β-AMILOIDE, EL PÉPTIDO QUE NO DEJA RECORDAR
Creado por: Lucía Fernández López, Paula de la Fuente Méndez y Andrea García Berrocal
Índice
1.Introducción
2.Origen y papel biológico
3.Isoformas, estructuras y mecanismo
4.Papel biomédico
5.Referencias
1.INTRODUCCIÓN
La β-amiloide es un péptido procedente del corte de la proteína APP. Se encuentra presente como un monómero, de peso molecular 4 kDal. En procesos normales se va a localizar circulando por el líquido cefalorraquídeo y en la sangre, pero en una situación patológica se encuentra formando depósitos en el cerebro, normalmente en la región del hipocampo o en el neocórtex.
Va a ser la responsable de una serie de enfermedades neurodegenerativas como son el Alzheimer o el Párkinson, entre otras.
Lo mencionado anteriormente junto con las funciones y la estructura del péptido será detallado en el artículo que se presenta a continuación.
2.ORIGEN DEL PÉPTIDO β-AMILOIDE
El péptido β-amiloide se forma a partir de la proteólisis de la proteína precursora amiloide, también conocida como APP. Esta proteína se encuentra insertada en la membrana plasmática, gracias a su dominio transmembrana, donde es procesada por las enzimas alfa, beta y gamma secretasa. Dependiendo de las enzimas que actúen se puede dar lugar a dos rutas:
RUTA NO AMILOIDOGÉNICA (predominante)
Esta ruta es la predominante y en ella no se produce el péptido β-amiloide.
La α-secretasa corta a la APP por los aminoácidos 687-688, generando dos fragmentos: uno llamado (s)APPα soluble (donde se encuentra el dominio N-terminal) y que queda en el espacio extracelular y otro fragmento carboxi-terminal llamado C83, con 83 aminoácidos, que quedará insertado en la membrana.
Posteriormente, la γ-secretasa realiza otro corte, en los aminoácidos 712/714/715, generando el péptido p3 (que saldrá al espacio extracelular) y el fragmento γ-CTF (que quedará en el intracelular).
RUTA AMILOIDOGÉNICA
En esta ruta, y gracias a la acción de dos secretasas, se produce el péptido β-amiloide.
La β-secretasa se encarga de cortar a la APP por los aminoácidos 671/672, produciendo dos fragmentos distintos a los anteriores: el fragmento (s)APPβ soluble (donde se encuentra el dominio N-terminal) que se libera al espacio extracelular y otro fragmento carboxi-terminal llamado C99, esta vez con 99 aminoácidos, que quedará insertado en la membrana, al igual que ocurría en la vía no amiloidogénica.
Este fragmento C99 será cortado por la enzima γ-secretasa, produciendo el péptido β-amiloide (Aβ), que saldrá al espacio extracelular y el fragmento γ-CTF, que será transportado al núcleo pudiendo actuar como factor activador de la expresión de determinados genes.
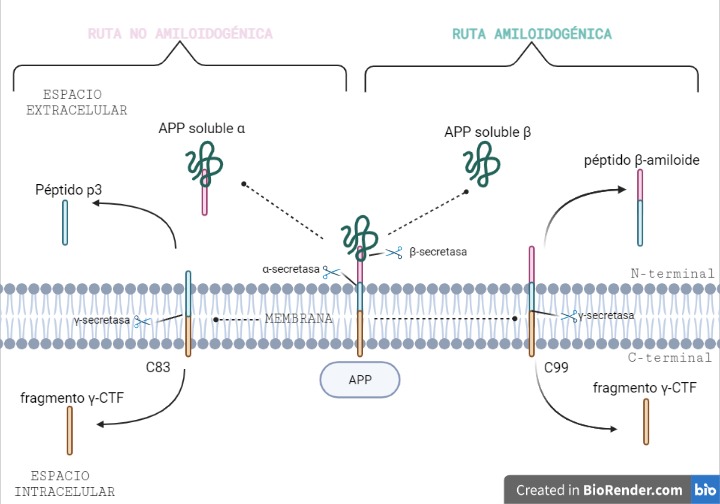
Figura 1. Procesamiento de la APP mediante la ruta no amiloidogénica y la ruta amiloidogénica. Imagen creada por Paula de la Fuente Méndez, a través de BioRender.
