El precio de la inmortalidad
Andrea Adrados Santa Elena, Alba Arranz Benayas y Laura Arranz Ortega, 3º Biología Sanitaria UAH
La muerte nos asusta a todos pero, ¿merecería la pena alcanzar la inmortalidad?
La esperanza de vida cada vez es mayor con el paso de los años, gracias a los avances médicos, y la mejora de las condiciones de vida; pero nuestras células siguen teniendo una fecha de caducidad.
Existen varias estrategias para frenar ese envejecimiento celular y así prolongar su duración. Esto se podría conseguir gracias a una enzima, la telomerasa, que permitiría alargar los telómeros acortados, que son los responsables de la muerte celular, lo que supondría un “rejuvenecimiento” de la célula. De esta manera se conseguiría incrementar nuestros años de vida.
No obstante, la telomerasa tiene su lado negativo, puesto que está presente en la mayoría de cánceres en los que las células tienen un crecimiento ilimitado. Por lo tanto, conseguir una activación constante de las células de nuestro cuerpo puede ser un gran peligro, ya que si no se controla, en vez de darnos más años de vida, nos los estaría quitando.
Telómeros
¿Qué son los telómeros?
Los telómeros son complejos nucleo-proteicos que constituyen las estructuras de los extremos de los cromosomas lineales permitiéndoles mantener su integridad ya que otorgan estabilidad y protección.
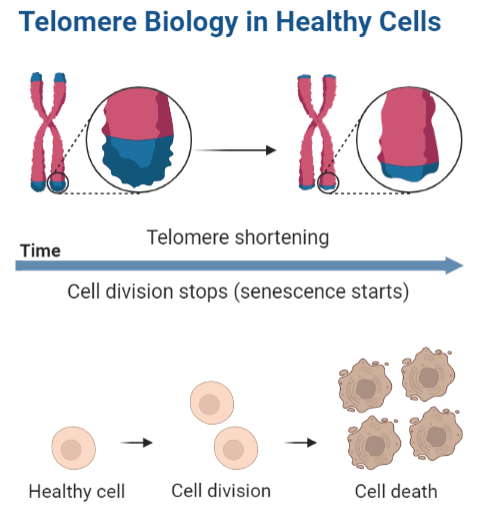
Las ADN polimerasas, enzimas encargadas de replicar el ADN, necesitan un extremo OH 3’ libre sobre el que ir añadiendo nucleótidos y rellenar el hueco que queda tras eliminar el cebador. Sin embargo, al tratarse de un cromosoma lineal dicho extremo no está presente y en cada ronda de replicación se pierde un determinado número de bases. Los telómeros desempeñan por tanto una función clave ya que evitan que se pierda información vital, en su lugar se perderán bases de su estructura.
Además, están a cargo del reloj mitótico y por consiguiente la senescencia celular, es decir, determinan el número de divisiones que tendrá la célula y la proliferación celular se frenará cuando la longitud de los telómeros sea crítica. Llegados a ese punto se dirige la célula a la muerte celular, que es lo que desemboca en degeneración tisular y se traduce, en lo que cotidianamente vemos como envejecimiento
Estructura de los telómeros
Su estructura difiere del resto de la cromatina, de manera que no se llevan a cabo procesos de degradación, recombinación o fusión, es decir, les permiten no ser reconocidos por los sistemas de reparación del ADN.
Podemos encontrar tres regiones:
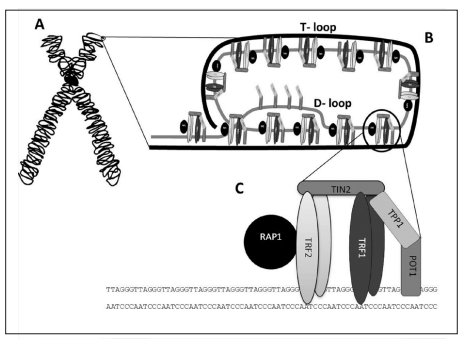
- Extremo saliente (extremo 3’ overhang): son las secuencias no replicadas que quedan libres en forma de cadena sencilla. Posibilita la formación de unas estructuras secundarias en forma de bucle (T-loop y D-loop) por inserción de dicho extremo 3’ overhang en la región de doble cadena y posterior hibridación por complementariedad. Esto evita que los extremos de los cromosomas sean confundidos con ADN dañado.
