CRISPR-Cas: mecanismo molecular, historia y aplicaciones en potencia.
escrito por Grupo3_B_4_nenana | 10 enero, 2022
Andrea Delgado Ruiz y Néstor Román Cueva Ramírez. Grado en Biología Sanitaria, Universidad de Alcalá.
El sistema CRISPR-Cas es un mecanismo de defensa presente en bacterias y archeas cuyo objetivo es degradar aquel material genético exógeno que trate de invadir el organismo.
Como su propio nombre indica, el complejo CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) consta de una región promotora encargada de regular la transcripción seguida de varias secuencias palindrómicas de pequeño tamaño que se repiten constantemente en el genoma y que, además, están interrumpidas por otras secuencias no repetidas y similares en tamaño que reciben el nombre de espaciadores (spacers). Dichos espaciadores se corresponden con fragmentos genéticos de origen extra cromosómico que el microorganismo adquiere tras entrar por primera vez en contacto con un patógeno, tratándose así de un curioso sistema de inmunidad adquirida con memoria.
Además del complejo CRISPR, también es necesaria la presencia de los genes Cas, cuya expresión dará lugar a las endonucleasas encargadas de cortar y degradar el material genético exógeno. En función de la endonucleasa que se exprese, podemos diferenciar tres tipos de sistema: CRISPR I activa a Cas3, CRISPR II activa a Cas 9 y CRISPR III activa a Cas6. De estos tres sistemas, el más empleado en ingeniería genética actualmente es CRISPR-Cas9 debido a la alta tasa de eficiencia de la endonucleasa Cas9.
El proceso inmune mediado por este sistema puede dividirse dos fases:
1. INMUNIZACIÓN: Incorporación de secuencias espaciadoras tras la exposición al patógeno.
Cuando la célula huésped (bacteria u archea) entra por primera vez en contacto con un organismo patógeno detecta su material genético y lo reconoce como exógeno. Este reconocimiento es gracias a la presencia de una secuencia específica en el DNA conocida como motivo adyacente del protoespaciador (PAM). Tras el reconocimiento de esta secuencia PAM, la célula incorporará los nucleótidos adyacentes a esta al genoma como un nuevo espaciador.
*Durante este proceso, una de las secuencias palindrómicas repetidas se duplica, y así el nuevo espaciador queda flanqueado en el genoma por una secuencia repetitiva a cada lado.
2. INMUNIDAD: Formación y actuación del complejo CRISPR-Cas.
La región CRISPR se transcribe dando lugar a un RNA largo y no codificante denominado pre-crRNA. En el caso del sistema CRISPR-Cas9, se transcribe adicionalmente otro RNA no codificante que es complementario a la secuencia palindrómica repetida y que recibe el nombre de tracrRNA. Este se va a unir a las secuencias repetidas del pre-crRNA formando un dímero crRNA/tracrRNA que será reconocido por una RNAsa III, la cual se encargará de procesar y generar un crRNA maduro.
Finalmente, la endonucleasa Cas, se asocia a el crRNA maduro formando el complejo CRISPR-Cas. Será este crRNA maduro el encargado de guiar al complejo hasta el blanco, es decir, hasta reconocer una secuencia complementaria que será degradada por la acción de Cas.
Como curiosidad, la gran variedad de elementos genéticos invasores ha favorecido que bacterias y archeas evolucionen creando distintos tipos de enzimas Cas para generar una respuesta inmune más efectiva, las cuales, además de la actividad de corte, poseen otras funciones auxiliares específicas de cada tipo.
Figura 1. Editada y traducida (2022) en BioRender.com. Reprinted from «CRISPR-Cas9 Adaptive Immune System of Streptococcus pyogenes Against Bacteriophages», by BioRender, July 2020.
Para poder llegar a describir con precisión todos los pasos de este curioso mecanismo molecular fueron necesarios muchos años de investigación en los que se vieron implicados numerosos científicos a nivel internacional. Sin embargo, el origen de este gran descubrimiento tiene lugar en España allá por el año 1987, cuando dos jóvenes microbiólogos, uno en Alicante (F. Mojica) y otro en Utrecht, se dedicaron a estudiar una serie de elementos genéticos repetitivos presentes en la bacteria Escherichia coli. Posteriormente, en el 2000, se encuentran estas mismas secuencias repetitivas en muchas otras especies bacterianas y se observó que junto a estas regiones repetitivas se encontraban cuatro genes distintos presentes en muchos otros procariotas también, situados de manera invariable respecto a las repeticiones. En el 2002 se hizo referencia por primera vez al nombre CRISPR, con el que se denominó a estas repeticiones por las características que tenían como ya se ha comentado previamente. Los genes asociados fueron denominados cas.