PAPEL BIOLÓGICO
FUNCIONES DEL PÉPTIDO β-AMILOIDE
El péptido β-amiloide además de ser uno de los responsables del Alzheimer, desarrolla una serie de funciones en el cuerpo que no están relacionadas con dicha enfermedad.
Se encarga de promover tanto la proliferación celular, como la adhesión entre células; interviene en vías de señalización tales como la activación de las MAP quinasas; en condiciones fisiológicas protege y evita la muerte de las neuronas; desarrolla una función autocrina y antimicrobiana. Además, se ha propuesto su posible labor ayudando al transporte del colesterol y en los canales de KCa.
ESTRÉS OXIDATIVO
El estrés oxidativo es un proceso debido al incremento excesivo de especies reactivas (conjunto de radicales libres) que tiene como efecto una pérdida de la funcionalidad en la célula por el daño producido por la oxidación.
La β-amiloide es responsable de generar radicales libres (15), que producen la oxidación de los elementos que se encuentran en el cerebro, muy sensible al estrés oxidativo debido a la gran cantidad de glucosa, ácidos grasos, enzimas y a la insuficiencia de antioxidantes en él. Tiene entre otras consecuencias que las ATPasas, las vías de señalización y los factores de transcripción no tengan una correcta actividad, dando lugar a la pérdida de la homeostasis.
Cabe destacar que los radicales libres aún con el daño que producen, desarrollan funciones en la célula como la protección inmunológica o la expresión de genes.
El estrés oxidativo promueve la formación de agregados (15), es decir, la unión de β-amiloides mal plegadas. Sin embargo, no se sabe con certeza si es uno de los responsables del inicio del Alzheimer o el resultado del mismo.
La importancia de la oxidación de los componentes del cerebro se ha demostrado sometiendo a neuronas a estrés oxidativo, dando como resultado una disminución en la producción de ATP (debido a las ATPasas de señalización) y un aumento de calcio (por la función de los canales Ca-K) (16). Esto desemboca en una situación excesivamente inestable, siendo la solución la apoptosis de la célula.
DAÑO TRAUMÁTICO
Al producirse un traumatismo se genera estrés oxidativo, produciendo una disminución en la eliminación del péptido β-amiloide. Al disminuir su eliminación, se va a tender a la formación de las placas amiloides. Por esta razón, aumenta de una manera más significativa la isoforma Aβ1-42 (la que crea dichas placas), aunque también se produce un aumento de la isoforma Aβ1-40 (15). Las placas van a tener una relación inversamente proporcional con el estrés oxidativo, van a hacer que este disminuya, y con ello disminuye el daño citoplasmático.
RECEPTORES β-AMILOIDE
LRP y el receptor Scanvenger (RS) son una serie de receptores que están relacionados con la toxicidad de la beta amiloide
LRP
El LRP1, regulado por la α2-macroglobulina, se encarga de endocitar y degradar el péptido β-amiloide. Sin embargo, en una situación patológica donde se produzca este péptido en exceso, LRP1 no va a poder llevar a cabo la endocitosis, por lo que se producen patologías.
Dentro de la familia de los LRP, también se encuentra la glicoproteína 330 (LRP2) que se encarga del transporte de un complejo, resultado de la unión de la beta amiloide y uno de sus ligandos: la ApoJ.
Receptor Scanvenger (RS)
Por último, resalta la posible importancia del receptor Scanvenger (RS). Se ha demostrado su incremento en los cerebros de las personas que sufren la enfermedad del Alzheimer y en un experimento realizado sobre ratones (1), donde se demostró la necesaria presencia de β-amiloide, sugiriendo que ayuda a la actividad neuronal hasta el momento en el que se produce la adhesión entre fibrillas.