- Repeticiones teloméricas: se trata de secuencias cortas repetidas y conservadas entre las especies. Son ricas en nucleótidos G y T (en el caso de los humanos las secuencias teloméricas son TTAGGG) y pueden formar los G-cuadruplexos, estructuras complejas donde 4 guaninas quedan unidas por puentes de hidrógeno de Hoogsten formando planos cuadrados. Estos están implicados en el mantenimiento de los telómeros, pero hay que regular su apertura para permitir la replicación del ADN.
- Áreas que están entre la primera secuencia de un gen y las repeticiones.
Interacción con proteínas
En los seres humanos, los telómeros interactúan con el complejo de la shelterina, formado por una serie de proteínas que incluyen a TRF1 y TRF2, las cuales interactúan con RAP1, TIN1, TPP1 y POT1. La función del complejo es impedir la activación del mecanismo de reparación del ADN en los extremos, protegiendo frente a la degradación, y regular la actividad de la telomerasa.
- TRF1: es la secuencia C-terminal. Reconoce específicamente el fragmento de ADN telomérico y actúa como regulador negativo de la longitud telomérica (represor de la telomerasa)
- TRF2: regulador negativo de la longitud telomérica, estabiliza la secuencia G repetitiva y previene de fusiones entre extremos teloméricos de distintos cromosomas
Por tanto, ambos restringen la actividad de la telomerasa, impidiendo la elongación telomérica
Reloj mitótico
A medida que los telómeros de las células se van acortando estas se vuelven senescentes, siendo la senescencia una situación en la que las células a pesar de ser viables y activas metabólicamente ya no proliferan, es irreversible, y conduce hacia la muerte celular. Cuando se detecta una longitud crítica de los telómeros se pone en marcha un mecanismo que bloquea el avance del ciclo celular y promueve la entrada en apoptosis gracias a la activación de las proteínas p53 y Rb (proteínas inhibidoras del ciclo celular).
