DESCUBREN QUE EL FÁRMACO PARA DEJAR DE FUMAR ES CANCERÍGENO. Pero, ¿es más cancerígeno que el propio tabaco?
Tabaquismo, Vareniclina y el fármaco definitivo para dejar de fumar.
Por Aitana López López y Marta de la Hoz Marchado. Grado en Biología Sanitaria, Universidad de Alcalá.
La vareniclina, comercialmente conocida como Champix®, es un potente fármaco para dejar de fumar (1). Sin embargo, en julio de 2021, la FDA solicitó su retirada voluntaria del mercado tras detectar la contaminación del fármaco con sustancias cancerígenas llamadas nitrosaminas (2) (Ver tabla 1). En concreto se detectaron impurezas de N-nitroso-vareniclina, sustancia con potencial mutagénico y cancerígeno (3). La farmacéutica Pfizer fue retirando progresivamente el fármaco. No obstante, el tratamiento solo dura 6 meses mientras que los efectos negativos se asocian al uso del fármaco a largo plazo (2,4). La FDA insistía en que los beneficios de dejar de fumar superaban el riesgo de cáncer por tomar vareniclina mientras la ingesta no superase los 37 ng de nitrosamina/día (2). Con lo cual comenzaba el siguiente debate: ¿Causa más cáncer fumar o el fármaco para dejar de fumar? En este blog enfrentaremos los riesgos del tabaco frente a los de la vareniclina y presentaremos un fármaco que podría ser más prometedor que el Champix para tratar a aquellos que quieren dejar de fumar, la citisina.
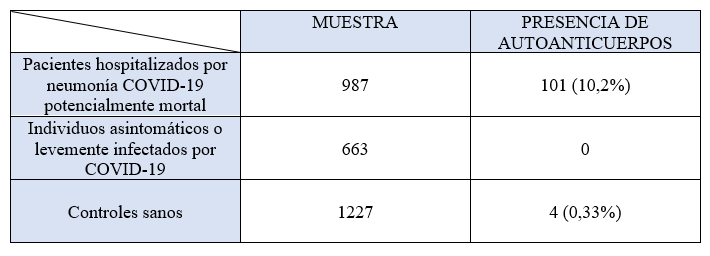
Tabla 1. Nitrosaminas cancerígenas. Tabla obtenida y adaptada de: Riesgos estimados de cáncer asociados con la contaminación por nitrosamina en medicamentos de uso común (2021). (3)
| Sustancia | Fórmula | Carcinogenicidad | Reconocidas por | Casos |
| NDMA | N[1]nitrosodimethylamine | Sustancia carcinógena | IARC, US EPA, NT y California´s Proposition | 40-126 casos adicionales de cancer por cada 100.000 individuos expuestos |
| NDEA | N-nitrosodiethylamine | Sustancia carcinógena | IARC, US EPA, NT y California´s Proposition | 12-48 casos adicionales de cancer por cada 100.000 individuos expuestos |
| NMBA | N[1]nitroso-N-methyl-4-aminobutyric acid | Sustancia que induce cáncer de vejiga y de riñón | ||
| N-nitroso-vareniclina | N-nitroso-vareniclina | Faltan datos concluyentes de que sea una sustancia carcinógena aunque tiene potencial mutagénico y cancerígeno |
1. TABACO
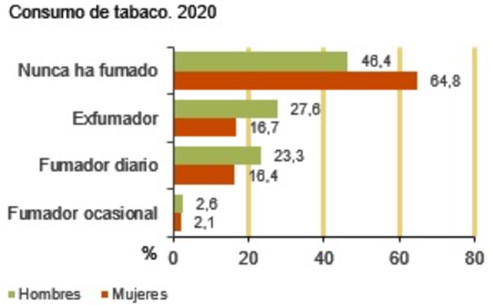
Fumar tabaco es un hábito extendido en todo el mundo. Según el INE el 16,4% de las mujeres y el 23,3% de los hombres fuman a diario (5). Las cifras siguen en aumento pese a que cada vez se sabe más sobre sus efectos negativos. Para estudiar por qué causa tanta adicción y a la vez tanto daño iremos desgranando sus componentes.