Este receptor facilita la adhesión de fibrillas y controla otros procesos relacionados con la β-amiloide, como su endocitosis y degradación. Además, se ha demostrado en otro estudio la relación de este receptor con el estrés oxidativo, observándose la reducción del RS. De esta manera, se produce la pérdida de la capacidad de endocitar el péptido, pudiendo ser el responsable de la acumulación de agregados.
3.ISOFORMAS, ESTRUCTURA Y MECANISMO
ISOFORMAS
Dependiendo del número de residuos de aminoácidos que tenga el péptido β-amiloide, se pueden generar distintas isoformas, siendo las más comunes la Aβ1-40, con 40 residuos, y la Aβ1-42 , con 42 residuos. Ambas isoformas son neurotóxicas, pero van a presentar diferencias con respecto a su metabolismo, funciones, toxicidad y a la forma de agregarse.
La isoforma Aβ1-40 se encuentra en mayor concentración en los vasos sanguíneos del cerebro, produciendo la angiopatía amiloide cerebral, de la que se hablará más tarde en este artículo.
La isoforma Aβ1-42 es menos soluble, con un fragmento C-terminal más estructurado y va a tender a agregarse de una manera más rápida para formar las placas amiloides, es decir, agregados que han precipitado en el espacio intercelular (entre neuronas). Esta isoforma, por tanto, se encuentra en el cerebro en mayor concentración que en la sangre.
Además de esas, podemos distinguir otras isoformas como pueden ser la β-amiloide-25, β-amiloide-35 y β-amiloide-38 con 25, 35 y 38 residuos respectivamente (13).
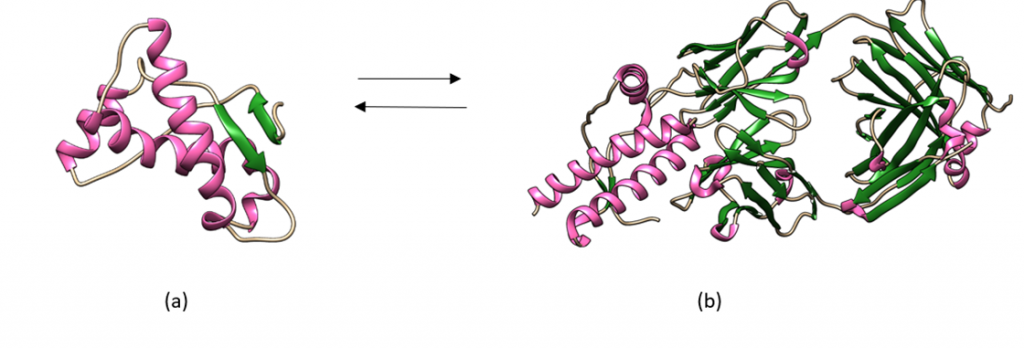
Aβ1-40
Figura 2. Vídeo de la estructura de la Aβ1-40 realizado con ChimeraX, basado en PDB 1BA4, obtenido de protein data banc. En el primer vídeo se observan en rosa las hélices α, mientras que en verde se ven los bucles (coils). Vídeo creado por Paula de la Fuente Méndez.
Aβ1-42
Figura 3. Vídeo de la estructura de la Aβ1-42 realizado con ChimeraX, basado en PDB 1IYT , obtenido de protein data banc. En el segundo vídeo se observan también las hélices α, mientras que en verde se ven los bucles (coils). Vídeo creado por Paula de la Fuente Méndez.
ESTRUCTURA
El péptido β-amiloide, cuando se encuentra aislado, no tiene estructura terciaria compacta, ya que es intrínsecamente desestructurado. Además, gracias a varios estudios realizados mediante Resonancia Magnética Nuclear y dinámica molecular (5), se sabe que tampoco tiene una estructura secundaria significativa, pero sí cuenta con una estructura en espiral.