Los telómeros pueden presentarse de dos formas: protegidos (capped) si están formando el T-loop, y desprotegidos (uncapped) si están de manera lineal, pudiendo haber transiciones entre ambas estructuras. El último caso se da cuando los telómeros son tan cortos que ya no son capaces de formar estructuras secundarias lo que hace que sean susceptibles de sufrir el ataque de nucleasas y fusión de extremos. Esto conduce a la inestabilidad cromosómica y senescencia, con la consecuente entrada en apoptosis ya mencionada.
Existe una teoría que sugiere que los telómeros del cromosoma 17 que es donde está codificada la proteína p53 sea el sitio donde se activa este mecanismo.
Telomerasa
Estructura de la telomerasa
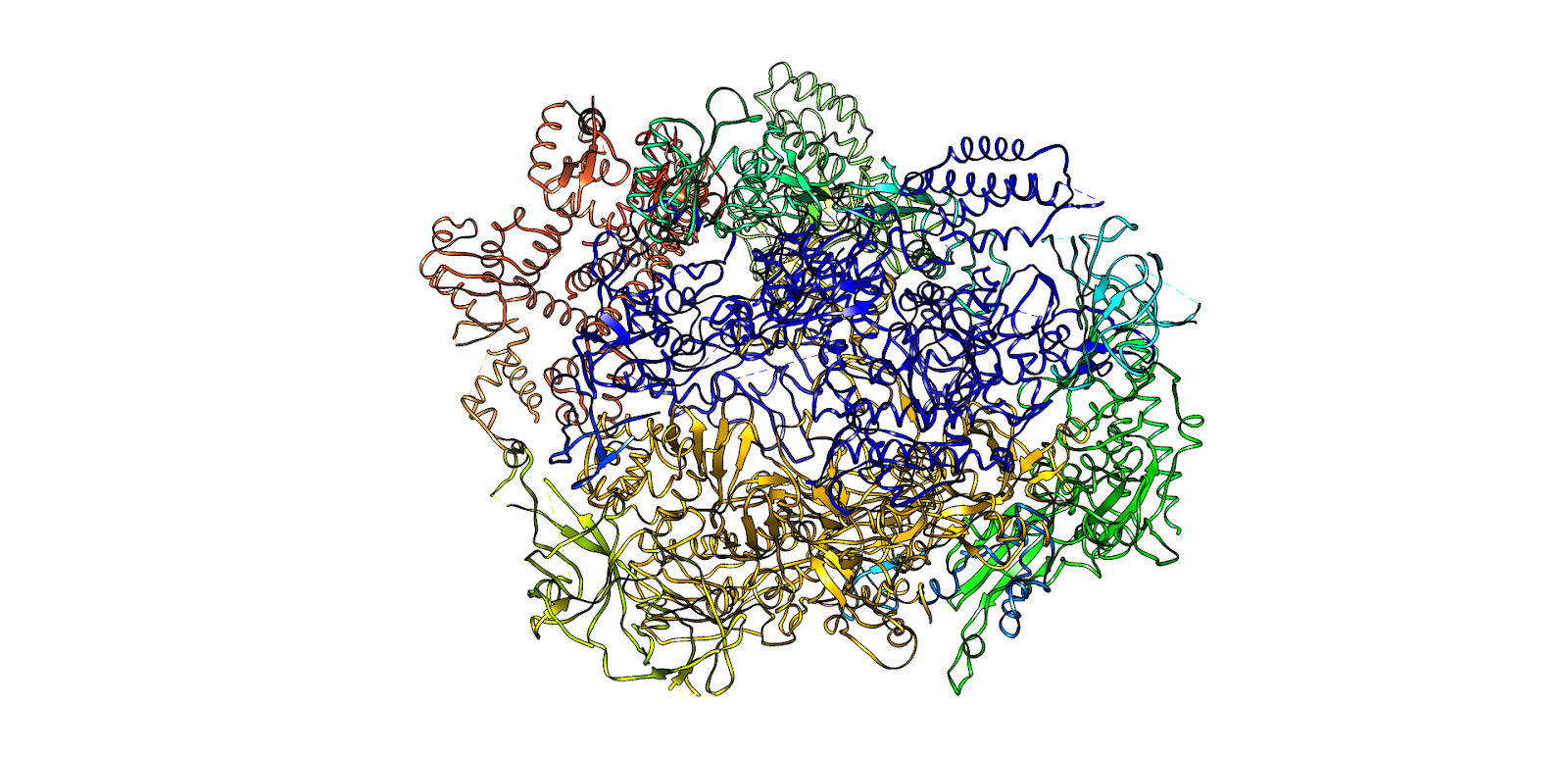
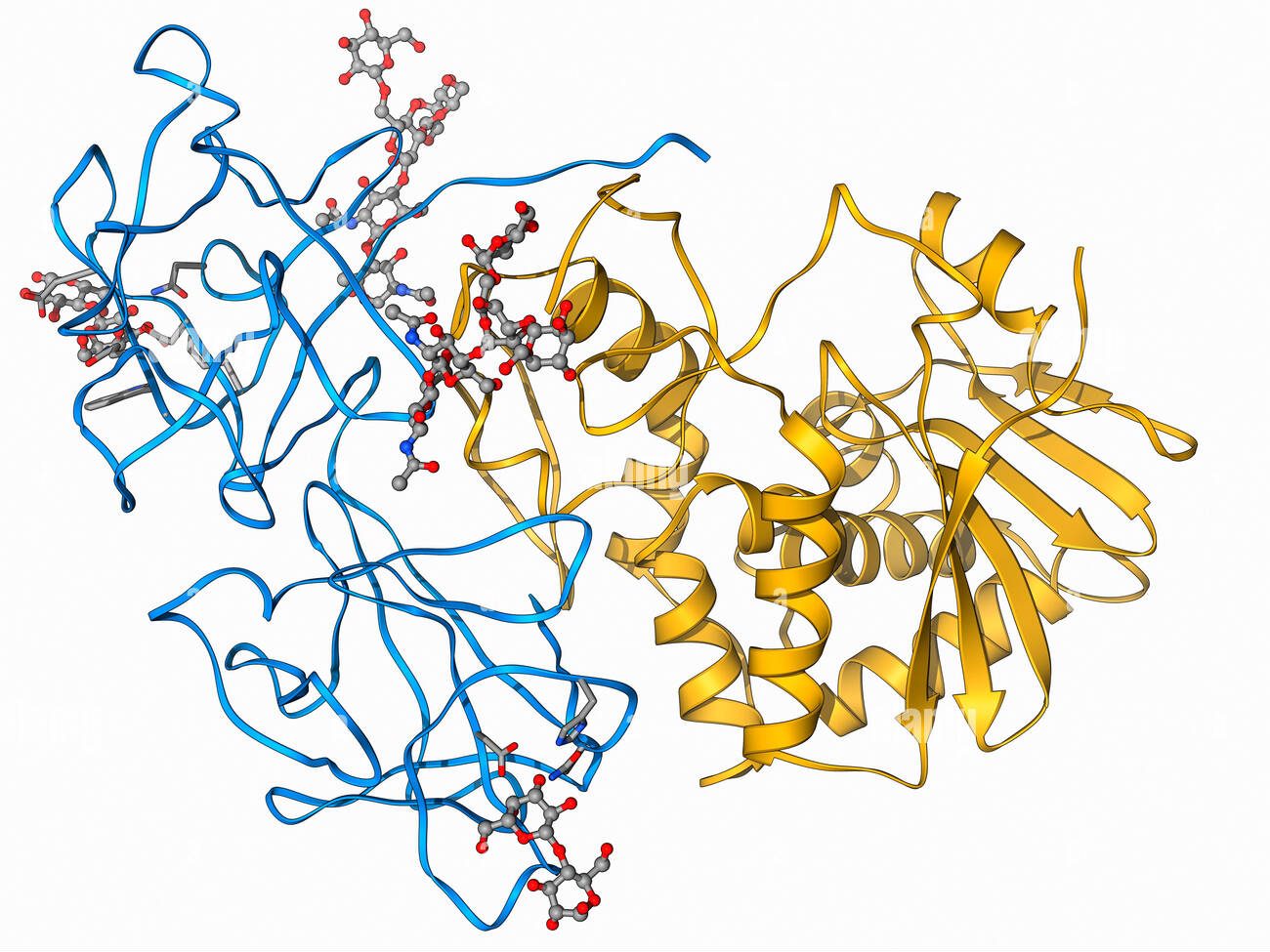
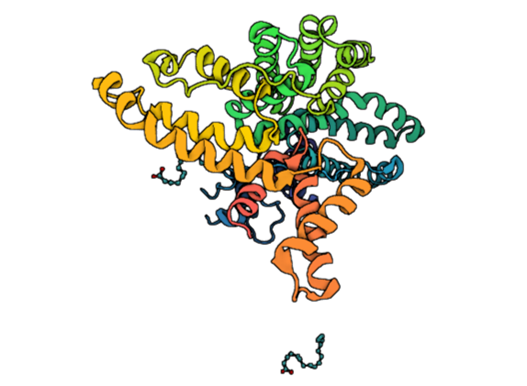
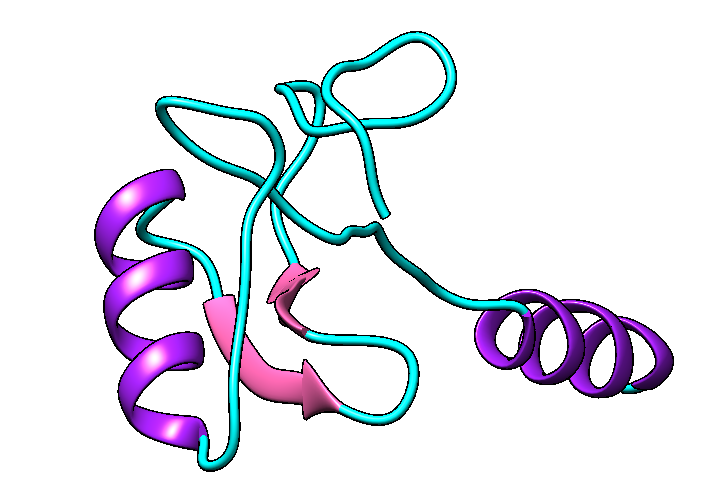
La telomerasas es una ribonucleoproteína formada por una subunidad catalítica llamada TERT, que es una retrotranscriptasa y un componente de ARN llamado TERC, que actúa como molde para la adición de secuencias teloméricas repetitivas en el extremo 3’.
