Figura 9)
La gran afinidad que tiene la unión de la espícula con el dominio extracelular de la ACE2, junto a la intervención de la proteína TMPRSS2, provoca la fusión del virus con la membrana y la entrada de éste por endocitosis. Una vez dentro, el virus libera su material genético para su respectiva replicación. El uso de la ACE2 por parte del virus hace que la ACE2 pierda su función. Por ello, la ACE2 no podrá degradar la Angiotensina II en angiotensina (1-7). Esto desencadenará, entre otras cosas, daño pulmonar vinculado con la enfermedad de la COVID-19 (21).
Por último, la ACE2, aunque en menor medida, se expresa también en el tracto gastrointestinal particularmente en los enterocitos absorbentes del íleon y el colon (15). Es por ello que, la ACE2 esta también implicada en el transporte de aminoácidos neutros del lumen intestinal al interior celular. Sin embargo, este transporte solo lo lleva acabo asociada al transportador de aminoácidos neutros dependiente de sodio (B 0 AT1) (10). (Figura 10)
Figura 10. Esquema de la absorción de triptófano a través del complejo ACE2-BºAT1 y el efecto que su metabolismo tiene sobre la flora intestinal. (11) Figura 11. Complejo ACE2-BºAT1. Imagen creada con ChimeraX, con el código PDB: 6M1D.
La razón de esta función es que esta proteína surgió por duplicación y fusión de genes siendo homóloga de ACE en el dominio catalítico y de colectrina en el dominio proximal de membrana. La colectrina es una proteína que se expresa en las células del riñón y que unida a B 0 AT1 actúa como transportador de aminoácidos. El problema es que esta proteína no es expresa en las células intestinales y por ello, es la ACE2 unida por su dominio colectrina a B 0 AT1 la que lleva a cabo esa función (10).
El complejo ACE2- B 0 AT1 se ensambla en forma de heterodímero, uniéndose el transportador al dominio colectrina de la ACE2. Sin embargo, el transporte de aminoácidos es solamente activo cuando se une dos heterodímeros del complejo anterior (2). (Figura 11)
Uno de los aminoácidos más importante absorbidos por el complejo ACE2- B 0 AT1 es el triptófano. Se trata de un aminoácido esencial que incorporamos con nuestra dieta y cuyo metabolismo regula la flora intestinal (10).
4. IMPLICACIONES BIOMÉDICAS
El sistema de la ACE2 protege contra infartos miocardios, la hipertensión, la diabetes… . Pero si la ACE2 interacciona con el SARS-COV-2, queda inactiva de su función principal, provocando un mayor riesgo de sufrir dichos problemas (6).
Dentro de los dos coronavirus (SARS-COV-1 y SARS-COV-2) ha existido una diferencia en cuanto a la gravedad que se ha producido de forma general entre las personas que han sufrido un tipo u otro. Aunque tanto la evolución del SARS-COV-1 como del SARS-COV-2 está bastante relacionada, el dominio de unión con el receptor (RBD) de uno y otro es distinto. La diferencia radica en una serie de residuos de aminoácidos que son esenciales para la interacción de la espícula que aportan una mayor afinidad con la ACE2 (2). Dentro de la alteración de dichos residuos, la más destacada es una sustitución de una Valina (SARS-COV-1) por una Lisina (SARS-COV-2) (14). Esta diferencia de residuos en los dominios de unión al receptor hace que la afinidad que tienen cada virus por el receptor ACE2 cambie, y esto demuestra la diferencia de patogenicidad de un virus y otro (6, 16).
Dentro del SARS-COV-2, tampoco ha sido la misma la gravedad del virus en todas las personas. Una mayor o menor infección causada por el virus se ha podido ver determinada por numerosos aspectos, algunos de los cuales se comentan a continuación.
Por un lado, aquellos pacientes con una enfermedad cardiovascular pueden haber sufrido una enfermedad causada por el SARS-COV-2 mucho más severa ya que dicha enfermedad cardiovascular podría estar asociada con una mayor presencia de ACE2, lo que va a promover a niveles superiores la recepción del virus, y con ello una mayor gravedad de la enfermedad. Además, en estos casos se podrá desarrollar linfopenia (escasez de glóbulos blancos en la sangre) o también linfopenia acompañada de inmunosupresión, lo que va a provocar que la enfermedad sea aún más grave. Asimismo, se podrán sufrir también por estas consecuencias, aunque es algo independiente de contraer el virus, infecciones bacterianas entre otras (3). El ejemplo más claro de enfermedad cardiovascular de riesgo frente al coronavirus es la hipertensión. Los pacientes hipertensos regulan su presión arterial a través de fármacos como IECA, un inhibidores de ACE o ARA-2, un inhibidor de angiotensina 2. Estos fármacos son los responsable de un aumento de la expresión de ACE2 en las células, lo que provoca una mayor entrada del virus y gravedad de la infección (19).
Figura 12. Esquema que representa la consecuencia que tiene sobre el sistema angiotensina-renina la unión del coronavirus a la ACE2. (8)
Además, la unión de SARS-COV-2 con ACE2 desregula el sistema angiotensina-renina al inhibir la función de esta proteína en el sistema e impedir que la angiotensina 2 se transforme en angiotensina (1-7). Esta inhibición deriva en una aumento de la angiotensina 2 en aquellos tejidos infectados por el coronavirus lo que se traduce, entre varias consecuencias, en un aumento de la presión arterial que en los pacientes hipertensos puede traer consecuencias letales (7). (Figura 12)
Por otro lado, la diferencia en la tasa de letalidad también puede explicarse por la posibilidad que tiene el SARS-COV-2 de recombinarse activamente, pudiendo generar variantes de sí mismo a la misma vez que está infectando al huésped. Alguna de las variantes pueden ser más patógenas que otras.
Otro determinante en la virulencia del SARS-COV-2 son los factores genéticos , clínicos y medioambientales. Por ejemplo, a edad avanzada, existe una mayor probabilidad de sufrir el virus con mayor gravedad (3).
Por otra parte, en (18) se realiza un estudio que determina la que puede ser la causa de que algunos hombres sufren la enfermedad con mayor infecciosidad que las algunas mujeres. Sin embargo, no se ha visto una gran diferencia en la cantidad de ACE2 en mujeres y hombres sin la enfermedad. No obstante, en caso de tener enfermedades como la hipertensión o insuficiencia cardíaca, sí que aumenta la cantidad de niveles séricos de ACE2. Además, este aumento es mayor en los hombres que en las mujeres, posiblemente a causa de la presencia de determinadas hormonas sexuales. Esta mayor expresión de ACE2 en los hombres puede ser la causa por la que, en algunas ocasiones, la patogenicidad del virus es mayor en hombres. No obstante, esta es una hipótesis que sigue siendo estudiada. (18)
Una vez comentados algunos aspectos relacionados con el grado de gravedad de la enfermedad en distintos colectivos, a continuación nos centramos más efectos y consecuencias que pueden derivarse de la enfermedad.
Como se ha mencionado anteriormente, la ACE2 puede encontrarse en células del tracto gastrointestinal. Por lo tanto, no es sorprendente que el sistema digestivo presente un riesgo de ser infectado por COVID provocando síntomas digestivos como diarrea, náuseas, vómitos o disminución del apetito. En el estudio realizado en (5) a pacientes infectados por SARS-CoV-2, se observó que los que presentaban estos síntomas (además de los síntomas más habituales como fiebre o tos) eran los pacientes de mayor gravedad , mientras que los casos más leves se correspondían con los síntomas más habituales.
Cabe señalar las siguiente posibles explicaciones de la aparición de estos síntomas digestivos. En el caso de las células intestinales infectadas por COVID, las ACE2 de estas pierden su función de transportador de aminoácidos al no poderse unir la proteína BºAT1 a esta. De esta forma, la absorción de aminoácidos se desregula provocando infección intestinal. En el caso del triptófano, su incorrecta absorción es lo que provoca en los pacientes de COVID la colitis grave. (10) (Figura 13)
Figura 13. Efectos que tiene sobre la flora intestinal la mala absorción del triptófano debido a la inactivación de la ACE2 por COVID. A la izquierda se representan la células intestinales sanas y a la derechas células intestinales infectadas por coronavirus. (26)
La inactividad de transporte de aminoácidos puede resultar también en una disminución de péptidos antimicrobianos y, en consecuencia, en una alteración de la flora intestinal provocando inflamación. La flora intestinal se ha encontrado alterada en algunos pacientes con COVID-19, como una disminución de la bacteria Lactobacillus. Además, diversos estudios , entre los que cabe mencionar (19), revelaron que ACE2 también se encuentra en las células epiteliales del esófago, lo que explicaría la esofagitis causada por COVID-19 (15).
Por otro lado, se ha encontrado una relación entre el SARS-COV-2 y la hiperglucemia, que está relacionada con la mayor o menor expresión de la ACE2.
Además de en las células alveolares, la ACE2 puede encontrarse expresada en las células del páncreas, concretamente en las de los islotes. El SARS-COV-2 normalmente se une a las ACE2 de las membranas de las células alveolares pero si hay una elevada expresión de ACE2 en el páncreas, el SARS-COV-2 podrá usarlos para entrar en las células del páncreas. Ingresará en dichas células provocando una disminución de producción y secreción de insulina, dando lugar finalmente a una hiperglucemia aguda. (Figura 14)
Figura 14. Daño pancreático causado por la interacción del SARS-COV-2 con las ACE2 de las células de los islotes del páncreas (13).
Para finalizar, queremos comentar que una posible solución podría ser la metformina, ya que activa la AMPK, que provoca la fosforilación de ACE2 de forma que la unión del SARS-COV-2 no sea tan efectiva. Por otro lado, indicar que todo aquello que pueda provocar que la expresión de la ACE2 aumente, será inadecuado para los pacientes diabéticos que contraigan el SARS-COV-2 como se puede leer en (13).
5. CONCLUSIÓN
En este trabajo, después de analizar algunas de las referencias más importantes sobre la ACE2, se ha visto que es una proteína transmembrana que requiere la presencia del cofactor zinc para funcionar como carboxipeptidasa. Asimismo, se observa que tiene un papel fundamental en el sistema de la renina-angiotensina, en el mecanismo de entrada del SARS-CoV-2 y en el transporte de aminoácidos. También, es una proteína relevante en el ámbito biomédico por su relación con la enfermedad del SARS-CoV-2, la hipertensión y la diabetes, entre otras patologías.