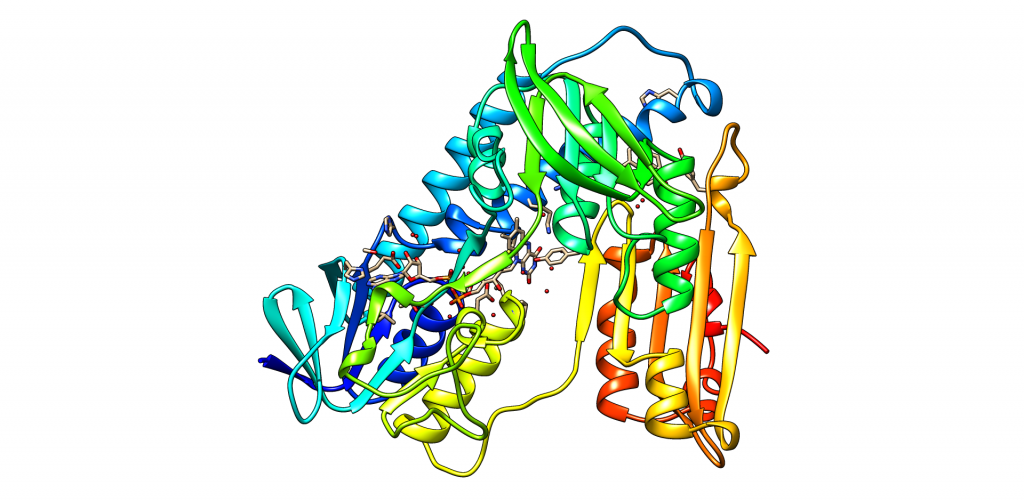
En el caso de que el péptido β-amiloide se encuentre formando agregaciones, es decir, que las hélices alfa hayan pasado a láminas beta y se hayan pegado unas a otras, los residuos 18 a 40 o 42 (dependiendo de la isoforma) van a formar un motivo β-lámina – giro – β-lámina a través de dos láminas beta paralelas, mientras que el resto de residuos se encuentran desordenados.
Figura 4. Vídeo del motivo β-lámina – giro – β-lámina realizado con ChimeraX, basado en PDB 2BEG, obtenido de protein data banc. Se observa el motivo formado entre los aminoácidos 18-42. Vídeo creado por Paula de la Fuente Méndez.
MECANISMO DE FORMACIÓN DE PLACAS
El péptido β-amiloide, puede plegarse de varias formas. En el caso de hacerlo correctamente, se pliega formando una α-hélice, que es una forma soluble y que se puede eliminar.
Sin embargo, este péptido tiene tendencia a plegarse de forma incorrecta. Esto se debe a que los anillos aromáticos de los residuos del péptido, orientados el uno al otro, tienden a solapar sus orbitales (“stacking”) y a pegarse entre ellos. Esto obliga a que cambie la estructura a β-láminas, en las que los anillos aromáticos estabilizan la estructura. Al cambiar la estructura de un péptido, por efecto amplificador, los β-amiloide cercanos también cambian su estructura a láminas beta.
Estos péptidos tienden, por tanto, a agregarse y polimerizarse, formando consecutivamente oligómeros, protofibrillas y fibrillas que darán lugar a las placas amiloides, unas estructuras insolubles que se depositan fuera de las células.
Figura 5. Vídeo de las fibrillas creadas por la Aβ1-42 realizado con ChimeraX, basado en PDB 2MPZ, obtenido de protein data banc. Vídeo creado por Paula de la Fuente Méndez.
También se pueden formar agregados no estructurados, donde las cadenas se unen unas con otras por la mala orientación de los anillos aromáticos de los residuos. Estos agregados no estructurados son estructuras insolubles, que precipitan, se acumulan y terminan destruyendo la célula.