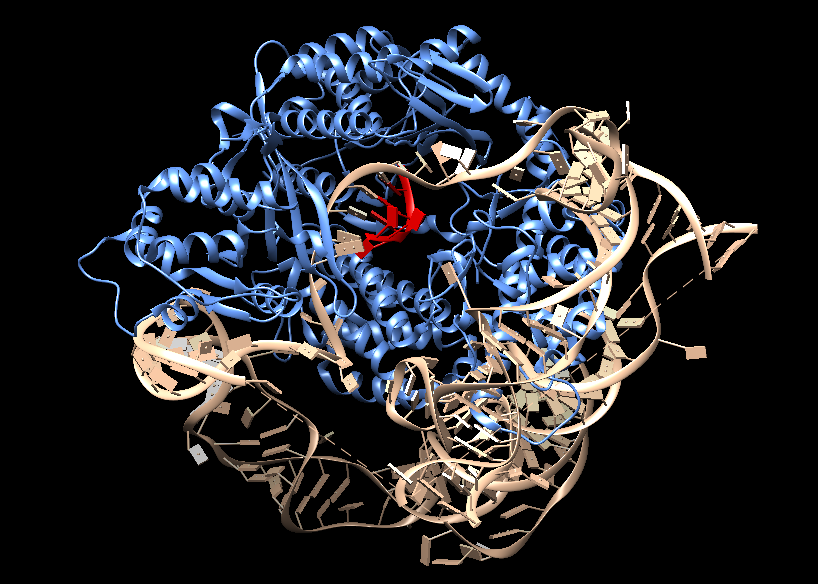
Imagen 3: Telomerasa humana: subunidad catalítica TERT (azul), componente ribonucleico TERC (beige) y unidad telomérica de DNA correspondiente a secuencia corta de repetición TTAGGG (rojo). Fuente: imagen elaborada con Chimera, código PDB 7TRD
Función de la telomerasa
La telomerasa es la encargada de alargar los telómeros, se encuentra activa en todos los tejidos durante la embriogénesis y tras esta únicamente permanece en células de líneas germinales productoras de gametos y en células cancerosas. Su función está regulada tanto por proteínas quinasas (añaden un grupo fosfato) que aumentan su actividad, como por fosfatasas (eliminan un grupo fosfato) que reducen su actividad, por ello el balance entre ambas juega un papel esencial en la tumorigénesis.
Esta enzima tiene una gran afinidad por las secuencias ricas en G, reconoce y se une al extremo 3’ overhang del telómero, al ser reclutada por complejo de la shelterina, y lo alarga mediante la adición de nucleótidos en sentido 5’ → 3’ usando como molde su propia fracción de RNA (TERC). Dicha unión es posible gracias a que TERC presenta una serie de bases complementarias al ADN telomérico de tal modo que se produce el apareamiento entre ellas.