1.1 NICOTINA
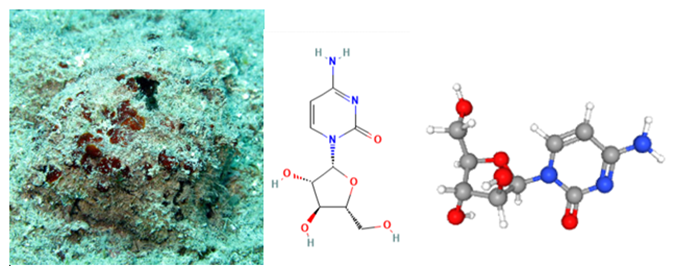
La nicotina es el alcaloide primario en los productos del tabaco y capaz de atravesar fácilmente las membranas biológicas y la barrera hematoencefálica. Es un agonista exógeno del receptor colinérgico nicotínico (6,7). Son canales iónicos dependientes de ligando. Aquellos que se encuentran en el cerebro y que están formados por 2 subunidades α4 y 3 subunidades β2 presentan mayor afinidad por la nicotina (6,8). La nicotina ejerce un efecto estimulante en el locus ceruleus y un efecto de recompensa en el sistema límbico (7). La respuesta ocurre en milisegundos provocando la liberación de acetilcolina, norepinefrina, serotonina y dopamina.
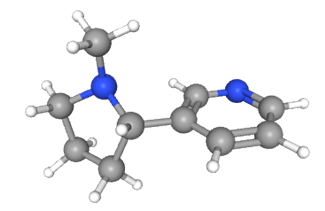
Figura 2. Molécula de nicotina. Imagen obtenida de PubChem (16).
1.2 ADICCIÓN Y TOLERANCIA
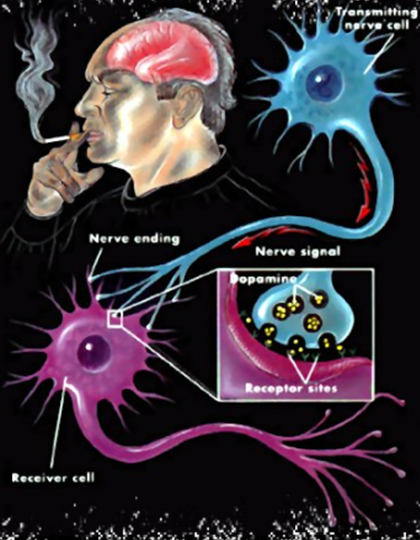
En el cerebro la nicotina actúa sobre las neuronas dopaminérgicas en las vías cortico-límbicas estimulando la liberación de dopamina. Esta activa el sistema dopaminérgico, en concreto las neuronas dopaminérgicas del núcleo accumbes, centro de la recompensa. Genera una sensación de euforia y placer. Por ello el fumador es reforzado a repetir el comportamiento que ha motivado la activación dopaminérgica, es decir, fumar (8). Debido al efecto ansiolítico de la nicotina (9) se ha demostrado que si el consumo de nicotina cesa provoca un cambio negativo en el comportamiento (10).
En los fumadores la exposición a nicotina es prologada. Además, la nicotina permanece más tiempo en la hendidura sináptica que la acetilcolina (agonista endógeno). Por ello, se produce una desensibilización de los receptores que conlleva a la tolerancia a la nicotina. Se requiere consumir cada vez más cantidad de nicotina para obtener los mismos efectos. (8)
1.3 TABACO Y CÁNCER
Se ha demostrado que la nicotina es capaz de inhibir la apoptosis celular, inducir estrés oxidativo en las células y estimular la proliferación celular. Por ello, podría estimular procesos de neoplasia aumentado el riesgo de padecer cáncer (6,11). Sin embargo, son el resto de las sustancias contenidas en el tabaco las que le hacen un potente cancerígeno (12).