En situaciones normales, la proteína ApoE actúa como chaperona, replegando correctamente el péptido, antes de que se formen las placas amiloides. Sin embargo, puede haber mutaciones en esta proteína, derivando en la formación de placas, ya que los péptidos no se pliegan correctamente.
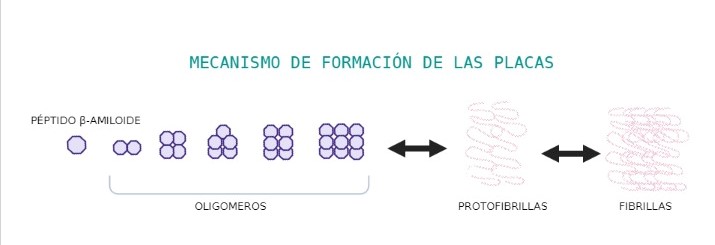
Figura 6. Proceso por el cual el péptido β-amiloide forma oligómeros, protofibrillas y fibrillas. Creado con BioRender por Paula de la Fuente Méndez.
4. PAPEL BIOMÉDICO
ENFERMEDADES
ALZHEIMER
El Alzheimer es un trastorno neurodegenerativo gradual. Esta enfermedad es la principal causa de demencia en personas mayores. Afecta, entre otras cosas, a la comprensión y a la memoria.
Está relacionada con el péptido β-amiloide, ya que altera la sinapsis al fragmentarse las mitocondrias del cerebro y desemboca en la muerte de las neuronas. Esto se produce debido a la excesiva producción de óxido nítrico (un radical libre) por parte de este péptido. El óxido nítrico, al reaccionar con la DRP1 (dynamin related protein 1), hace que las mitocondrias del cerebro se fragmenten.
El péptido β-amiloide, además, es el que forma las placas amiloides o las placas seniles, localizadas en el cerebro de las personas que padecen el Alzheimer. Estas placas también se encuentran en personas que padecen la enfermedad del Párkinson y la enfermedad de los cuerpos de Lewy, ambas resultando en demencias.
Es probablemente la producción de radicales libres en exceso la causa del Alzheimer, mientras que la agregación de las β-amiloide es una de las consecuencias. Hay mutaciones en la proteína APP que pueden provocar un aumento de la concentración de β-amiloide (un 10% son las personas que presentan la enfermedad de forma hereditaria). También puede haber mutaciones que favorezcan la vía amiloidogénica, de forma que se crea más péptido β-amiloide.
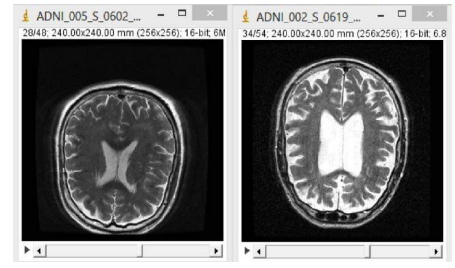
Los tratamientos existentes para el Alzheimer no frenan la enfermedad, únicamente disminuyen los síntomas que esta produce, mejorando la calidad de vida de las personas que la padecen.
Figura 7. Comparación entre un cerebro sano (izquierda) y uno afectado por Alzheimer (derecha). ISBN: 978-84-608-3354-3
Angiopatía amiloide cerebral
El péptido β-amiloide, además de formar las placas seniles (Aβ1-42), puede acumularse cubriendo las paredes de los vasos sanguíneos del cerebro (Aβ1-40) y producen la angiopatía amiloide cerebral. Esta afección, además de demencia, causa un sangrado del cerebro.
Además, la isoforma Aβ1-40 se encuentra en el miocardio de las personas que padecen Alzheimer e insuficiencia cardiaca, por lo que hay una correlación entre la concentración de este péptido y los factores de riesgo de esta última enfermedad. Esto quiere decir que un aumento de la concentración de Aβ40 implica un aumento de los factores de riesgo. (11)
FÁRMACOS (INHIBIDORES)
Se ha intentado llevar a cabo una serie de fármacos cuyo objetivo sea la inhibición de la formación del péptido β-amiloide e incluso la inhibición de la agregación del mismo.