En el 2005 Mojica encuentran similitudes entre los espaciadores asociados a cispr y el material genético de ciertos virus que afectan a bacterias, sospechando vinculación de estas secuencias con la inmunidad de los virus, lo que sientan las bases para el posterior desarrollo de las aplicaciones; hipótesis que fue demostrada experimentalmente dos años después. En ese momento empezó a investigarse la manera en la que actuaban sistemas inmunes CRISPR; en 2012 se descubrió que se genera un corte asociado a CRISPR para fragmentar el DNA y que el factor estrella de este proceso es la proteína cas9. El hecho del descubrimiento de la actividad endonucleasa de este sistema y su alta especificidad para los sitios de corte hizo replantearse la posibilidad de usarlo como sistema de edición génica. De hecho, este mismo año, se hicieron los primeros cortes con este sistema; en un tubo de ensayo, el primero de todos, realizado por un equipo liderado por Doudna y Charpentier, y en una célula viva de mamífero más tarde.
A partir de este momento se empieza a entender este sistema como un conjunto (CRISPR/Cas9). En 2013, tras estos últimos experimentos, surge el boom de esta técnica y se le da empieza a dar real importancia; en los años anteriores prácticamente había pasado desapercibida. El motivo de tanto alboroto es que esta técnica suplía las carencias de otras técnicas de edición genética anteriores, como la complejidad, alto coste y escasa eficacia. Aunque hoy en día sea considerado uno de los sistemas más eficientes y precisos, su potencial aún es una incógnita.
Figura 2. Timeline de la historia de CRISPR-CAS9, creada por los autores de la entrada (2021).
Está técnica revolucionaria en la edición genética ofrece oportunidades prácticamente ilimitadas. Solo con conocer la secuencia objetivo y que el gen que se desea modificar se encuentre junto a un motivo PAM, se puede cortar, pegar o editar. Por tanto, esta herramienta se utiliza en casi cualquier ser vivo, así como en campos muy dispares.
En humanos se utiliza en medicina, para prevenir y tratar enfermedades de distintos tipos; genéticas, virales, mentales e incluso cáncer, para realizar diagnósticos o para modificar animales y conseguir tejidos parecidos a los humanos que puedan usarse en trasplantes. En animales se puede usar en la industria cárnica, para mejorar las carnes de consumo, así como para erradicar especies. Se han conseguido también plantas con diversas resistencias (a plagas, enfermedades..) así como mejoradas genéticamente para aumentar su consumo o mejorarlo.
Todos estos ejemplos son solo una pequeña demostración de todo lo que se puede conseguir con esta herramienta y para hacernos a la idea de todo lo que aún está por conseguir.
En los últimos años, gracias a ella, se han conseguido verdaderos progresos. En 2019, mediante la genética dirigida, se consiguió erradicar una población en cautividad del mosquito Anopheles, vector transmisor de la malaria. Esta técnica consiste en alterar la transmisión mendeliana mediante la modificación de genes sexuales de una especie. De esta manera, se consigue una mayor y más rápida propagación de un gen de interés. Para ello se copia un gen del cromosoma sexual beneficioso de uno de los progenitores en el cromosoma sexual del otro progenitor. Así, en la población femenina de este mosquito se introdujo una mutación que produce esterilidad femenina, consiguiendo eliminar la población completa en 10 generaciones. Esta técnica no ha obtenido resultados tan exitosos en mamíferos.
Por supuesto, esto conlleva una serie de implicaciones éticas importantes. En el caso anteriormente expuesto, erradicar una población de una especie puede tener importantes consecuencias en el medio ambiente. Por otro lado, editar genes ofrece la posibilidad de diseñar humanos. Por tanto, todas las aplicaciones de CRISPR-Cas necesitan estar reguladas y llevar un control de seguridad y eficacia.
REFERENCIAS:
Montoliu, L. M. (2021). Prólogo. En F. M. Mojica (Ed.), Editando genes: recorta, pega y colorea (3.a ed., pp. 22–27).