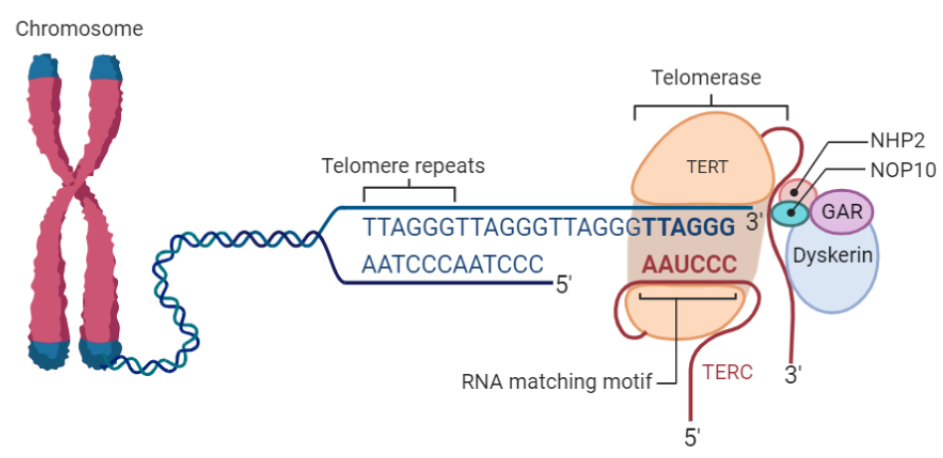
A continuación, se recluta la primasa y la ADN polimerasa α que sintetizan el primer, un pequeño fragmento mixto de ARN y ADN que proporciona el extremo 3’ OH necesario para que intervenga la ADN polimerasa δ y complete el fragmento. Finalmente se da la eliminación del cebador y el ligado de los extremos, quedando nuevamente un extremo 3’ overhang.
También hay que destacar que TERT es el componente limitante de la telomerasa. Mientras que TERC tiene una expresión constitutiva en la mayoría de los tejidos, será la expresión de TERT la que conduzca hacia una activación de la función de la telomerasa por lo que su transcripción estará reprimida en células somáticas.
El lado oscuro de la telomerasa: cáncer
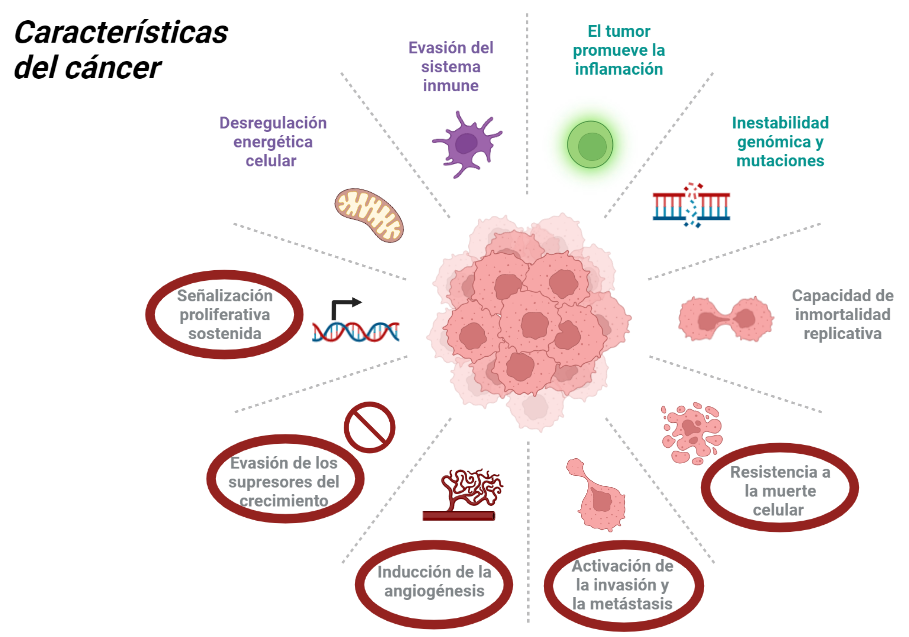
El cáncer es una enfermedad originada a partir de la transformación maligna de una célula que comienza a dividirse sin control y escapa de la muerte celular programada dando lugar a gran cantidad de células hijas, las cuales presentan también alteraciones en los mecanismos de proliferación, diferenciación y apoptosis.
Varios estudios han demostrado la participación de la telomerasa en el proceso de carcinogénesis, puesto que se ha visto que varias líneas celulares cancerosas la presentan y además la inmortalización de las células in vitro ocurre a la vez que la activación de la enzima.
Las células somáticas no presentan telomerasa, por lo que tienen una capacidad limitada para replicarse, siendo esto una barrera de la proliferación. Sin embargo, los tumores malignos tienen una proliferación infinita, gracias a que sus células poseen la telomerasa activa que permite esa replicación sin límites.