INHIBIDORES DE LA β- SECRETASA (BACE1)
La enzima β-secretasa va a comenzar una de las rutas del procesamiento de la APP, la ruta amiloidogénica. A la hora de desarrollar inhibidores para la misma, hay que tener en cuenta que tiene otros muchos sustratos, por lo que su inhibición inespecífica puede tener los efectos contrarios. Otro factor que dificulta la creación de estos inhibidores es la propia estructura de la proteína: la β-secretasa forma parte de las aspartilproteasas, por lo que la molécula que se encargue de inhibirla tendría que ser grande e hidrofóbica, dificultando su paso por la barrera hematoencefálica.
A pesar de esto, unos estudios recientes muestran que hay 2 inhibidores (E2609 y MK-8931) capaces de disminuir la producción de los niveles de β-secretasa en el líquido cefalorraquídeo entre un 80 y 90 %. (10)
INHIBIDORES Y MODULADORES DE LA γ- SECRETASA
La γ-secretasa, además de procesar la APP, se encarga de procesar múltiples proteínas más. Por lo que, al igual que sucede con la β-secretasa, a la hora de desarrollar inhibidores, hay que tener en cuenta que tiene otros muchos sustratos, por lo que su inhibición inespecífica puede tener los efectos contrarios.
Se desarrollaron una serie de fármacos como posibles inhibidores de la γ-secretasa como semagacestat (LY450139), (10) comprobándose que reducía los niveles del péptido β-secretasa en la sangre y en el líquido cefalorraquídeo. Sin embargo, no disminuía la progresión lenta de la enfermedad del Alzheimer.
Se desarrollaron también una serie de moduladores selectivos de esta enzima, para poder impedir los efectos adversos que podrían causar los inhibidores. Estos moduladores bloquean a la enzima con respecto al procesamiento de la APP, pero no influye en otras vías de señalización que pueda llevar a cabo como es la Notch.
Uno de estos moduladores es el NIC5-15 (10) que va a inhibir a la γ-secretasa sin interferir en Notch. Además, es un sensibilizador bastante potente de la insulina y se está investigando también su posible papel en la inhibición del proceso inflamatorio, más concretamente inhibiendo la activación de la microglía.
Otro modulador de la γ-secretasa es CHF5074 (10) que es un derivado antiinflamatorio no esteroideo.
ACTIVACIÓN DE LA α-SECRETASA
Mediante la activación de la α-secretasa se pretende estimular la vía no amiloidogénica para que, de esta manera, no se lleve a cabo la vía amiloidogénica, evitando así que se produzca la formación del péptido β-amiloide. Aunque la mayoría de estos inhibidores están siendo probados aún en estudios clínicos.
El galato de epigalocatequina (EGCG) (10) es un flavonoide polifenólico que va a inhibir la producción de oligómeros tóxicos malformados de la β-amiloide y, además, va a activar a la α-secretasa.
Etazolato (EHT 0202) (10) impulsa la acción neurotrófica de esta enzima inhibiendo, además, la muerte neuronal producida por el péptido β-amiloide.
La acitretina, (10) una de las funciones que tiene es activar la α-secretasa, activando la ruta no amiloidogénica de la APP en células de neuroblastoma y, además, va a reducir los niveles de β-amiloide en ratones transgénicos.
INHIBIDORES DE LA AGREGACIÓN
El objetivo del desarrollo de los inhibidores de las agregaciones de la β-amiloide es encontrar un péptido que se parezca a la isoforma Aβ1-40 y que consiga retardar la agregación de la Aβ1-42, ya que se ha demostrado que entre ambas isoformas va a haber una interacción que tiene la capacidad de regular el proceso de agregación de dicho péptido.
En base a esto se han creado muchos inhibidores basados en la secuencia 17-21 de este péptido (considerado el centro de nucleación) ya que es considerada como una diana sobre la cual se van a dirigir los potenciales inhibidores de la agregación.