Un cigarrillo contiene 250 sustancias nocivas, de las cuales, 50 son cancerígenas. Estos agente químicos carcinógenos están incluidos en el “Grupo I de carcinógenos humanos” por la International Agency for Research on Cancer (IARC) y son : Benceno, cadmio, arsénico, níquel, cromo, 2-naftil-amino, clorovinil, 4 aminobifenil y Berilio. Pero las sustancias cancerígenas del tabaco más potentes son los hidrocarburos aromáticos policíclicos y las nitrosaminas. (12,13,15)
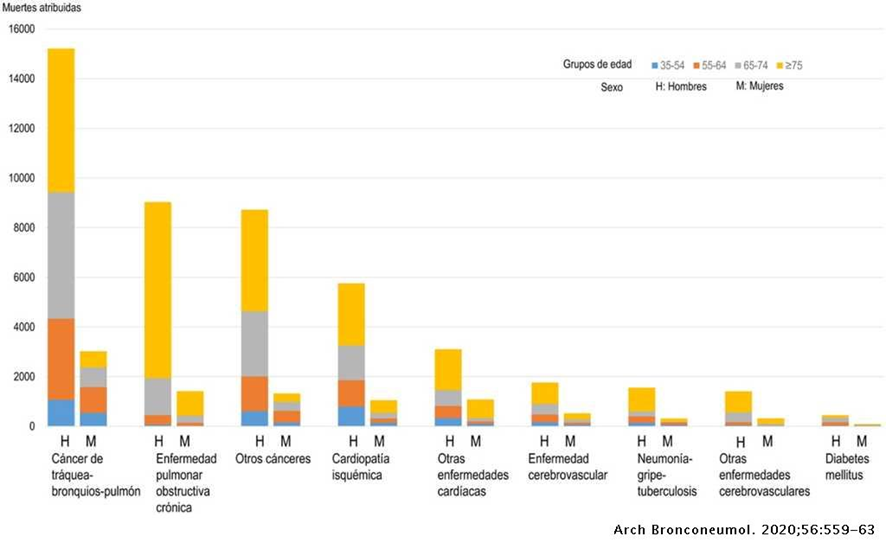
El tabaquismo causa un tercio de las muertes por cáncer (tracto aerodigestivo, esófago, estomago, hígado, riñón, cervix, colorrectal) en los países occidentales. Se estima que el tabaco provoca el 25% de los cánceres en hombres y el 4% en mujeres. Además, es causa del 16% de los canceres en países desarrollados y del 10% en países subdesarrollados. Sin embargo, fumar causa más muertes por patologías cardiovasculares y respiratorias que por cáncer. Por ello, el tabaquismo se considera una epidemia. (14)
2. VARENICLINA
Como hemos mencionado arriba, la vareniclina, comercializada como Champix®, es una droga que contiene tartrato de vareniclina. Es un agonista parcial de los receptores nicotínicos (20).
Los receptores nicotínicos pasan por tres fases:
- Reposo: canal iónico cerrado por ausencia de ligando.
- Activo: la unión del agonista produce la apertura del canal y la correspondiente despolarización.
- Desensibilizado: periodo refractario en el que el canal permanece cerrado aunque haya ligando.
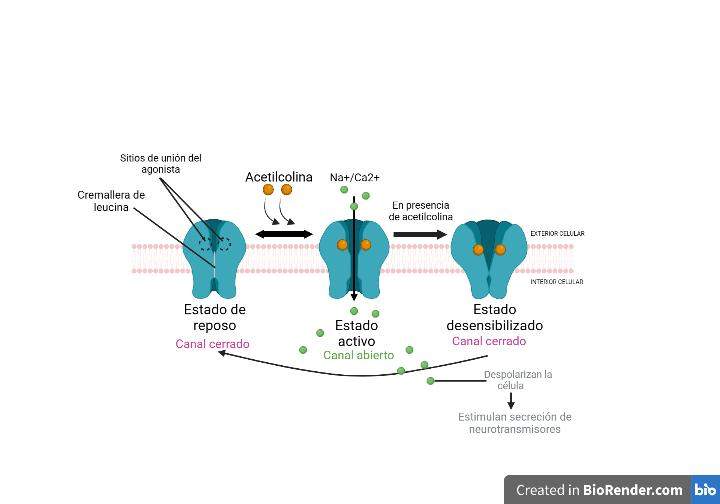
No obstante, cuando hay bajas concentraciones de nicotina, se puede producir el paso directo de estado de reposo a desensibilizado, mientras que, altas dosis de la droga inducen el paso de desensibilizado a activado.