Un posible tratamiento contra el cáncer
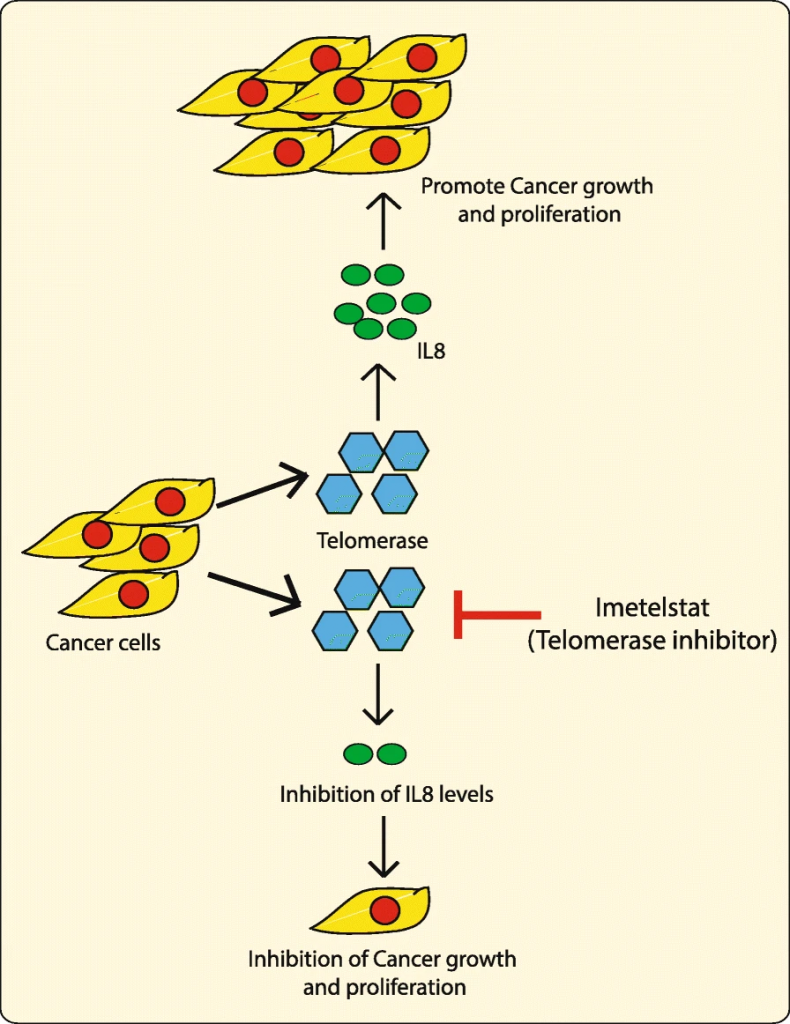
Últimamente ha habido investigaciones enfocadas en el acortamiento de los telómeros de las células cancerosas, sin embargo, esto es algo impreciso ya que tendría que ser específico para la longitud de estos en cada una de las células a tratar y podría demorarse demasiado tiempo. Las estrategias más prometedoras son aquellas que inhiben la protección de los telómeros atacando a la telomerasa, de este modo se podría lograr entrar en apoptosis en pocos días e incluso podría funcionar con telómeros largos.
Los estudios estiman que la telomerasa se detecta en un 80-90% de los tumores cancerosos, por lo que podría ser considerada un marcador de malignidad en tumores. Esto la convierte en un blanco ideal para la terapia contra el cáncer, pero como su regulación es muy compleja se han estudiado diferentes estrategias:
- Nivel transcripcional: la clonación de las regiones promotoras de los genes que codifican para las subunidades TERT y TERC han permitido identificar reguladores positivos y negativos, conociendo estos se puede aumentar o inhibir su transcripción
- Nivel postranscripcional: se está buscando bloquear el ARNm de las subunidades TERT y TERC mediante el uso de ribozimas con actividad ribonucleasa
- Nivel post-traduccional se ha demostrado que para que la enzima funcione necesita el ensamblaje de todos los constituyentes, por lo que bloqueando alguna de las proteínas que forman parte del complejo se podría bloquear su acción
El lado bueno de la telomerasa: la inmortalidad
Como ya hemos mencionado las células germinales expresan la enzima telomerasa mientras que las somáticas no, por lo que estas últimas en cada división van acortando los telómeros entrando así en senescencia.