6. REFERENCIAS
(1). Bosso, M., Thanaraj, T. A., Abu-Farha, M., Alanbaei, M., Abubaker, J., & Al-Mulla, F. (2020). The two faces of ACE2: the role of ACE2 receptor and its polymorphisms in hypertension and COVID-19. Molecular Therapy-Methods & Clinical Development, 18, 321-327.
(2). Carrillo, C. J. A. (2021). La enzima convertidora de angiotensina 2 en hipertensión, diabetes y obesidad, y su participación en la vulnerabilidad ante el virus SARS-COV-2. Revista de Educación Bioquímica, 39(4), 121-130.
(3). de León, J. D. L. P., Cárdenas-Marín, P. A., Giraldo-González, G. C., & Herrera-Escandón, Á. (2020). Coronavirus–COVID 19: Más allá de la enfermedad pulmonar, qué es y qué sabemos del vínculo con el sistema cardiovascular. Revista Colombiana de cardiología, 27(3), 142-152.
(4). Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000 Sep 1;87(5):E1-9.
(5). Febres-Ramos, R. J. (2022). Características clínicas del sistema digestivo en COVID-19. Horizonte Médico (Lima), 22(3)
(6). Gheblawi, M., Wang, K., Viveiros, A., Nguyen, Q., Zhong, J. C., Turner, A. J., Raizada, M.K., Grant, M. B. & Oudit, G. Y. (2020). Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circulation research, 126(10), 1456-1474.
(7). González, M. A. (2020). Bloqueadores del sistema renina-angiotensina: enemigos o amigos en pacientes con COVID-19. Revista Venezolana de Endocrinología y Metabolismo, 18(1), 1-3
(8). González-Rayas, J. M., Rayas-Gómez, A. L., García-González, J. J., González-Yáñez, J. M., Hernández-Hernández, J. A., & López-Sánchez, R. D. C. (2020). COVID-19, ACE-inhibitors and angiotensin receptor blockers: The need to differentiate between early infection and acute lung injury. Revista Colombiana de Cardiología, 27(3), 129-131.
(9). Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, & van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A frst step in understanding SARS pathogenesis. J Pathol. 2004; 203(2): 631–7.
(10). Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, Sigl V, Hanada T, Hanada R, Lipinski S, Wild B, Camargo SM, Singer D, Richter A, Kuba K, Fukamizu A, Schreiber S, Clevers H, Verrey F, Rosenstiel P, Penninger JM. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature. 2012 Jul 25;487(7408):477-81.
(11). He, Y., Wang, J., Li, F., & Shi, Y. (2020). Main clinical features of COVID-19 and potential prognostic and therapeutic value of the microbiota in SARS-CoV-2 infections. Frontiers in Microbiology, 11, 1302.
(12). Li W, Moore MJ, Vasilieva N, & Sui J. Angiotensinconverting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003; 426:450-4
(13). Lima-Martínez, M. M., Boada, C. C., Madera-Silva, M. D., Marín, W., & Contreras, M. (2021). COVID-19 y diabetes mellitus: una relación bidireccional. Clinica E Investigacion En Arteriosclerosis, 33(3), 151-157.
(14). Lubbe, L., Cozier, G. E., Oosthuizen, D., Acharya, K. R., & Sturrock, E. D. (2020). ACE2 and ACE: structure-based insights into mechanism, regulation and receptor recognition by SARS-CoV. Clinical Science, 134(21), 2851-2871.
(15). Ma, C., Cong, Y., & Zhang, H. (2020). COVID-19 y el sistema digestivo
(16). Pastrian-Soto, G. (2020). Presencia y expresión del receptor ACE2 (Target de SARS-CoV-2) en tejidos humanos y cavidad oral. Posibles rutas de infección en órganos orales. International journal of odontostomatology, 14(4), 501-507.
(17). Patel VB, Parajuli N, & Oudit GY. Role of angiotensin-converting enzyme 2 (ACE2) in diabetic cardiovascular complications. Clin. Sci. 2014; 126(7): 471–82.
(18). Salah, H. M., & Mehta, J. L. (2021). Hypothesis: sex-related differences in ACE2 activity may contribute to higher mortality in men versus women with COVID-19. Journal of Cardiovascular Pharmacology and Therapeutics, 26(2), 114-118.
(19). Salazar, M., Barochiner, J., Espeche, W., & Ennis, I. (2020). COVID-19, hipertensión y enfermedad cardiovascular. Hipertensión y riesgo vascular, 37(4), 176-180.
(20). Soler, M. J., Lloveras, J., & Batlle, D. (2008). Enzima conversiva de la angiotensina 2 y su papel emergente en la regulación del sistema renina-angiotensina. Medicina clinica, 131(6), 230-236.
(21). South, A. M., Diz, D. I., & Chappell, M. C. (2020). COVID-19, ACE2, and the cardiovascular consequences. American Journal of Physiology-Heart and Circulatory Physiology.
(22). Tipnis, S. R., Hooper, N. M., Hyde, R., Karran, E., Christie, G., & Turner, A. J. (2000). A human homolog of angiotensin-converting enzyme: cloning and functional expression as a captopril-insensitive carboxypeptidase. Journal of Biological Chemistry, 275(43), 33238-33243.
(23). Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA, Patane MA, Pantoliano MW. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem. 2004 Apr 23;279(17):17996-8007.
(24). Turner, A. J., & Nalivaeva, N. N. (2022). Angiotensin-converting enzyme 2 (ACE2): Two decades of revelations and re-evaluation. Peptides, 170766.
(25). Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002 Apr 26;277(17):14838-43.
(26). Wang, B., Zhang, L., Wang, Y. et al. Alterations in microbiota of patients with COVID-19: potential mechanisms and therapeutic interventions. Sig Transduct Target Ther7, 143 (2022).
(27). Xia H, & Lazartigues E. Angiotensin-converting enzyme 2: Central regulator for cardiovascular function. Curr Hypertens Rep. 2010; 12(3): 170–5.
(28). Yan, R., Zhang, Y., Li, Y., Xia, L., Guo, Y., & Zhou, Q. (2020). Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science, 367(6485), 1444-1448.
(29). Zhou Angela. ACE2: Targeting a Potentially Important Receptor in Disease Pathogenesis (2022). Disponible en este enlace.
ATPsintasa: el motor del organismo
escrito por nereaadalialucia_1B | 20 febrero, 2023
Por Adalía Gómez, Lucía Devidé y Nerea Ercoreca
INTRODUCCIÓN
La ATP sintasa, también conocida como F0F1-ATP sintasa, es uno de los complejos enzimáticos más importantes presentes en las células de los seres vivos, ya que lleva a cabo el 90% de la síntesis de ATP en condiciones aerobias (en presencia de O2) de toda la célula. Es el encargado de proporcionar la energía necesaria para desarrollar todos los procesos vitales, y lo hace mediante la síntesis de ATP a partir de la unión de ADP y fosfatos inorgánicos. Su nombre se debe a las dos subunidades que lo componen, que son la F0 y la F1. Ambas están unidas por un tallo central perteneciente a la F1. (Cano-Estrada y González-Halphen, 2011)
En la imagen de la izquierda observamos la subunidad F0 de la Atp sintasa. Elaboración propia a partir de PDB 1c17. En la imagen de la derecha observamos la subunidad F1 de la ATP-sintasa. Elaboración propia a partir de PDB 1e79
En general, su tamaño ronda los 371 kDa, por lo que se podría considerar bastante grande. La síntesis de ATP es posible gracias al gradiente electroquímico que se genera entre los dos compartimentos que separa la membrana en la que está insertada la proteína. Los protones van pasando desde uno de los espacios hacia otro gracias a los complejos enzimáticos pertenecientes a la cadena de transporte de electrones, de manera que cuando el proceso acaba, en uno de los dos espacios la concentración de iones es mucho mayor, generando la diferencia de potencial necesaria. Los protones, al tener carga, no pueden atravesar las membranas, así que solo pueden pasar por el camino que les deja la ATP sintasa (1).
En cuanto a su localización, podemos encontrarla en las mitocondrias, cloroplastos y en bacterias. En las mitocondrias, se encuentra insertada en la membrana interna, en las crestas, de manera que la subunidad catalítica, es decir, la F1, queda en contacto con la matriz mitocondrial. El NADH y del FADH2, que han sido sintetizados durante todas las rutas del catabolismo anteriores, llegan a la mitocondria y ceden tanto sus electrones como los protones, de manera que estos pasan desde la matriz mitocondrial hacia el espacio intermembrana, donde aumenta la concentración de estos iones. Una vez necesitan pasar de nuevo a la matriz, atraviesan la ATP sintasa. Su mecanismo se explicará más adelante.
Además, cabe destacar que en las mitocondrias la ATP sintasa aparece formando un dímero en el que las subunidades aparecen muy pegadas, lo que ayuda a la formación de la membrana interna que está tan replegada en este orgánulo. Hay unas 320.000 ATP sintasas en cada mitocondria.
En el caso de los cloroplastos, los protones pasan del estroma al lumen de los tilacoides, y una vez pasan por la ATP sintasa vuelven al estroma. En las células vegetales, se trata del paso final de la fase luminosa o dependiente de la luz de la fotosíntesis, que culmina con la síntesis del ATP que se empleará más tarde en la fase oscura o no dependiente de la luz. En las bacterias. Los protones pasan desde el citoplasma al espacio entre la membrana y la pared bacteriana, y una vez atraviesan la ATP sintasa vuelven al citoplasma. Esto significa que el ATP, una vez sintetizado, se libera a la matriz mitocondrial, al estroma del cloroplasto y al citoplasma de la bacteria.
En esta imagen, de elaboración propia, se ve la posición de la ATP-sintasa tanto en la mitocondria, cloroplasto y bacteria, indicando con que superficies están en contacto.
En 1997, Paul Boyer y John Walker fueron galardonados con el premio Nobel de Química debido a la explicación que dieron sobre la síntesis de ATP gracias a la ATP sintasa. Boyer y Walker determinaron con precisión el proceso a través del cual el ADP recupera la unión con el fosfato por un enlace covalente, que es rico en energía, para formar nuevamente el ATP, proceso promovido por la presencia de H+.
En la década de los años 70, se creó un modelo para el funcionamiento de esta enzima, que establece su estructura como tres proteínas ensambladas. Una simula una rueda, alojada en la membrana interna de la mitocondria, otra una varilla con un extremo fijo al centro de la rueda, y otra un gran cilindro que envuelve y gira alrededor del otro extremo de la varilla. El ATP se forma en tres sitios sobre el cilindro y la varilla juega un papel fundamental en la actividad catalítica.