La vareniclina inhibe competitivamente la unión de nicotina a los receptores. Esta, al unirse, activa los receptores nicotínicos y produce liberación de dopamina, por lo que se produce una sensación placentera similar a la generada por la nicotina, pero mantiene los receptores en estado desensibilizado por más tiempo cuando hay una sobreexposición a la droga. De este modo, se logra reducir la urgencia por fumar, los síntomas de la abstinencia y disminuye la recompensa ante la nicotina porque la vareniclina está bloqueando a los receptores. Además, no altera la farmacodinámica de otros fármacos al no ser metabolizado por el citocromo p450.
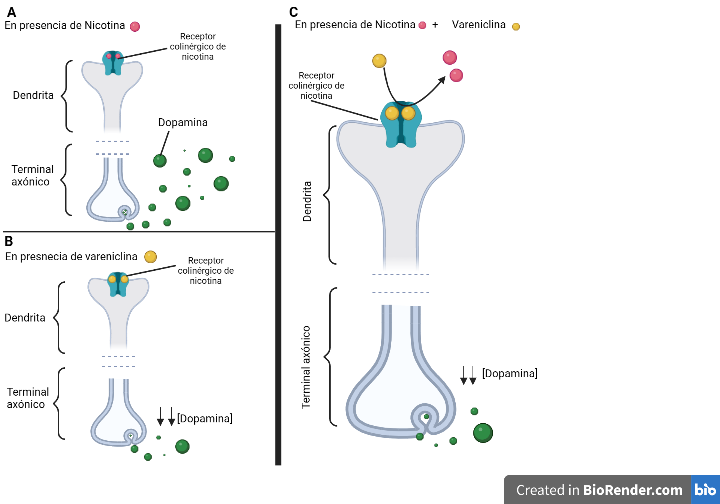
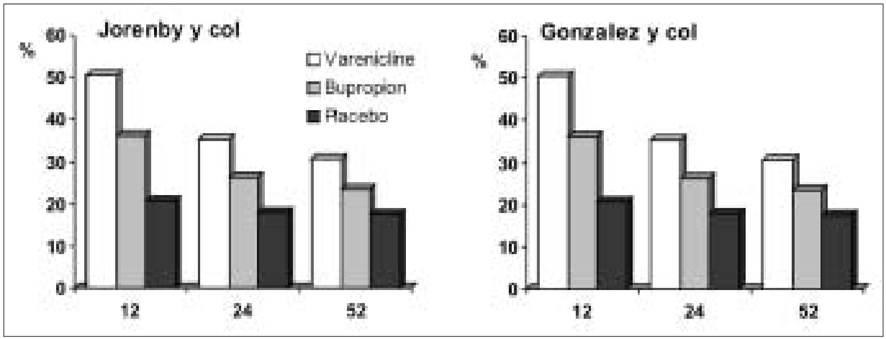
Mediante ensayos in vitro en ratas se descubrió que la vareniclina tiene alta afinidad por los receptores α4β2, pero no por otros receptores nicotínicos. Además mediante ensayos clínicos se verificó su gran efecto contra la abstinencia puntual, comparado con otros fármacos como el bupropión, pero no se observaron resultados homogéneos en cuanto a disminuir la urgencia puntual. Por este motivo, Pfizer comercializó la vareniclina como el principal fármaco para dejar de fumar porque, a pesar de poseer efectos secundarios, como náuseas, no eran suficientes para condicionar su retirada.
2.2 LA CARA OSCURA DE LA VARENICLINA.
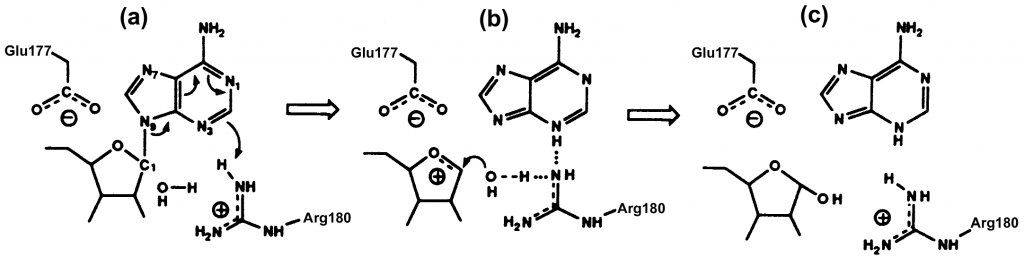
Recientemente se ha descubierto que este fármaco posee nitrosaminas (21), en concreto, N-nitroso-vareniclina. Las nitrosaminas son compuestos orgánicos cancerígenos que se forman mediante reacciones químicas entre nitritos con aminas. Son los metabolitos de las nitrosaminas los que interaccionan con el ADN y producen los efectos tóxicos. Las nitrosaminas no solo se encuentran en fármacos sino que también las podemos encontrar en productos de látex, caucho, maquillaje o en los conservantes usados para conservar alimentos cárnicos. Como no podía ser de otra forma, también se encuentran en la nicotina, esta, en concreto origina tanto el carcinógeno más potente, NNK, como el más débil, NNN. Estos compuestos se han visto implicados en el desarrollo de distintos cánceres, como el de estómago o faringe, por lo que se ha establecido una cantidad diaria máxima para evitar estos efectos nocivos. Champix® contiene una dosis más alta de lo permitido diariamente. Por ello se decidió retirarlo del mercado.