UTILIZACIÓN DE AMINOÁCIDOS SINTÉTICOS
Una de las numerosas técnicas que se han probado para reducir la toxicidad de la beta amiloide es el cambio de aminoácidos en el tramo hidrofóbico del péptido.
Existen una serie de residuos (G25 a G37) que promueven la agregación. El cambio por leucinas o glicinas previene la formación de dímeros, trímeros y fibrillas, además de la pérdida de la estabilidad de la estructura.
De igual manera, la Metionina 35 es un residuo que interviene en el estrés oxidativo. Si es sustituido por valina, aumenta la toxicidad, mientras que si lo es por isoleucina o cisteína, disminuye.
Cabe destacar que en el laboratorio usualmente se utiliza el péptido beta amiloide 25-35, muy similar a las isoformas 40 y 42 del péptido, ya que comparten el mismo grado de toxicidad y capacidad de agregación pero es posible su producción de una manera menos costosa.
5.REFERENCIAS
- Abramov E, Dolev I, Fogel H, Ciccotosto , Ruff E y Slutsky I. (2009). “La proteína beta amiloide es esencial para la transmisión de la información entre neuronas”. Nat Neurosci 2009; 12: 1567-1576 https://neurologia.com/noticia/1916/la-proteina-beta-amiloide-es-esencial-para-la-transmision-de-la-informacion-entre-neuronas
- Aguirre-Rueda, Diana. (2014). “Daño inflamatorio y estrés oxidativo en la Enfermedad de Alzheimer. Efecto de polifenoles y cannabinoides”. Tesis doctoral. DOI 10.13140/RG.2.1.5099.6080
- Alvaro Barrera-Ocampo, Francisco Lopera. (2016). “Inmunoterapia beta-amiloide: ¿la esperanza para la enfermedad de Alzheimer?”. Colomb. Med. vol.47 no.4 Cali Oct./Dec. 2016. Disponible en: http://www.scielo.org.co/scielo.php?pid=S1657-95342016000400203&script=sci_arttext&tlng=es Download PDF: http://www.scielo.org.co/pdf/cm/v47n4/1657-9534-cm-47-04-00203.pdf
- Ana Esther Estrada Rodríguez y Viviana Chantal Zomosa Signoret. (2017). “Papel de la agregación del péptido Beta Amiloide en la enfermedad de Alzheimer”. Revista de Educación Bioquímica (REB) 36(1):2-11 Download PDF: https://www.medigraphic.com/pdfs/revedubio/reb-2017/reb171b.pdf
- Chen, Gf., Xu, Th., Yan, Y. et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin 38, 1205–1235 https://doi.org/10.1038/aps.2017.28 Download PDF: file:///C:/Users/Admin/Downloads/aps201728-1.pdf
- Cho D-H, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z y Lipton SA. (2004). “Las placas de proteína beta amiloide dispersan radicales libres que pueden dañar las neuronas”. Science 2009; 324: 102-105. Disponible en: https://neurologia.com/noticia/1485/las-placas-de-proteina-beta-amiloide-dispersan-radicales-libres-que-pueden-danar-las-neuronas
- Cruz Gatell, Montserrat (2010). “Diseño, síntesis y evaluación de inhibidores de la proteína beta-amiloide. Desarrollo de un modelo de fibrilogénesis”. Tesis del Departament de Química Orgànica de la Universitat de Barcelona (UB). Disponible en: http://hdl.handle.net/10261/22990 Download Pdf: https://digital.csic.es/bitstream/10261/22990/3/Cruz_Montse_3.pdf
- Dra. Silvia Gra Menéndez, Dr. Noel Padrón Pérez y Dr. Juan de Jesús Llibre Rodríguez (2002). “Péptido beta amiloide, proteína Tau y enfermedad de Alzheimer”. Rev Cubana Invest Bioméd v.21 n.4 Ciudad de la Habana oct.-dic. 2002. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-03002002000400006 Download PDF: http://scielo.sld.cu/pdf/ibi/v21n4/ibi06402.pdf