Paul Boyer es quien elaboró la teoría sugiriendo que los H+ hacen girar a la rueda, al pasar a través de la membrana interna de la mitocondria hacia la matriz del orgánulo. Esta rotación de la rueda implica cambios conformacionales en la proteína, y altera la conformación de los sitios en los que se produce la unión y la síntesis del ATP. Este bioquímico observó que en el caso de la ATP sintasa, se usa la energía para unir los elementos necesarios a la enzima y soltar las moléculas de ATP una vez han sido formadas.
En 1994, Walker confirmó la teoría y mediante cristalografía se consiguió crear un mapa en escala atómica de la porción catalítica de la enzima. Esta visualización de la estructura tridimensional permitió observar mejor el funcionamiento de la enzima, y propusieron las tres conformaciones por las que pasan las subunidades catalíticas que se explicarán más adelante.
ESTRUCTURA Y MECANISMO
Esta proteína se caracteriza por estar compuesta por dos grandes subunidades, cada una determinada por un dominio estructural distinto. Como su nombre indica, estas subunidades son la F0 y la F1.
La primera de ellas es la subunidad encargada de la translocación de los protones, es decir, la encargada de la rotación de la proteína de manera que los protones pueden atravesarla. Tiene dominio transmembrana, ya que está insertada en las membranas correspondientes que ya se han explicado antes, por lo que está compuesta por moléculas hidrófobas. En función de la especie, se pueden determinar la clase de subunidades que a su vez presenta este dominio, al igual que el número de las mismas que está presente, pero en general tenemos los siguientes.
En primer lugar, un anillo con entre 10 y 14 subunidades de tipo C, que son proteolípidos organizados en dos cadenas hélices alfa asociadas. Destaca en su composición la presencia de un aminoácido de tipo aspartato en posición 61, que es imprescindible para el bombeo de protones. Además, encontramos dos subunidades de tipo B, que se organizan formando un puente entre las dos grandes subunidades de la proteína, y dos otras subunidades, una delta y una A, que unen el dímero de subunidades B con el resto de la proteína, la A con la unidad bombeadora y la delta con la unidad sintetizadora. Cabe destacar que los protones pasan a través de la proteína por un conducto formado en este dominio, entre las subunidades C y A. (Domínguez-Ramírez & Gómez Puyou, 2005)
En segundo lugar, la subunidad catalítica es la encargada de la síntesis del ATP propiamente dicho, ya que se encarga de unir el fosfato inorgánico a la ADP. Este dominio está compuesto por cinco tipos distintos de subunidades (alfa, beta, gamma, delta y épsilon), que son solubles e hidrofílicas. Está en contacto con el medio interno de la mitocondria (la matriz mitocondrial), con el estroma del cloroplasto y con el citoplasma en el caso de las bacterias.
Estructura de la ATP-sintasa, con todas sus partes indicadas. Elaboración propia a partir de PDB 5ari
El anillo principal está constituido por tres dímeros con subunidades alfa y beta. En el caso de la ATP sintasa, son las subunidades beta las encargadas de llevar a cabo la síntesis del ATP, no las alfa. Las subunidades beta están codificadas cada una de las tres por un gen distinto, pero una vez formadas se puede observar que los aminoácidos que las componen son equivalentes. En cuanto a las otras subunidades, gamma, delta y épsilon son las encargadas de formar el tallo que une las dos subunidades principales, y además gamma interviene en la rotación de la subunidad sintetizadora, por lo que como se explicará más adelante, modifica a las subunidades beta para que acepten el ADP.
La acción catalizadora de esta proteína se debe tanto a un cambio conformacional en los dímeros de alfa y beta como a la rotación de la subunidad sintetizadora. Existen tres conformaciones entre las que se alternan a lo largo del proceso de síntesis de ATP. La primera se denomina E (empty, vacía), aunque también se puede denominar O (open, abierta), y es la primera, ya que todavía no tiene nada unido. Una vez se une el tanto el ADP como el fosfato, se pasa a la conformación DP o L. Cuando se está catalizando la formación de ATP, se pasa a la conformación TP o T. Una vez formado, se libera y se pasa de nuevo a la conformación E, para que pueda volver a comenzar el ciclo. (Sánchez-Vásquez, L., & González-Halphen, D, 2017)
ATP sintasa dimérica deYarrowia lipolytica. Están marcadas las subunidades F0 y F1 , el eje y el puente. Elaboración propia a partir de PDB 6b8h
FUNCIÓN
ATP-sintasa con gradiente de protones. Elaboración propia
Las ATP sintasas funcionan gracias a la diferencia de potencial eléctrico entre los dos lados de la membrana en la que se encuentran. Encontramos una mayor concentración de protones en el espacio intermembrana y una menor concentración en la matriz mitocondrial. En los cloroplastos, los protones de hidrógeno se acumulan dentro de los tilacoides, en lo denominado espacio mitocondrial espacio tilacoidal o lumen; y hay una menor concentración de protones en el estroma, por lo que pasarán por la atp sintasa, hacia ese espacio. A medida que los protones pasan por la atp sintetasa se produce ATP.
La Atp sintetasa permite el paso de protones del espacio intermembrana a la matriz. Cuando entra un protón en la atp sintetasa se une a una de las proteínas de C12 y saldrá cuando esta haya completado una vuelta entera. Las proteínas gama y espriol tmb giran y se produce la catálisis rotatoria. Esto produce la síntesis de atp
Cuando entra un protón en la ATP sintasa se une a las proteínas C12 (de f0). Cuando el protón atraviesa el anillo c, la F0 gira 120 grados. Las proteínas gama y épsilon también giran y se produce la catálisis rotatoria. Al producirse esta rotación, esto hace que cambie la conformación de la subunidad beta. Como ya se ha mencionado anteriormente en la estructura, la atp sintasa cuenta con 3 estados (el estado L, T y O). En el estado L (libre), en la subunidad beta se encuentran tanto ADP como el P; pero al pasar el protón, se produce la rotación y la atp sintasa pasa al estado T (tensa). En el estado T aumenta la afinidad de la proteína beta con el atp por lo que se produce una fosforilación de ADP y el P. Cuando entra otro protón en la atp sintasa, se produce una rotación, y pasa del estado T al estado O (open, abierto). El estado O o E tiene menos afinidad por el ATP y lo suelta a la matriz mitocondrial en el caso de las mitocondrias; al estroma en el caso de los cloroplastos y al citoplasma en el caso de las bacterias.(García Bermúdez, 2015)
Funcionamiento de la ATP-sintasa y rotación de sus subunidades. Imagen cedida por César Menor Salván.
Cuando el estado O libera el ATP, al mismo tiempo se produce la unión de un nuevo ADP y P. En una rotación de 120 grados se produce un ATP y en una rotación entera se producen 3 ATP (120×3=360)
En este proceso se ha podido observar que la ATP sintetasa transforma la energía del gradiente de protones en la energía química que se acumula al formar los enlaces del ATP. La ATP sintetasa permite que se mantenga el equilibrio eléctrico y se obtiene energía.
EL PAPEL BIOMÉDICO DE LA ATP-SINTASA
Como ya se ha mencionado, la ATP-sintasas desempeña un papel fundamental en la producción de ATP. Pero ¿Qué pasaría si su funcionamiento se viera alterado?, lo primero en lo que pensaríamos sería que no produciríamos tanto ATP y ¿esto cómo afectaría a nuestro organismo? Para empezar, se reducirían nuestros niveles de energía y no podríamos realizar funciones básicas, por ejemplo, no podríamos crear nuevas proteínas, el corazón no podría bombear la sangre, tampoco seriamos capaces de caminar o hablar, etc.
Desde un punto de vista bioquímico, una disminución del ATP provoca acidosis láctica, ya que la disminución de ATP hace que la relación de NADH: NAD aumente y, según el principio de Le Châtelier, el equilibrio de la reacción se desplaza hacia el lactato. La acidosis láctica es conocida por provocar una disminución de la tensión arterial y del número de moléculas de O2 que llegan al cuerpo.
Una solución que se han investigado con el objetivo de que los niveles de ATP no se reduzcan es la conocida cómo: ‘la técnica de los tres padres’. Con este mecanismo se busca evitar que se transmitan mutaciones que pueden afectar a la producción de ATP. Su funcionamiento es el siguiente, se mezcla el núcleo del ovulo de una portadora con el óvulo de una donadora sana y se obtendría un hijo sano. Se emplean óvulos y no espermatozoide porque la mitocondria, orgánulo en el que se encuentran las ATP-sintasas, procede del ADN materno.
No obstante, la ATP-sintasas también está relacionada con la medicina de otras maneras que no implican una reducción de los niveles de ATP.Una de ellas es el envejecimiento. Durante la respiración mitocondrial, una parte de los electrones que van desde los sustratos hasta el oxígeno en la cadena respiratoria reacciona con el oxígeno molecular produciendo O2. También se produce NO a partir deóxido nítrico sintetasa, un electrón no apareado y un radical libre de oxígeno y nitrógeno. Una gran acumulación de estos dos radicales es lo que acelera el envejecimiento, pues produce daños en grandes moléculas, ya que estos dos compuestos dan lugar al radical HO, el cual es un oxidante muy reactivo, capaz de extraer un átomo de hidrógeno de los ácidos grasos insaturados de las membranas celulares y de las bases púricas y pirimidínicas de los ácidos nucleicos. Esto da lugar a radicales alquilo y, en el caso de los ácidos nucleicos, estos radicales se dimerizan. No obstante, en el caso de los ácidos grasos, los radicales dan lugar a reacciones que adicionan O2, que originan radicales peroxilo y, cuando hay una nueva extracción de hidrogeno, se produce un hidroperóxido y un nuevo radical alquilo. A este proceso se le conoce como peroxidación lipídica y lleva a una pérdida de función de las membranas. »Envejecimineto mitocondrial» (Boveris et al., 2000)
Otra relación que tiene la ATP-sintasa con las enfermedades es el cáncer, pues estudios recientes confirman que en la gran mayoría de células tumorales se encuentra un compuesto conocido como ALDH1A3 (se trata de un biomarcador de este tipo de células y guarda una relación con la resistencia a quimioterapia y la metástasis).El mismo estudio ha descubierto que al ALDH1A3 está unido el ATP y que este compuesto actúa como un inhibidor reversible de ALDH1A3.De esto deducíamos que una determinada cantidad de ATP sintetizado se relaciona con la cantidad de células neoplásicas. «Structural and biochemical evidence that ATP inhibits the cancer biomarker human aldehyde dehydrogenase 1A3.» (Castellví, Albert, et al. 2022)
Referencias
Atanasio, R. V. (2012). ¿ ATP-sintasas ectópicas?. MoleQla: revista de Ciencias de la Universidad Pablo de Olavide, (7), 40-43.