Por un lado la senescencia, sirve como mecanismo de supresión celular, ya que las células senescentes no son capaces de replicarse, por tanto, no se replicarán cromosomas anormales. Uso que se daría en un posible tratamiento contra células cancerosas.
Sin embargo, se ha propuesto que la reconstitución de la actividad de la telomerasa en distintos tejidos podría ser empleada como terapia para enfermedades asociadas al envejecimiento y que están caracterizadas por una disminución de la capacidad proliferativa y regeneración celular.
Se ha buscado usar la telomerasa como diana terapéutica en medicina regenerativa frente a enfermedades crónicas, por ejemplo, frente a enfermedades de la piel: estimulando células madre para que expresen el componente de ARN de hTERT de tal modo que se active la telomerasa reemplazando la piel perdida. También serviría para enfermedades cardiovasculares y neurodegenerativas asociadas al envejecimiento e incluso el propio envejecimiento.
La última estrategia, desarrollada por un grupo del CNIO en 2012, se basa en una terapia génica que activa el gen de la telomerasa durante unas pocas horas, por lo que la enzima puede ejercer su función reparadora un tiempo limitado, y así se disminuye los riesgos.
Podríamos pensar que una activación continua de la telomerasa implicaría que nuestras células no murieran, lo que se podría considerar conseguir la inmortalidad. Sin embargo, no sería tan fácil porque la actividad constante de esta enzima es muy probable que derive en un cáncer como hemos comentado.
Conclusiones
El acortamiento de los telómeros es el mecanismo fisiológico de nuestro cuerpo que explica el envejecimiento. La naturaleza ha sido capaz de evitar esa muerte celular, mediante la activación de la telomerasa en células somáticas que derivan en células cancerígenas. Sin embargo, lo que encontramos hoy en día es que la ciencia quiere aprovechar esa idea a nuestro favor. Si se consigue activar la telomerasa de una manera regulada, se podrá extender los años de vida; lo que sería un paso más cerca de esa idea ficticia que tenemos de la inmortalidad.
Bibliografía
- Mengual Gómez, D. L., Armando, R. G., Farina, H. G., & Gómez, D. E. Telomerasa y telómero: su estructura y dinámica en salud y enfermedad. MEDICINA (Buenos Aires), 74(1), 69-76 (2014).
- Cascales Angosto, M., Álvarez Gómez, J. A. Anales de la Real Academia de Doctores de España. Volumen 14, pp. 49-70 (2010).
- Isnais Luna Rodríguez(1), Odania Mondeja Ortiz(2), Maritza Roque Tarife(3). Telomerasa. Enzima del futuro. Revista médica electrónica de ciego de Ávila, Vol.11, No. 1 (2005)
- Arvelo, F., & Morales, A. Telómero, telomerasa y cáncer. Acta Científica Venezolana, 55, 288-303. (2004).
- Greider, C. W., & Blackburn, E. H. Telómeros, telomerasa y cáncer. Investigación y Ciencia, 235, 20-26.(1996).
- Figueroa, E. F., & Mayani, H. Cromosomas, control celular y cáncer: una cuestión de telomerasas. Revista Ciencia, julio – septiembre (2003)
- Dias, J. Proliferación celular y regulación de la telomerasa en cáncer de mama (Doctoral dissertation, Universidad de Málaga). (2017).
- Hernández Fernández, R. A. Telómeros y telomerasas. Revista Cubana de Investigaciones Biomédicas, 18(2), 121-129. (1999).
- Saretzki G. Telomerase inhibition as cancer therapy. Cancer Lett;194(2):209-19 (2003)
- Sarborit, A. & Muñiz, C. Telomerasa: Salud y Envejecimiento. Morfovirual (2020)