- Grégory A. García, Sergio Hernández V., Ómar Ramón M., Segundo A. Baez y Ananías García C. (2007). “Biología y patobiología humanas del complejo de absorción y transporte epitelial megacubam”. Revista MED 15 (1): 94-104 Download PDF: http://www.scielo.org.co/pdf/med/v15n1/v15n1a11.pdf
- J. Folch, M. Ettcheto, D. Petrov, S. Abad, I. Pedrós, M. Marin, J. Olloquequi, A. Camins (2015) “Review of the advances in treatment for Alzheimer disease: strategies for combating β-amiloide protein: Neurología (English Edition), Volume 33, Issue 1, January–February 2018, Pages 47-58”. Disponible en: https://www.sciencedirect.com/science/article/pii/S021348531500064X Download PDF
- Kimon Stamatelopoulos, Konstantinos Stellos (2017). “El amiloide beta (1-40) circulante predice eventos en pacientes con insuficiencia cardiaca”. Revista Española de Cardiología vol. 70, issue 11,pp: 905-906 Published by Elsevier. DOI: 10.1016/j.recesp.2017.05.019. Download PDF: https://www.revespcardiol.org/es-pdf-S0300893217303512
- Qiu T, Liu Q, Chen YX, Zhao YF, Li YM. “Aβ42 and Aβ40: similarities and differences”. J Pept Sci. 2015 Jul;21(7):522-9. DOI: 10.1002/psc.2789. Epub 2015 May 28. PMID: 26018760. Disponible en: https://pubmed.ncbi.nlm.nih.gov/26018760
- Fuente: ABC, M.Lopez (2017). “Hallan cómo detectar precozmente el alzhéimer a partir de un simple análisis de sangre”. Disponible en: https://www.diagnosticsnews.com/noticias/27107-la-medicion-de-las-isoformas-de-la-proteina-beta-amiloide-en-la-sangre-hallan-como-detectar-precozmente-el-alzheimer-a-partir-de-un-simple-analisis-de-sangre
- Muñoz López, F. J. (2001). “El péptido β-amiloide: mecanismos de neurotoxicidad. Neuroprotección por antioxidantes y estrógenos” Rev Esp Geriatr Gerontol 36(2):109-116. Disponible en: https://www.elsevier.es/es-revista-revista-espanola-geriatria-gerontologia-124-articulo-el-peptido-amiloide-mecanismos-neurotoxicidad–S0211139X0174694X Download PDF: https://www.elsevier.es/es-revista-revista-espanola-geriatria-gerontologia-124-pdf-S0211139X0174694X
- Natalia Manzano-León y Jaime Mas-Oliva. (2006). “Estrés oxidativo, péptido β-amiloide y enfermedad de Alzheimer”: Instituto de Fisiología Celular, Universidad Nacional Autónoma de México. México D.F., México. Download PDF: https://www.scielo.org.mx/pdf/gmm/v142n3/v142n3a9.pdf
- Pacheco Jenkins, C. (2009).”Efecto del stress metabólico sobre la susceptibilidad celular frente a la formación de canales iónicos y efectos citotóxicos generados por los péptidos amiloides”. Disponible en: https://repositorio.uchile.cl/handle/2250/133177 Dowload PDF: https://repositorio.uchile.cl/bitstream/handle/2250/133177/Efecto-del-stress-metab%c3%b3lico-sobre-la-susceptibilidad-celular-frente-a-la-formaci%c3%b3n-de-canales-i%c3%b3nicos-y-efectos-citot%c3%b3xicos-generados-por-los-p%c3%a9ptidos-amiloides.pdf?sequence=1&isAllowed=y
QUIMERA → Molecular graphics and analyses performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. Protein Sci. 2021 Jan;30(1):70-82








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}