Boveris, A., Costa, L. E., & Junqueira, V. B. (2000). Envejecimiento mitocondrial. Ciencia e Investigación, 3(1), 13-23.
Cano-Estrada, A., & González-Halphen, D. (2011). F1F0-ATP Sintasa y sus diferencias estructurales. Revista de Educación Bioquímica, 30(3), 98-108.
Castellví, Albert, et al. «Structural and biochemical evidence that ATP inhibits the cancer biomarker human aldehyde dehydrogenase 1A3.» Communications Biology 5.1 (2022): 354.
Domínguez-Ramírez, L., & Gómez-Puyou, M. T. D. (2005). La F 1 F 0 ATP sintasa: un complejo proteico con gran versatilidad estructural y funcional. TIP. Revista especializada en ciencias químico-biológicas, 8(1), 18-27.
García Bermúdez, J. (2015). Regulación de la expresión y actividad de IF1, el inhibidor fisiológico de la H+-ATP sintasa de la mitocondria.
Sánchez-Vásquez, L., & González-Halphen, D. (2017). Topología y función de las subunidades intrínsecas de la membrana de las F 1 FO-ATP sintasa mitocondriales. TIP. Revista especializada en ciencias químico-biológicas, 20(2), 29-47.
El Fotosistema I
escrito por luwel1C | 20 febrero, 2023
Pawel Kocieda Aleksandrowicz, Luna María Ortiz García. Biología Sanitaria. UAH
Estructura biologica del Fotosistema I. Forma un trímero con tres centros de reacción. Imagen editada a partir de PDB 1JB0.
INTRODUCCIÓN
La fotosíntesis oxigénica es el principal método de conversión de la luz solar en energía química en la Tierra, y es catalizada por cuatro complejos de proteínas de membrana distintos: el fotosistema I, fotosistema II, el complejo citocromo b6f y la F-ATPasa. Aquí nos vamos a centrar en el primero de estos.
El fotosistema I, conocido también por P700 o PSI, es uno de los dos fotosistemas presentes en la membrana tilacoidal de los cloroplastos que se encargan de absorber los fotones de la luz y transformar la energía luminosa en energía química para llevar a cabo la fotosíntesis. Funciona como una plastocianina/ferredoxina oxidorreductasa dependiente de la luz, y genera un complejo molecular extremadamente reductor.
ESTRUCTURA
Pigmentos Fotosintéticos:
Estructuras de la Clorofila A y el Beta Caroteno. Imagen creada a partir de PDB 1JB0.
Antes de comenzar a explicar las unidades funcionales que componen el fotosistema, es importante mencionar el papel indispensable de los pigmentos fotosintéticos (que son los componentes básicos ambos fotosistemas existentes). Estos son lo que hacen posible la fotosíntesis, estando encargados de captar la energía luminosa.
¿Y cómo actúan los pigmentos?
Cuando un fotón impacta con un electrón de un pigmento fotosintético, éste electrón se excita a un nivel de energía superior; a una posición más alejada del núcleo. Cuando la molécula de pigmento vuelve a su estado de reposo, cede la energía a otra molécula de pigmento vecina. Esto es lo que ocurre en el pigmento antena (que detallaremos más adelante).
La energía recogida se transmite de un pigmento vecino a otro hasta que llega a una clorofila (primer dador de electrones), cuyo electrón no volverá a su estado de reposo sino que será dado a otra molécula (denominada primer aceptor de electrones).
Los pigmentos más importantes de los fotosistemas son las clorofilas (clorofilas a y b) que le otorgan su caracteristico color verde; aunque encontramos también pigmentos secundarios como los carotenoides como β-caroteno (de color rojo), xantofilas (de color amarillo), etc. Éstos absorben luz de longitud de onda diferente a las clorofilas (lo que a su vez explica su distinta coloración).
Complejo Central:
El complejo central es la localización de la gran mayoría de moléculas de pigmento, que se encuentran formando una red de pigmentos antena que absorben la energía de la luz y la transfieren al centro de reacción.
Está compuesto por 14 subunidades, de las cuales PsaA y PsaB son las que se unen al complejo de reacción P700; es decir, las dos moléculas especiales de clorofila a del centro de reacción se sitúan en el heterodímero PsaAB.
Además del par especial, el dímero PsaAB contiene una serie de portadores electrónicos individuales: A0 , A1 y Fx . A0 es una molécula específica de clorofila a que acepta un electrón energizado de P700 y lo transfiere a A1 , que ya se identificó como una filoquinona (vitamina K1). Luego, el electrón se transfiere de A1 a una serie de centros de hierro-azufre (Fx , FA y FB ).
Este proceso lo volveremos a mencionar de nuevo cuando desarrollemos el centro de reacción P700, y de manera muy breve en la fotofosforilación acíclica.
Complejo LHC (Light Harvesting Complex):
Localización del Complejo LHC (forma de Media Luna)
Además de las clorofilas presentes en el interior del complejo central, el fotosistema está equipado con un sistema antena periférico, que consiste en un conjunto de proteínas y pigmentos fotosintéticos adicionales que contribuyen a la absorción eficiente del espectro visible de la luz y transfiere energía al centro de reacción.
A nivel estructural, este complejo está conformado por cuatro subunidades que se repiten, denominadas LHCa 1-4, que son polipéptidos que se unen al cuerpo central del fotosistema por los pigmentos de éste y se disponen formando como una media luna.
Centro de reacción P700:
Estructura del Fotosistema I (sin algunas proteínas), con zoom en el centro de reacción P700. Creado a partir de PDB 1JB0.
Denominado así porque capta la luz cuya longitud de onda es menor o igual a 700 nanómetros, está constituido por un par de moléculas de clorofila a que se excitan cuando la energía procedente de los pigmentos antena llegan a ellas.
Cuando esto ocurre, tiene lugar la formación de un estado excitado extremadamente reductor, P700*. Al tener un potencial electroquímico tan negativo (aprox. -1.4 V), es capaz de transferir electrones a través de una cadena de transportadores compuesta por los portadores del dímero PsaAB( A0 , A1 y Fx).
A0 transfiere un electrón a una molécula de filoquinona (Qk o vitamina K1), provocando que ésta se reduzca y forme quinol, que se reoxida transfiriendo el electrón a una cadena de centros hierro-azufre (Fe-S). Al final éste electrón es cedido a la ferredoxina, una pequeña proteína con hierro-azufre externo al complejo PSI.
Este proceso de transferencia de electrones termina con la actuación de esta última molécula, que llevará a cabo la reducción del NADP+ a NADPH.
Sacado de https://chemevol.web.uah.es/wp/estructuras-moleculares/el-fotosistema-i-y-los-colores-del-otono/
PROCESOS DE LA FOTOSÍNTESIS EN LOS QUE INTERVIENE PSI
Conocida la estructura y los procesos internos que se llevan a cabo en este complejo supramolecular, el cual es esencial para que se lleve a cabo la fotosíntesis oxigénica (dada en plantas superiores y algas verdes, entre otros organismos), pensamos: ¿cómo y cuándo interviene PSI en la fotosíntesis?
El fotosistema I solamente interviene en la fase luminosa de la fotosíntesis. A su vez, dependiendo del tipo de fase luminosa que se lleve a cabo, el complejo actúa de diferente manera:
Fotofosforilación Acíclica
En este proceso interviene el fotosistema II acoplado al fotosistema I. No nos vamos a parar en la finalidad de este tipo de fotofosforilación, sino únicamente en la función realizada aquí por el complejo que estamos estudiando.
Cuando incide la luz sobre el PSI, los pigmentos antena de éste se excitan a un estado energético mayor y transmiten esa energía por resonancia hasta llegar al centro de reacción, que contiene un pigmento diana (la clorofila a P700), la cual pierde dos electrones. Estos electrones pasan por una cadena de cofactores: primero son transmitidos a la filoquinona, que queda totalmente reducida. Ésta va a entregar estos electrones a los diferentes complejos de hierro-azufre de forma escalonada, para que ellos los transfieran a la ferredoxina (el electrón se cede a la ferredoxina por el último centro Fe4S4), una pequeña enzima que cataliza la reducción del NADP+ para formar NADPH.
El pigmento diana recupera esos electrones perdidos (para llevar a cabo este proceso múltiples veces) por la plastocianina, que cede al fotosistema los dos electrones procedentes del citocromo b6f.
Mostración de la cadena general de transporte de electrones. Se puede observar el Fotosistema I en este proceso, que proporciona los recursos de energía para el ciclo de Calvin. Sacado de https://chemevol.web.uah.es/wp/estructuras-moleculares/el-fotosistema-i-y-los-colores-del-otono/
Fotofosforilación Cíclica
En este proceso solo interviene el PSI. Como no interviene el fotosistema II, únicamente se obtiene ATP del flujo de electrones y no se forman ni NADPH ni O2.
Se lleva a cabo de la siguiente manera: los electrones son elevados desde la clorofila P700 hasta el aceptor de electrones primario del fotosistema, pero no siguen la serie de portadores electrónicos que llevan al NADP+, sino que se desvían hacia la cadena de transporte de electrones que conecta a los fotosistemas I y II, y continúan cuesta abajo por esa cadena volviendo a la clorofila P700. En este recorrido se produce ATP.
El fotosistema I capta un fotón que permite la liberación de un electrón del centro de reacción P700, el cual es cedido a la clorofila A0, luego a la filoquinona (Q), y por último a la ferredoxina (Fd). Esta última molécula cede el electrón al complejo b6f, donde se produce suficiente energía para transportar los H+ en contra del gradiente electroquímico, y producir ATP como en la fotofosforilación acíclica. El complejo b6f cede los electrones a la plastocianina, que los lleva hasta el hueco dejado en el P700, para que puedan volver a ser excitados por un fotón y reiniciar la fotofosforilación cíclica.
Representación del flujo de electrones en la fosforlicación cíclica. Creado en Biorender.
IMPLICACIONES BIOTECNOLÓGICAS
En el mundo moderno hay una gran necesidad de nuevas formas de nuevas fuentes energéticas de orígenes limpios y renovables. Uno de los métodos más comunes es la utilización de la luz solar, que produce 175.000 TJ (terajulios) de energía fotónica al año, por medio de paneles solares. Sin embargo, estos tienen otros efectos medioambientales, pues la obtención de los materiales necesarios para producirlos es muy contaminante.