La cuestión es, ¿qué resulta más cancerígeno, el tabaco per se o el fármaco para dejar de fumar? ¿Merece la pena asumir los riesgos de tomar vareniclina con tal de poner fin a la adicción por la nicotina? Bien pues, en primer lugar debemos comparar la toxicidad de las nitrosaminas producidas por la nicotina con las N-nitroso-vareniclinas del Champix®.Las nitrosaminas de la vareniclina se incluyen según la IARC dentro del grupo II (“Probablemente cancerígeno para los seres humanos”). Mientras que todos los componentes cancerígenos del tabaco se encuentran dentro del grupo 1 (“Cancerígeno para los seres humanos”) (Ver tabla 2). Podemos concluir que el tabaco es más cancerígeno, que la vareniclina en sí (15). Así mismo, la gran efectividad de la vareniclina en lograr el cese del tabaquismo en tiempos desde tres meses a un año (22) marca la diferencia, ya que ningún otro fármaco anterior había logrado tasas del 80% de éxito con efectos secundarios tolerables. Además, se han conseguido resultados similares disminuyendo la dosis del fármaco. En conclusión, el Champix® resulta tan eficaz en la erradicación del tabaquismo, incluso previniendo la recaída (20) (primer fármaco que logra esto), que realmente merece la pena asumir los riesgos de ingerir las nitrosaminas que contiene la vareniclina con tal de dejar de introducir todos los carcinógenos presentes en el tabaco.
Tabla 2. Comparación agentes carcinógenos del tabaco y del fármacos según la IARC. Tabla adaptada de Lista de Clasificaciones de sutancias cancerígenas de la IARC (13, 14)
| Dónde se encuentra | Agente carcinógeno | Grupo IARC |
| Tabaco | 4-aminobifenil | I |
| Tabaco | Arsénico | I |
| Tabaco | Benceno | I |
| Tabaco | Berilio | I |
| Tabaco | Cadmio | I |
| Tabaco | Cromo y derivados | I |
| Tabaco | 2-naftilamina | I |
| Tabaco | Níquel | I |
| Tabaco | Cloruro de vinilo | I |
| Fármaco | N-nitrosodimetilamina | 2A |
| Fármaco | N-nitrosodietilamina | 2A |
| Fármaco | Ácido N-nitroso-N-metil-4-aminobutírico | No evaluado |
3. CITISINA
Actualmente hay en vigor un nuevo fármaco para dejar de fumar, la citisina (23), comercializada como Tabex® o Todacitan®. Es un alcaloide natural derivado de la familia de plantas Fabaceae, en la que se incluyen los géneros Laburnum y Cytisus. Es un tratamiento contra el tabaquismo no solo más efectivo que otros sino también más económico. La citisina, también conocida como sophorina o baptitoxina, actúa, al igual que la vareniclina, es un agonista parcial de los receptores nicotínicos, especialmente el α4β2. Este fármaco, de igual manera que el Champix®, previene la completa activación del sistema nicotínico-dependiente de la liberación de dopamina en el sistema mesolímbico (disminuye el placer generado al fumar) y reduce los principales síntomas de la abstinencia tanto centrales, al aumentar ligeramente la liberación de dopamina en el cerebro, como periféricos, estimulando la liberación de catecolaminas por la glándula suprarrenal, evitando recaídas.