Paneles solares
Una alternativa que está en desarrollo es la utilización de procesos fotosintéticos para obtener energía, para lo cual las propiedades del Fotosistema I son muy útiles. No sólo presenta capacidad de producir gran poder reductor (el mayor que se puede encontrar en la naturaleza); sino también se puede encontrar en grandes cantidades, es fácil de purificar y no es tóxico en ninguna manera. Además, debido a la diversidad de especies fotosintéticas, algunas presentan variaciones del Fotosistema I con diferentes propiedades que pueden ser beneficiosas. Por ejemplo, el Fotosistema I de T. elongatus es muy termoestable.
Sin embargo, existen problemas a la hora de utilizar FSI en producción energética. Pese a que su eficiencia ha estado aumentando continuamente, esta es aún relativamente pequeña comparada a la de métodos fotovoltaicos tradicionales. Esto se debe a factores propios del FSI, como su limitación en el espectro de luz que puede absorber o la poca superficie que cubre de los electrodos, pero también a que es una rama de investigación relativamente reciente y poco desarrollada.
Aún así, cabe esperar que con el tiempo la utilización del fotosistema I será más y más viable conforme se perfeccione el proceso, permitiéndonos producir energía verdaderamente verde.
BIBLIOGRAFÍA
Nelson, N. y Yocum, CF (2006). Estructura y función de los fotosistemas I y II. año Rev. Plant Biol. , 57 , 521-565.
Fotosíntesis. McKee T, & McKee J.R.(Eds.), (2016). Bioquímica. Las bases moleculares de la vida,5e.McGrawHill.https://accessmedicina.mhmedical.com/Content.aspx?bookid=1960§ionid=148096471.
Ho, Thi Thu Hoai, et al. «Photosystem I light-harvesting proteins regulate photosynthetic electron transfer and hydrogen production.» Plant Physiology 189.1 (2022): 329-343.
Caffarri, Stefano, et al. «A comparison between plant photosystem I and photosystem II architecture and functioning.» Current Protein and Peptide Science 15.4 (2014): 296-331
Amunts, Alexey, and Nathan Nelson. «Plant photosystem I design in the light of evolution.» Structure 17.5 (2009): 637-650.
Yang, Huixia, et al. «Molecular mechanism of photosystem I assembly in oxygenic organisms.» Biochimica et Biophysica Acta (BBA)-Bioenergetics 1847.9 (2015): 838-848.
Teodor, Alexandra H., and Barry D. Bruce. «Putting photosystem I to work: truly green energy.» Trends in biotechnology 38.12 (2020): 1329-1342.
https://chemevol.web.uah.es/wp/estructuras-moleculares/el-fotosistema-i-y-los-colores-del-otono/. consultado el 10-01-2023.
β-AMILOIDE, EL PÉPTIDO QUE NO DEJA RECORDAR
escrito por PauladelaFuente_1B | 20 febrero, 2023
Creado por: Lucía Fernández López, Paula de la Fuente Méndez y Andrea García Berrocal
Índice
1.Introducción
2.Origen y papel biológico
3.Isoformas, estructuras y mecanismo
4.Papel biomédico
5.Referencias
1.INTRODUCCIÓN
La β-amiloide es un péptido procedente del corte de la proteína APP. Se encuentra presente como un monómero, de peso molecular 4 kDal. En procesos normales se va a localizar circulando por el líquido cefalorraquídeo y en la sangre, pero en una situación patológica se encuentra formando depósitos en el cerebro, normalmente en la región del hipocampo o en el neocórtex.
Va a ser la responsable de una serie de enfermedades neurodegenerativas como son el Alzheimer o el Párkinson, entre otras.
Lo mencionado anteriormente junto con las funciones y la estructura del péptido será detallado en el artículo que se presenta a continuación.
2.ORIGEN DEL PÉPTIDO β-AMILOIDE
El péptido β-amiloide se forma a partir de la proteólisis de la proteína precursora amiloide, también conocida como APP. Esta proteína se encuentra insertada en la membrana plasmática, gracias a su dominio transmembrana, donde es procesada por las enzimas alfa, beta y gamma secretasa. Dependiendo de las enzimas que actúen se puede dar lugar a dos rutas:
RUTA NO AMILOIDOGÉNICA (predominante)
Esta ruta es la predominante y en ella no se produce el péptido β-amiloide.
La α-secretasa corta a la APP por los aminoácidos 687-688, generando dos fragmentos: uno llamado (s)APPα soluble (donde se encuentra el dominio N-terminal) y que queda en el espacio extracelular y otro fragmento carboxi-terminal llamado C83, con 83 aminoácidos, que quedará insertado en la membrana.
Posteriormente, la γ-secretasa realiza otro corte, en los aminoácidos 712/714/715, generando el péptido p3 (que saldrá al espacio extracelular) y el fragmento γ-CTF (que quedará en el intracelular).
RUTA AMILOIDOGÉNICA
En esta ruta, y gracias a la acción de dos secretasas, se produce el péptido β-amiloide.
La β-secretasa se encarga de cortar a la APP por los aminoácidos 671/672, produciendo dos fragmentos distintos a los anteriores: el fragmento (s)APPβ soluble (donde se encuentra el dominio N-terminal) que se libera al espacio extracelular y otro fragmento carboxi-terminal llamado C99, esta vez con 99 aminoácidos, que quedará insertado en la membrana, al igual que ocurría en la vía no amiloidogénica.
Este fragmento C99 será cortado por la enzima γ-secretasa, produciendo el péptido β-amiloide (Aβ), que saldrá al espacio extracelular y el fragmento γ-CTF, que será transportado al núcleo pudiendo actuar como factor activador de la expresión de determinados genes.
Figura 1. Procesamiento de la APP mediante la ruta no amiloidogénica y la ruta amiloidogénica. Imagen creada por Paula de la Fuente Méndez, a través de BioRender.
PAPEL BIOLÓGICO
FUNCIONES DEL PÉPTIDO β-AMILOIDE
El péptido β-amiloide además de ser uno de los responsables del Alzheimer, desarrolla una serie de funciones en el cuerpo que no están relacionadas con dicha enfermedad.
Se encarga de promover tanto la proliferación celular, como la adhesión entre células; interviene en vías de señalización tales como la activación de las MAP quinasas; en condiciones fisiológicas protege y evita la muerte de las neuronas; desarrolla una función autocrina y antimicrobiana. Además, se ha propuesto su posible labor ayudando al transporte del colesterol y en los canales de KCa.
ESTRÉS OXIDATIVO
El estrés oxidativo es un proceso debido al incremento excesivo de especies reactivas (conjunto de radicales libres) que tiene como efecto una pérdida de la funcionalidad en la célula por el daño producido por la oxidación.
La β-amiloide es responsable de generar radicales libres (15), que producen la oxidación de los elementos que se encuentran en el cerebro, muy sensible al estrés oxidativo debido a la gran cantidad de glucosa, ácidos grasos, enzimas y a la insuficiencia de antioxidantes en él. Tiene entre otras consecuencias que las ATPasas, las vías de señalización y los factores de transcripción no tengan una correcta actividad, dando lugar a la pérdida de la homeostasis.
Cabe destacar que los radicales libres aún con el daño que producen, desarrollan funciones en la célula como la protección inmunológica o la expresión de genes.
El estrés oxidativo promueve la formación de agregados (15), es decir, la unión de β-amiloides mal plegadas. Sin embargo, no se sabe con certeza si es uno de los responsables del inicio del Alzheimer o el resultado del mismo.
La importancia de la oxidación de los componentes del cerebro se ha demostrado sometiendo a neuronas a estrés oxidativo, dando como resultado una disminución en la producción de ATP (debido a las ATPasas de señalización) y un aumento de calcio (por la función de los canales Ca-K) (16). Esto desemboca en una situación excesivamente inestable, siendo la solución la apoptosis de la célula.
DAÑO TRAUMÁTICO
Al producirse un traumatismo se genera estrés oxidativo, produciendo una disminución en la eliminación del péptido β-amiloide. Al disminuir su eliminación, se va a tender a la formación de las placas amiloides. Por esta razón, aumenta de una manera más significativa la isoforma Aβ1-42 (la que crea dichas placas), aunque también se produce un aumento de la isoforma Aβ1-40 (15). Las placas van a tener una relación inversamente proporcional con el estrés oxidativo, van a hacer que este disminuya, y con ello disminuye el daño citoplasmático.
RECEPTORES β-AMILOIDE
LRP y el receptor Scanvenger (RS) son una serie de receptores que están relacionados con la toxicidad de la beta amiloide
LRP
El LRP1, regulado por la α2-macroglobulina, se encarga de endocitar y degradar el péptido β-amiloide. Sin embargo, en una situación patológica donde se produzca este péptido en exceso, LRP1 no va a poder llevar a cabo la endocitosis, por lo que se producen patologías.
Dentro de la familia de los LRP, también se encuentra la glicoproteína 330 (LRP2) que se encarga del transporte de un complejo, resultado de la unión de la beta amiloide y uno de sus ligandos: la ApoJ.
Receptor Scanvenger (RS)
Por último, resalta la posible importancia del receptor Scanvenger (RS). Se ha demostrado su incremento en los cerebros de las personas que sufren la enfermedad del Alzheimer y en un experimento realizado sobre ratones (1), donde se demostró la necesaria presencia de β-amiloide, sugiriendo que ayuda a la actividad neuronal hasta el momento en el que se produce la adhesión entre fibrillas.
Este receptor facilita la adhesión de fibrillas y controla otros procesos relacionados con la β-amiloide, como su endocitosis y degradación. Además, se ha demostrado en otro estudio la relación de este receptor con el estrés oxidativo, observándose la reducción del RS. De esta manera, se produce la pérdida de la capacidad de endocitar el péptido, pudiendo ser el responsable de la acumulación de agregados.
3.ISOFORMAS, ESTRUCTURA Y MECANISMO
ISOFORMAS
Dependiendo del número de residuos de aminoácidos que tenga el péptido β-amiloide, se pueden generar distintas isoformas, siendo las más comunes la Aβ1-40, con 40 residuos, y la Aβ1-42, con 42 residuos. Ambas isoformas son neurotóxicas, pero van a presentar diferencias con respecto a su metabolismo, funciones, toxicidad y a la forma de agregarse.
La isoforma Aβ1-40 se encuentra en mayor concentración en los vasos sanguíneos del cerebro, produciendo la angiopatía amiloide cerebral, de la que se hablará más tarde en este artículo.
La isoforma Aβ1-42es menos soluble, con un fragmento C-terminal más estructurado y va a tender a agregarse de una manera más rápida para formar las placas amiloides, es decir, agregados que han precipitado en el espacio intercelular (entre neuronas). Esta isoforma, por tanto, se encuentra en el cerebro en mayor concentración que en la sangre.
Además de esas, podemos distinguir otras isoformas como pueden ser la β-amiloide-25, β-amiloide-35 y β-amiloide-38 con 25, 35 y 38 residuos respectivamente (13).
Aβ1-40
Figura 2. Vídeo de la estructura de la Aβ1-40 realizado con ChimeraX, basado en PDB 1BA4, obtenido de protein data banc. En el primer vídeo se observan en rosa las hélices α, mientras que en verde se ven los bucles (coils). Vídeo creado por Paula de la Fuente Méndez.
Aβ1-42
Figura 3. Vídeo de la estructura de la Aβ1-42 realizado con ChimeraX, basado en PDB 1IYT , obtenido de protein data banc. En el segundo vídeo se observan también las hélices α, mientras que en verde se ven los bucles (coils). Vídeo creado por Paula de la Fuente Méndez.
ESTRUCTURA
El péptidoβ-amiloide, cuando se encuentra aislado, no tiene estructura terciaria compacta, ya que es intrínsecamente desestructurado. Además, gracias a varios estudios realizados mediante Resonancia Magnética Nuclear y dinámica molecular (5), se sabe que tampoco tiene una estructura secundaria significativa, pero sí cuenta con una estructura en espiral.
En el caso de que el péptido β-amiloide se encuentre formando agregaciones, es decir, que las hélices alfa hayan pasado a láminas beta y se hayan pegado unas a otras, los residuos 18 a 40 o 42 (dependiendo de la isoforma) van a formar un motivo β-lámina – giro – β-lámina a través de dos láminas beta paralelas, mientras que el resto de residuos se encuentran desordenados.
Figura 4. Vídeo del motivo β-lámina – giro – β-lámina realizado con ChimeraX, basado en PDB 2BEG, obtenido de protein data banc. Se observa el motivo formado entre los aminoácidos 18-42. Vídeo creado por Paula de la Fuente Méndez.
MECANISMO DE FORMACIÓN DE PLACAS
El péptido β-amiloide, puede plegarse de varias formas. En el caso de hacerlo correctamente, se pliega formando una α-hélice, que es una forma soluble y que se puede eliminar.
Sin embargo, este péptido tiene tendencia a plegarse de forma incorrecta. Esto se debe a que los anillos aromáticos de los residuos del péptido, orientados el uno al otro, tienden a solapar sus orbitales (“stacking”) y a pegarse entre ellos. Esto obliga a que cambie la estructura a β-láminas, en las que los anillos aromáticos estabilizan la estructura. Al cambiar la estructura de un péptido, por efecto amplificador, los β-amiloide cercanos también cambian su estructura a láminas beta.
Estos péptidos tienden, por tanto, a agregarse y polimerizarse, formando consecutivamente oligómeros, protofibrillas y fibrillas que darán lugar a las placas amiloides, unas estructuras insolubles que se depositan fuera de las células.
Figura 5. Vídeo de las fibrillas creadas por la Aβ1-42 realizado con ChimeraX, basado en PDB 2MPZ, obtenido de protein data banc. Vídeo creado por Paula de la Fuente Méndez.
También se pueden formar agregados no estructurados, donde las cadenas se unen unas con otras por la mala orientación de los anillos aromáticos de los residuos. Estos agregados no estructurados son estructuras insolubles, que precipitan, se acumulan y terminan destruyendo la célula.
En situaciones normales, la proteína ApoE actúa como chaperona, replegando correctamente el péptido, antes de que se formen las placas amiloides. Sin embargo, puede haber mutaciones en esta proteína, derivando en la formación de placas, ya que los péptidos no se pliegan correctamente.
Figura 6. Proceso por el cual el péptido β-amiloide forma oligómeros, protofibrillas y fibrillas. Creado con BioRender por Paula de la Fuente Méndez.
4. PAPEL BIOMÉDICO
ENFERMEDADES
ALZHEIMER
El Alzheimer es un trastorno neurodegenerativo gradual. Esta enfermedad es la principal causa de demencia en personas mayores. Afecta, entre otras cosas, a la comprensión y a la memoria.
Está relacionada con el péptido β-amiloide, ya que altera la sinapsis al fragmentarse las mitocondrias del cerebro y desemboca en la muerte de las neuronas. Esto se produce debido a la excesiva producción de óxido nítrico (un radical libre) por parte de este péptido. El óxido nítrico, al reaccionar con la DRP1 (dynamin related protein 1), hace que las mitocondrias del cerebro se fragmenten.
El péptido β-amiloide, además, es el que forma las placas amiloides o las placas seniles, localizadas en el cerebro de las personas que padecen el Alzheimer. Estas placas también se encuentran en personas que padecen la enfermedad del Párkinson y la enfermedad de los cuerpos de Lewy, ambas resultando en demencias.
Es probablemente la producción de radicales libres en exceso la causa del Alzheimer, mientras que la agregación de las β-amiloide es una de las consecuencias. Hay mutaciones en la proteína APP que pueden provocar un aumento de la concentración de β-amiloide (un 10% son las personas que presentan la enfermedad de forma hereditaria). También puede haber mutaciones que favorezcan la vía amiloidogénica, de forma que se crea más péptido β-amiloide.
Los tratamientos existentes para el Alzheimer no frenan la enfermedad, únicamente disminuyen los síntomas que esta produce, mejorando la calidad de vida de las personas que la padecen.
Figura 7. Comparación entre un cerebro sano (izquierda) y uno afectado por Alzheimer (derecha). ISBN: 978-84-608-3354-3
Angiopatía amiloide cerebral
Elpéptido β-amiloide, además de formar las placas seniles (Aβ1-42), puede acumularse cubriendo las paredes de los vasos sanguíneos del cerebro (Aβ1-40) y producen la angiopatía amiloide cerebral. Esta afección, además de demencia, causa un sangrado del cerebro.
Además, la isoforma Aβ1-40 se encuentra en el miocardio de las personas que padecen Alzheimer e insuficiencia cardiaca, por lo que hay una correlación entre la concentración de este péptido y los factores de riesgo de esta última enfermedad. Esto quiere decir que un aumento de la concentración de Aβ40 implica un aumento de los factores de riesgo. (11)
FÁRMACOS (INHIBIDORES)
Se ha intentado llevar a cabo una serie de fármacos cuyo objetivo sea la inhibición de la formación del péptido β-amiloide e incluso la inhibición de la agregación del mismo.
INHIBIDORES DE LA β- SECRETASA (BACE1)
La enzima β-secretasa va a comenzar una de las rutas del procesamiento de la APP, la ruta amiloidogénica. A la hora de desarrollar inhibidores para la misma, hay que tener en cuenta que tiene otros muchos sustratos, por lo que su inhibición inespecífica puede tener los efectos contrarios. Otro factor que dificulta la creación de estos inhibidores es la propia estructura de la proteína: la β-secretasa forma parte de las aspartilproteasas, por lo que la molécula que se encargue de inhibirla tendría que ser grande e hidrofóbica, dificultando su paso por la barrera hematoencefálica.
A pesar de esto, unos estudios recientes muestran que hay 2inhibidores(E2609 y MK-8931) capaces de disminuir la producción de los niveles de β-secretasa en el líquido cefalorraquídeo entre un 80 y 90 %. (10)
INHIBIDORES Y MODULADORES DE LA γ- SECRETASA
La γ-secretasa, además de procesar la APP, se encarga de procesar múltiples proteínas más. Por lo que, al igual que sucede con la β-secretasa, a la hora de desarrollar inhibidores, hay que tener en cuenta que tiene otros muchos sustratos, por lo que su inhibición inespecífica puede tener los efectos contrarios.
Se desarrollaron una serie de fármacos como posibles inhibidores de la γ-secretasa como semagacestat (LY450139), (10) comprobándose que reducía los niveles del péptido β-secretasa en la sangre y en el líquido cefalorraquídeo. Sin embargo, no disminuía la progresión lenta de la enfermedad del Alzheimer.
Se desarrollaron también una serie de moduladores selectivos de esta enzima, para poder impedir los efectos adversos que podrían causar los inhibidores. Estos moduladores bloquean a la enzima con respecto al procesamiento de la APP, pero no influye en otras vías de señalización que pueda llevar a cabo como es la Notch.
Uno de estos moduladores es el NIC5-15 (10) que va a inhibir a la γ-secretasa sin interferir en Notch. Además, es un sensibilizador bastante potente de la insulina y se está investigando también su posible papel en la inhibición del proceso inflamatorio, más concretamente inhibiendo la activación de la microglía.
Otro modulador de la γ-secretasa es CHF5074 (10) que es un derivado antiinflamatorio no esteroideo.
ACTIVACIÓN DE LA α-SECRETASA
Mediante la activación de la α-secretasa se pretende estimular la vía no amiloidogénica para que, de esta manera, no se lleve a cabo la vía amiloidogénica, evitando así que se produzca la formación del péptido β-amiloide. Aunque la mayoría de estos inhibidores están siendo probados aún en estudios clínicos.
El galato de epigalocatequina (EGCG) (10) es un flavonoide polifenólico que va a inhibir la producción de oligómeros tóxicos malformados de la β-amiloide y, además, va a activar a la α-secretasa.
Etazolato (EHT 0202) (10) impulsa la acción neurotrófica de esta enzima inhibiendo, además, la muerte neuronal producida por el péptido β-amiloide.
La acitretina, (10) una de las funciones que tiene es activar la α-secretasa, activando la ruta no amiloidogénica de la APP en células de neuroblastoma y, además, va a reducir los niveles de β-amiloide en ratones transgénicos.
INHIBIDORES DE LA AGREGACIÓN
El objetivo del desarrollo de los inhibidores de las agregaciones de la β-amiloide es encontrar un péptido que se parezca a la isoforma Aβ1-40 y que consiga retardar la agregación de la Aβ1-42, ya que se ha demostrado que entre ambas isoformas va a haber una interacción que tiene la capacidad de regular el proceso de agregación de dicho péptido.
En base a esto se han creado muchos inhibidores basados en la secuencia 17-21 de este péptido (considerado el centro de nucleación) ya que es considerada como una diana sobre la cual se van a dirigir los potenciales inhibidores de la agregación.
UTILIZACIÓN DE AMINOÁCIDOS SINTÉTICOS
Una de las numerosas técnicas que se han probado para reducir la toxicidad de la beta amiloide es el cambio de aminoácidos en el tramo hidrofóbico del péptido.
Existen una serie de residuos (G25 a G37) que promueven la agregación. El cambio por leucinas o glicinas previene la formación de dímeros, trímeros y fibrillas, además de la pérdida de la estabilidad de la estructura.