En definitiva, centrándonos en su modo de acción, la citisina parece ser una sustituta ejemplar de la vareniclina, pero, ¿resulta igual de eficaz? Bien pues, no solo existen estudios que corroboran que esta droga proporciona resultados competentes respecto a la vareniclina sino que, además, resulta extremadamente más productiva al presentar un coste de producción significativamente menor (24). Por lo tanto, podemos concluir que, en efecto, la citisina se lleva el podio de los fármacos contra el tabaquismo.

4. CONCLUSIÓN
Resulta evidente que debemos poner fin al problema del tabaquismo en nuestra sociedad, en vista de la gran cantidad de eventos nocivos que tiene para nuestro organismo al contener potentes carcinógenos. Por ello, en un principio, merecía la pena asumir el riesgo que presentan las N-nitroso-vareniclinas cancerígenas que presenta el Champix®, el fármaco más eficaz que existía hasta la fecha para dejar de fumar. No obstante, con la aparición de la citisina, una droga con un modo de acción idéntico a la vareniclina, que no solo presenta la misma eficacia, sino que también es más barata, el Champix® queda totalmente excluido como tratamiento de elección a la hora de abandonar este hábito nocivo tan socialmente aceptado como es el tabaquismo.

BIBLIOGRAFÍA
- Fouz Rosón, N. (2016). Impacto del tratamiento con vareniclina a bajas dosis vs estándar, ambas en pautas cortas, en la tasa de abstinencia tabáquica, adherencia al tratamiento y efectos adversos. (Tesis doctoral inédita). Universidad de Sevilla, Sevilla.
- US FDA. FDA Alerts Health Care Professionals and Patients to a Voluntary Recall of Varenicline (Chantix) to the Warehouse Level. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-alerts-health-care-professionals-and-patients[1]voluntary-recall-varenicline-chantix-warehouse?utm_medium=email&utm_source=govdelivery (accessed on 2 July 2021).
- Li, K., Ricker, K., Tsai, F. C., Hsieh, C. J., Osborne, G., Sun, M., … & Sandy, M. S. (2021). Riesgos estimados de cáncer asociados con la contaminación por nitrosamina en medicamentos de uso común. Revista internacional de investigación ambiental y salud pública, 18(18), 9465.
- CHAMPIX (varenicline) — Potential Risk Posed by Long-Term Exposure to Nitrosamine Impurity, N-nitrosovarenicline, Exceeding Acceptable Intake Limit – Canada.ca. (s. f.). Recuperado 7 de octubre de 2022, de https://recalls-rappels.canada.ca/en/alert-recall/champix-varenicline-potential-risk-posed-long-term-exposure-nitrosamine-impurity-n.
- Instituto Nacional de Estadística. (2020). Determinantes de Salud (Consumo de Tabaco). INE.es. https://www.ine.es/ss/Satellite?L=es_ES&c=INESeccion_C&cid=1259926698156&p=1254735110672&pagename=ProductosYServicios%2FPYSLayout
- Yildiz, D. (2004). La nicotina, su metabolismo y una visión general de sus efectos biológicos. Toxicon, 43(6), 619-632.2.
- Nicotine: Uses, Interactions, Mechanism of Action | DrugBank Online. (2022, diciembre). DrugBank. https://go.drugbank.com/drugs/DB00184
- Treviño, L. J., Fernández, M. T. B., GONZÁLEZ, M. P. G., Martínez, P. A. S., & BOUSOÑO, M. (2004). La nicotina como droga. Monografía Tabaco, 16(suplemento 2), 143.
- Salas, R., Pieri, F., Fung, B., Dani, J. A., & De Biasi, M. (2003). Respuestas alteradas relacionadas con la ansiedad en ratones mutantes que carecen de la subunidad β4 del receptor nicotínico. Revista de Neurociencia, 23(15), 6255-6263.
- al’Absi, M., Amunrud, T., & Wittmers, L. E. (2002). Efectos psicofisiológicos de la abstinencia de nicotina y los desafíos conductuales en fumadores habituales. Farmacología Bioquímica y Comportamiento, 72(3), 707-716.
- Sasco, A. J., Secretan, M. B., & Straif, K. (2004). Tabaquismo y cáncer: una breve revisión de la evidencia epidemiológica reciente. Cáncer de pulmón, 45, S3-S9.