De igual manera, la Metionina 35 es un residuo que interviene en el estrés oxidativo. Si es sustituido por valina, aumenta la toxicidad, mientras que si lo es por isoleucina o cisteína, disminuye.
Cabe destacar que en el laboratorio usualmente se utiliza el péptido beta amiloide 25-35, muy similar a las isoformas 40 y 42 del péptido, ya que comparten el mismo grado de toxicidad y capacidad de agregación pero es posible su producción de una manera menos costosa.
Aguirre-Rueda, Diana. (2014). “Daño inflamatorio y estrés oxidativo en la Enfermedad de Alzheimer. Efecto de polifenoles y cannabinoides”. Tesis doctoral. DOI 10.13140/RG.2.1.5099.6080
Ana Esther Estrada Rodríguez y Viviana Chantal Zomosa Signoret. (2017). “Papel de la agregación del péptido Beta Amiloide en la enfermedad de Alzheimer”. Revista de Educación Bioquímica (REB) 36(1):2-11 Download PDF: https://www.medigraphic.com/pdfs/revedubio/reb-2017/reb171b.pdf
Chen, Gf., Xu, Th., Yan, Y. et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin 38, 1205–1235 https://doi.org/10.1038/aps.2017.28 Download PDF: file:///C:/Users/Admin/Downloads/aps201728-1.pdf
Grégory A. García, Sergio Hernández V., Ómar Ramón M., Segundo A. Baez y Ananías García C. (2007). “Biología y patobiología humanas del complejo de absorción y transporte epitelial megacubam”. Revista MED 15 (1): 94-104 Download PDF: http://www.scielo.org.co/pdf/med/v15n1/v15n1a11.pdf
J. Folch, M. Ettcheto, D. Petrov, S. Abad, I. Pedrós, M. Marin, J. Olloquequi, A. Camins (2015) “Review of the advances in treatment for Alzheimer disease: strategies for combating β-amiloide protein: Neurología (English Edition), Volume 33, Issue 1, January–February 2018, Pages 47-58”. Disponible en: https://www.sciencedirect.com/science/article/pii/S021348531500064XDownload PDF
Kimon Stamatelopoulos, Konstantinos Stellos (2017). “El amiloide beta (1-40) circulante predice eventos en pacientes con insuficiencia cardiaca”. Revista Española de Cardiología vol. 70, issue 11,pp: 905-906 Published by Elsevier. DOI: 10.1016/j.recesp.2017.05.019. Download PDF: https://www.revespcardiol.org/es-pdf-S0300893217303512
Qiu T, Liu Q, Chen YX, Zhao YF, Li YM. “Aβ42 and Aβ40: similarities and differences”. J Pept Sci. 2015 Jul;21(7):522-9. DOI: 10.1002/psc.2789. Epub 2015 May 28. PMID: 26018760. Disponible en: https://pubmed.ncbi.nlm.nih.gov/26018760
Natalia Manzano-León y Jaime Mas-Oliva. (2006). “Estrés oxidativo, péptido β-amiloide y enfermedad de Alzheimer”: Instituto de Fisiología Celular, Universidad Nacional Autónoma de México. México D.F., México. Download PDF: https://www.scielo.org.mx/pdf/gmm/v142n3/v142n3a9.pdf
QUIMERA → Molecular graphics and analyses performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. Protein Sci. 2021 Jan;30(1):70-82
PROTEÍNA P53: UNA MUERTE NECESARIA
escrito por AlexLeyre_3C | 20 febrero, 2023
Realizado por Leyre Navajo Gómez y Alejandro Muñoz Benito
INTRODUCCIÓN
La proteína p53 es conocida como el “guardián del genoma” por su papel en la regulación del ciclo celular, pero también participa en el mantenimiento de la homeostasis o en la respuesta al estrés. P53 es un factor de transcripción que en su forma activa va a unirse a secuencias del ADN para inducir o reprimir la expresión de genes, produciendo la inhibición del ciclo celular y la activación de la apoptosis.
De forma fisiológica, p53 solo se activa cuando se ha producido un daño en el ADN que los sistemas de reparación de ADN no pueden solucionar y conduce a la célula a procesos de muerte celular para evitar que los daños sean heredados por las células hija, de esta manera está involucrado en el mantenimiento de la integridad génica. Cuando las células no presentan daño en el ADN, p53 va a ser degradada por el proteosoma para impedir que células sanas entren en apoptosis y puedan continuar el ciclo celular.
P53 desarrolla funciones muy importantes dentro de las células, por eso cualquier fallo puede derivar en enfermedades graves como el cáncer. Se ha visto que el 50% de los tumores presentan mutada esta proteína lo que evidencia su importancia y el gran interés que hay en su estudio. Las mutaciones en p53 hacen que pierda su actividad supresora y comience a actuar como una oncoproteína cuya acción conduce a la aparición de células tumorales que escapan de la muerte celular y pierden la regulación del ciclo celular. Generalmente, los tumores con mutación en p53 tienen peor pronóstico, porque aunque se trate al paciente con radioterapia o quimioterapia la vía que conduce a la apoptosis está inhibida y las células pese a la acumulación de tumores seguirán proliferando. Por ello, existen muchos estudios centrados en esta proteína con el fin de entender su estructura, regulación y función para poder desarrollar mecanismos que puedan impedir el progreso de los tumores.
ESTRUCTURA
El gen TP53 se encuentra en el brazo corto del cromosoma 17 (17 p13) y codifica para la proteína nuclear p53, se trata de un homotetrámero donde cada monómero lo constituyen 393 aminoácidos con distintos dominios funcionales. En los tejidos humanos se encuentran 9 isoformas distintas de esta proteína siendo la expresión de cada una de ellas dependiente del tejido en el que se encuentra.
Las cuatro regiones que constituyen cada monómero son:
Dominio aminoterminal. p53 contienen dos dominios de transactivación (AD1, AD2) que son necesarios para activar la transcripción de los genes diana, una región rica en prolinas que participa en la regulación de la apoptosis Y una señal de exportación nuclear (NES). Ante diferentes señales, la proteína sufre modificaciones postraduccionales (fosforilaciones) que conducen a su separación de la proteína MDM2 permitiendo su activación. (2)
Dominio de unión de ADN (Core). Localizado entre los aminoácidos 94-294, es un dominio resistente a proteasas y con una secuencia conservadas en otros miembros de su familia (p63 y p73) Este dominio se encarga de reconocer y unirse de manera específica a secuencias promotoras del ADN para regular la transcripción de los genes diana. Su estructura la constituyen dos láminas β antiparalelas, dos grandes “loops” (L2 y L3) que se estabilizan con Zinc y un motivo loop lámina-hélice al final del dominio. Este dominio tiende a ser rico en aminoácidos básicos que permiten la unión al ADN. (3)
Dominio de tetramerización (Tet). Se encuentra entre los aminoácidos 320-360 y participa en la formación del tetrámero. Lo forma un dímero de dímeros, con dos laminas-β y dos α-hélices entrecruzadas (4). Consta de una región flexible de 30 aminoácidos aproximadamente, que conecta este dominio con cada uno de los dominios centrales.
Dominio carboxiloterminal. Localizado entre los aminoácidos 363-393, presenta muchos residuos básicos (arginina y lisina) siendo capaz de unirse al ADN de forma inespecífica por interacciones electroestáticas. Ante diversas señales de estrés sufre modificaciones postraduccionales (glicosilación, fosforilación o ubiquitinación). (1)
Figura 2. Esquema de los dominios presentes en la estructura de p53.
REGULACIÓN
P53 sufre modificaciones postraduccionales que incluyen fosforilación, ubiquitinación, acetilación y metilación; todas ellas están implicadas en la estabilidad, conformación, localización e interacciones de p53. La desregulación de alguna de estas rutas aparecen entre las principales causas de enfermedades del desarrollo asociados con p53, especialmente en cánceres.
La estructura multimodular de p53 hace que presente múltiples sitios donde pueden realizarse modificaciones que influirán en su función, estas modificaciones pueden ser reversibles por la acción de otras enzimas.
Algunos ejemplos de modificación postraduccional son (5):
Ubiquitinación. La ubiquitina es una pequeña proteína que se une a las proteínas que van a ser degradadas, se trata de una cascada de reacciones enzimáticas donde participan ligasas (E1: activación, E2: ligación, y E3: conjugación) cuyo objetivo es generar proteínas marcadas con una cola de ubiquitinas que se dirigirán al proteosoma para ser degradadas. Mdm2 es la principal ubiquitina ligasa E3, puede modificar p53 en seis residuos de lisina dentro de CTD. Cuando en la célula hay niveles altos de mdm2 se conduce a la poliubiquitinación y degradación nuclear de p53; mientras que si los niveles son bajos se promueve la monoubiquitinación y exportación nuclear de p53, en el citoplasma p53 realiza funciones diferentes a las de un factor de transcripción. Por tanto, mdm2 es un regulador negativo de p53.
Fosforilación. En p53 aparecen muchos sitios ricos en treonina y serina que pueden fosforilarse por acción de Ser-Thr quinasas, la mayoría se acumulan en el extremo N-terminal y se modifican como respuesta al estrés celular para iniciar la reacción de p53. Algunos sitios (como T55, S376 y S378) se fosforilan constitutivamente en células no estresadas, en condiciones normales de crecimiento celular el factor TAF1 fosforila p53 en T55 promoviendo su degradación a través del proteosoma. En respuesta al daño del ADN, la fosfatasa 2A puede desfosforilar T55, mejorando la estabilidad de p53 para promover la detención del ciclo celular. Cuándo ocurre la fosforilación (antes o después de la activación de p53) y qué efectos tiene la fosforilación en p53 (activación o represión) son muy variables en diferentes condiciones. Hay fosforilaciones que impiden la unión a mdm2 a p53 para mejorar su estabilidad, otras modificaciones como fosforilaciones en el dominio C-terminal que promueven la unión de p53 al ADN
Acetilación. CBP/p300 podría acetilar p53 en varias lisinas C-terminales conduciendo a la unión de p53 con el gen diana para activar vías posteriores como detención del ciclo celular, senescencia o apoptosis. Además, la acetilación puede impedir la ubiquitinación de p53 y aumentar su estabilidad. Dos sitios modificados por CBP/p300 son K164 y K101, la acetilación de K164 promueve la inducción de la detención del ciclo celular mediante la expresión de p21 y la acetilación de K101 es fundamental para la regulación de la ferroptosis. En respuesta al daño del ADN, también se puede acetilar p53 en K320 para impedir la fosforilación de serinas en el extremo N-terminal de p53 y permitir la expresión de genes específicos que promueven la supervivencia celular, no la apoptosis . La p53 acetilada puede ser desacetilada por varias desacetilasas como la histona desacetilasa-1 (HDAC1), el cual puede ser reclutado por mdm2.