- Ruiz, A. M., Gómez, I. R., Rubio, C., Revert, C., & Hardisson, A. (2004). Efectos tóxicos del tabaco. Revista de toxicología, 21(2-3), 64-71.
- Smith CJ, Livingston SD, Doolittle DJ. Una encuesta de literatura internacional de «carcinógenos del Grupo I de la IARC» reportados en el humo del cigarrillo convencional. Alimentos Químicos Toxicol. 1997 Oct-Nov;35(10-11):1107-30. doi: 10.1016/s0278-6915(97)00063-x. PMID: 9463546.
- Sasco, A. J., Secretan, M. B., & Straif, K. (2004). Tabaquismo y cáncer: una breve revisión de la evidencia epidemiológica reciente. Cáncer de pulmón, 45, S3-S9.
- Sustancias cancerígenas. (1974). IARC. https://doi.org/10.1111/j.2041-5370.1977.tb01333.x
- PubChem. (s/f). Nicotine. Nih.gov. Recuperado el 29 de diciembre de 2022, de https://pubchem.ncbi.nlm.nih.gov/compound/89594
- .PDB. (s/f). 6PV7. Receptor nicotínico de acetilcolina alfa3beta4 humano en complejo con nicotina. Banco de Datos de Proteínas. Recuperado el 29 de diciembre de 2022, de https://www.rcsb.org/structure/6PV7.
- LOS EFECTOS DE LA NICOTINA EN EL CEREBRO. (s/f). Diadmbe.es. Recuperado el 29 de diciembre de 2022, de https://www.diadmbe.es/los-efectos-de-la-nicotina-en-el-cerebro/
- Pérez-Ríos, M., Schiaffino, A., Montes, A., Fernández, E., López, M. J., Martínez-Sánchez, J. M., Sureda, X., Martínez, C., Fu, M., García Continente, X., Carretero Ares, J. L. y Galán, I. (2020). Mortalidad atribuible al consumo de tabaco en España 2016. Archivos de Bronconeumologia, 56(9), 559–563. https://doi.org/10.1016/j.arbres.2019.11.021
- GONZÁLES D, RENNARD SI, NIDES M, ONCKEN C, AZOULAY S, BILLING CB, et al. Vareniclina, un agonista parcial del receptor de acetilcolina de nicotina α4β2 versus bupropión de liberación sostenida y placebo para dejar de fumar. JAMA 2006; 296: 47-55.
- Schuller HM. Nitrosamines as nicotinic receptor ligands. Life Sci. 2007 May 30;80(24-25):2274-80. doi: 10.1016/j.lfs.2007.03.006. Epub 2007 Mar 19. PMID: 17459420; PMCID: PMC1987356.
- Fouz Rosón, N. (2016). Impacto del tratamiento con vareniclina a bajas dosis vs estándar, ambas en pautas cortas, en la tasa de abstinencia tabáquica, adherencia al tratamiento y efectos adversos.
- Cytisine. Uses, Interactions, Mechanism of Action | DrugBank Online. (n.d.). Retrieved December 31, 2022, from https://go.drugbank.com/drugs/DB09028
- Leaviss, J., Sullivan, W., Ren, S., Everson-Hock, E., Stevenson, M., Stevens, JW, … y Cantrell, A. (2014). ¿Cuál es la eficacia clínica y la rentabilidad de la citisina en comparación con la vareniclina para dejar de fumar? Una revisión sistemática y evaluación económica. Evaluación de tecnologías sanitarias (Winchester, Inglaterra) , 18 (33), 1-120.
- Orozco, Á. (2013). Caracterización farmacológica de los efectos del péptido amiloide β sobre los receptores nicotínicos neuronales. Memoria de Tesis UAM. https://repositorio.uam.es/bitstream/handle/10486/14161/66342_orozco%20alarcon%20angela.pdf?sequence=1
- Zabert, G. (2006). Vareniclina: Nuevo agonista parcial del receptor nicotínico. Revista Argentina de Medicina Respiratoria. https://www.ramr.org/articulos/volumen_6_numero_3/actualizacion/actualizacion_vereniclina.pdf
- Tabex [Photograph]. Gintarinė Vaistinė. https://quitwithtabex.co.uk/wp-content/uploads/2017/01/tabex-x1.jpg



















{kind=link}
{kind=link}