Metilación. Al igual que la acetilación, es una marca epigenética importante en la lisina de las colas de histonas, también destaca la metilación de arginina. Esta modificación puede estabilizar p53 y restringirlo en el núcleo. También se asocia con una transcripción mejorada de algunos genes diana como p21. Destaca la proteína arginina metiltransferasa 5 (PRMT5) que se encarga de la metilación de p53 en R333, R335 y R337, esta reacción afecta a la especificidad del gen diana de p53 haciéndola más sensible a ciertos promotores o potenciadores.
FUNCIÓN
La proteína p53 en condiciones normales está presente en su forma inactiva a muy baja concentración, mientras que en situaciones de estrés celular, falta de nutrientes o de daño en el ADN, se produce su activación y estabilización. Si el daño es leve detiene la progresión del ciclo celular para permitir su reparación y posteriormente reiniciarlo. Ante un daño grave o irreparable, activa el proceso de apoptosis, reduciendo de esta manera la posibilidad de que células con mutaciones puedan sobrevivir y contribuir al proceso de carcinogénesis. (6)(7)
Por ello, dentro de las funciones más importantes de p53 encontramos la detención del ciclo celular y activación de la apoptosis además de otras funciones secundarias como regular ciertos procesos metabólicos, la respuesta antioxidante y la reparación del ADN. Todas estas funciones confluyen en la idea de que p53 tiene como misión principal ser un supresor de tumores. (6)(7)
DETENCIÓN DEL CICLO CELULAR
La detención del ciclo celular mediada por p53(7)
Está mediada principalmente por la activación transcripcional de p21/WAF1. P53 se une a dos sitios aguas arriba del promotor de P21. El sitio 5′ en el promotor p21 es uno de los sitios de unión a p53 más fuertes analizados, con una constante de disociación muy baja (nM).
El ARNm de p21 es altamente inducido después de la activación de p53. p21 se une a los complejos ciclina E/Cdk2 y ciclina D/Cdk4 para causar la detención de G1 en el ciclo celular. La inhibición de Cdk2 y Cdk4 por p21 bloquea la fosforilación de pRb, promueve la unión de pRb a E2F1 y promueve el silenciamiento de la transcripción de los objetivos E2F1 imprescindibles para la replicación del ADN y la progresión del ciclo celular. p21 también interactúa con el antígeno nuclear celular proliferante (PCNA) e inhibe la replicación del ADN, lo que puede contribuir a su actividad de detención del ciclo celular. Se ha demostrado que la ausencia del factor p21 causa deficiencias en la detención del ciclo celular después del lo que indica que es un mediador importante por p53.
Sin embargo, estas células nulas de p21 no son completamente defectuosas para la detención de G1, lo que se sugiere que otros genes diana de p53 también contribuyen a la detención del ciclo celular.
La activación de p53 también detiene las células en las fases G2/M. Aunque p21 también puede inhibir la ciclina B/Cdc2 para inhibir la progresión del ciclo celular a través de la mitosis, otros genes diana de p53, como el 14-3-3σ, pueden participar en el bloqueo de la transición G2/M.
También se ha demostrado que la represión p53 del promotor cdc25C promueve la detención de G2 / M después del daño del ADN. Estudios recientes ex vivo sugieren que la inducción de micro ARN mir34a contribuye a la detención del crecimiento por p53. Sin embargo, la eliminación de la familia de genes mir34 en ratones no afectó la detención y la apoptosis mediadas por p53, lo que sugiere que la función de mir34 in vivo es limitada o redundante.
En las células de mamíferos, la detención del ciclo celular mediada por p53 proporciona importantes funciones como punto de control:
Las roturas de doble cadena de ADN que se producen por acción de la radiación ionizante activa la (ATM) quinasa que inhibe la actividad de la ligasa MDM2 E3 a través de la fosforilación, causando una rápida acumulación de p53 e inducción de p21. Al detener las células en la fase G1 se da tiempo a la reparación de roturas de doble cadena potencialmente letales.
P53 mantiene la integridad cromosómica y mejora la supervivencia de las células dañadas ya que además de forzar un punto de control del ciclo celular, p53 también regula un grupo de genes implicados en la recombinación y reparación del ADN.
P53 regula los genes implicados en la formación de heterocromatina para facilitar la reparación oportuna de las roturas de la cadena de ADN.
PROMOVER APOPTOSIS
La apoptosis se define como una forma de muerte celular programada en la que las células «se suicidan» fragmentándose en trozos empaquetados de membrana denominados cuerpos apoptóticos. (9)
En la activación de p53, ciertos tipos de células (leucemia o fibroblastos transformados) sufren predominantemente apoptosis en lugar de detención del ciclo celular. (7)
Las limitadas anomalías del desarrollo asociadas con la mayoría de los ratones p53-nulos indican que la proteína p53 puede ser prescindible, en gran medida, para los procesos apoptóticos que ocurren durante el desarrollo normal.(9)
Sin embargo, en condiciones de estrés celular que ponen en peligro la integridad del genoma, p53 se vuelve crucial para restringir la proliferación celular inapropiada. De esta manera, p53 evita la expansión clonal de las células que poseen defectos genéticos peligrosos, restringiendo así la neoplasia. Las situaciones de estrés celular que pueden inducir la apoptosis mediada por p53 incluyen daño en el ADN, hipoxia, escasez de ribonucleótidos y estrés oxidativo. (9)
Además, p53 puede inducir apoptosis sin detención aparente del crecimiento, por ejemplo, en células que tienen expresión desregulada de los protooncogenes c-myc o adenovirus 12S E1A, o el factor de transcripción E2F-1 entre otros, provocan una acumulación y estabilización de p53. (8)
La separación de las vías dependientes de p53 para la detención del crecimiento y la apoptosis puede basarse en la inducción selectiva de productos génicos específicos para cada vía. (8)
Se ha demostrado que p53 regula al alza genes proapoptóticos como Bax y suprime genes antiapoptóticos como BCL-2, mediante la activación transcripcional o represión, alterando así las proporciones relativas de Bax a Bcl-2 y cambiando el equilibrio hacia la apoptosis. (8)
p53 también induce directamente la transcripción de IGF-bp3, una proteína que puede inhibir la actividad antiapoptótica de IGF-1, a través de un motivo de consenso p53 en la región promotora. Esta proteína también puede inducir apoptosis, a través del secuestro de IGF-1. (8)
Por otro lado, Se sugiere que podría existir una interacción funcional entre p53 y una proteína llamada RUNX3 en respuesta al daño del ADN, mejorando así la actividad proapoptótica de p53. Cabe destacar que RUNX3 recluta formas fosforiladas de ATM a p53 en SER-15 y por lo tanto activa p53 (10)
Figura 5: Interacción entre RUNX3 y p53 ante un daño en el ADN y la consecuente activación de p53. Imagen obtenida de (10)
La demostración de que la apoptosis mediada por p53 puede ocurrir en ausencia de síntesis de proteínas o ARN implicó que la transactivación específica de secuencias SST no siempre es imprescindible para la apoptosis. Posteriormente, las investigaciones sobre la capacidad apoptótica de los mutantes p53 deficientes en SST dieron lugar a resultados contradictorios. (9)
Se ha propuesto por tanto que la apoptosis mediada por p53 ocurre a través de una combinación de mecanismos dependientes de SST e independientes de SST, actuando de forma sinérgica. En condiciones fisiológicas la apoptosis óptima requiera una combinación de ambos mecanismos. (9)
Es pertinente señalar un importante gen diana de p53 que aparentemente no contribuye a la apoptosis. El gen p21 inducible por p53 codifica un inhibidor de quinasas dependientes de ciclina. Los estudios de fibroblastos deficientes en p21 revelaron la contribución significativa de la inducción de p21 a la detención del ciclo celular en G1. Sin embargo, los mismos estudios también mostraron claramente que p21 no era necesaria para la apoptosis mediada por p53. (9)
Algunos mutantes de p53 pueden transactivar p21 e inducir la detención del ciclo celular, pero no pueden inducir IGF-bp3 o Bax, y por lo tanto muestran una actividad pro-apoptótica deteriorada. Esto indica que la selectividad del promotor de los mutantes p53 puede determinar si p53 induce la detención del crecimiento o la apoptosis. (8)
Se ha sugerido que la inhibición de la apoptosis inducida por p53 está mediada por MDM2, que está regulada al alza por p300, pero que inhibe la función de p53. MDM2 se une al dominio de activación transcripcional de p53, induce una rápida degradación de p53 y bloquea su capacidad para regular genes diana y ejercer efectos antiproliferativos y proapotóticos. (8)
Figura 6: Esquema de funciones de p53 y su papel en cáncer. Imagen obtenida de (10)
RELACIÓN P53 Y CÁNCER
Ante un daño grave o irreparable en el ADN, p53 activa procesos de apoptosis para disminuir la posibilidad de que las células mutadas sobrevivan y transmitan esos daños a las células hija. Cualquier mutación en p53 u otras proteínas que regulen su función inhiben la actividad de p53 condiciendo a carcinogénesis.
La inactivación de p53 produce un desequilibrio entre la división celular y la muerte celular, que provoca la proliferación continua de la célula. Además, las células con ADN dañado no pueden ser eliminadas y las mutaciones serán heredadas e irán acumulándose en las nuevas células. Como consecuencia, las células tumorales presentan un nuevo fenotipo que las diferencian de las células normales del tejido donde se encuentran, son células que presentan una función y morfología diferentes a las células de las que proceden.
BIBLIOGRAFÍA
1. Appella, E, Anderson, CW (2001.a). Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem., 268, 10:2764-72.
2. Candau, R, Scolnick, DM, Darpino, P, Ying, CY, Halazonetis, TD, Berger, SL (1997). Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene, 15, 7:807-16
3. Cho, Y., Gorina, S., Jeffrey, P. D. & Pavletich, N. P (1994). Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 265, 346-55
4. Jeffrey, PD, Gorina, S, Pavletich, NP (1995). Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science, 267, 5203:1498-502
5. Liu, Y., Tavana, O. & Gu, W. (2019). p53 modifications: exquisite decorations of the powerful guardian. Journal of Molecular Cell Biology, 11(7), 564-577.
6. Liu, J., Zhang, C., Hu, W. & Feng, Z. (2018). Tumor suppressor p53 and metabolism. Journal of Molecular Cell Biology, 11(4), 284-292.
7. Chen, J. (2016). The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harbor Perspectives in Medicine, 6(3), a026104.