Herrera-Cabrera, B.E.; Salgado-Garciglia, R.; López-Valdez, L.G.; Reyes, C.; Montiel-Montoya, J.; Zaragoza-Martínez, F.; Guillermo Lucho-Constantino, G. y Barrales-Cureño, H. J. Edición genómica con CRISPR/Cas9: Premio Nobel de Química 2020. Revista de Química, 2021, 35(1), 22-30 http://revistas.pucp.edu.pe/index.php/quimica/article/view/23324
Joung, J., Konermann, S., Gootenberg, J. S., Abudayyeh, O. O., Platt, R. J., Brigham, M. D., Sanjana, N. E., & Zhang, F. (2017). Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nature protocols, 12(4), 828–863. https://doi.org/10.1038/nprot.2017.016
María Fernanda Lammoglia-Cobo, Ricardo Lozano-Reyes, César Daniel García-Sandoval, Cynthia Michelle Avilez-Bahena, Violeta Trejo-Reveles, Rodrigo Balam Muñoz-Soto, César López-Camacho (2016). The revolution in genetic engineering: CRISPR/Cas system. https://www.medigraphic.com/pdfs/invdis/ir-2016/ir162e.pdfç
Desde que en 1971 Alfred Knudson propuso la hipótesis de que en la oncogénesis debían ocurrir mutaciones en el ADN, se ha invertido mucho esfuerzo en la investigación de la genética del cáncer. Pero igualmente importante es la epigenética de éste. Por eso, también salen al mercado medicamentos que tratan de restablecer el epigenoma normal, lo que induciría la muerte de las células malignas. Además, también pueden llevar a efectos sinergistas al combinarse con otros medicamentos. En este contexto, existen actualmente dos tipos de fármacos: los inhibidores de DNA-metiltransferasas y los inhibidores de desacetilasas de histonas (HDAC), y nosotros vamos a desarrollar un ejemplo de estos últimos: la romidepsina.
La romidepsina es un fármaco con actividad antineoplásica inhibidor de las desacetilasas de histonas (HDAC). En este post hablaremos de su naturaleza molecular, su mecanismo de acción y sus propiedades farmacológicas y clínicas. A grandes rasgos, la romidepsina es un depsipéptido bicíclico que interfiere en la expresión génica inhibiendo las HDAC de clase I y II, induciendo la detención del ciclo celular y apoptosis. Este medicamento también se conoce como FR901228 o FK228, y se comercializa como Istodax®.
La romidepsina fue aislada en 1993 como producto de fermentación de una cepa de Chromobacterium violaceum, en un programa de investigación japonés que buscaba compuestos bacterianos con propiedades antimicrobianas y antitumorales. El medicamento fue descubierto por Fujisawa Corporation y mostró grandes propiedades citotóxicas contra distintas células tumorales. La cepa se obtuvo de una muestra del suelo de la prefectura de Yamagata, en Japón (1,2). Posteriormente, el National Cancer Institute (NCI) de EEUU confirmó que era un potente anticancerígeno. Comenzaron los ensayos clínicos, y en 2009 la FDA aprobó el fármaco para el tratamiento contra el linfoma cutáneo de células T (CTCL), y en 2011 contra el linfoma periférico de células T (PTCL).
2. ESTRUCTURA
La romidepsina tiene estructura de depsipéptido bicíclico formado por la unión cíclica de cuatro aminoácidos y un ácido heptenoico con un grupo tiol, y presenta un puente disulfuro intramolecular. Un depsipéptido es aquel en el que algún enlace amida peptídico se reemplaza por un enlace tipo éster. Se encuentran mayoritariamente en productos naturales de origen marino y microbiano.
La romidepsina se compone de los aminoácidos L-Val, D-Val, Z-dehidrobutirina y D-Cys, y el ácido (3S,4E)-3-hidroxi-7-mercapto-4-heptenoico. Este ácido forma el puente disulfuro con la D-Cys y el enlace éster con la L-Val.
La estructura fue determinada usando una combinación de técnicas espectroscópicas, resonancia magnética nuclear y cristalografía por rayos X (3).
Figura 1. Estructura plana de la romidepsina. Estructura obtenida de PubChem y modificada con Chimera y Biorender
Figura 2. Modelo tridimensional de la romidepsina natural. Estructura obtenida de Drugbank. Creado con Sketchfab, clicar en la imagen para interactuar con el modelo.
3. EPIGENÉTICA
Los mecanismos epigenéticos son aquellos que regulan la expresión génica sin modificar la secuencia de bases nitrogenadas del DNA. Estos mecanismos se pueden clasificar en 3 grupos: metilación de bases, interferencia con RNA no codificante y modificaciones de la cromatina (4).
La metilación de bases consiste en la adición de un grupo metilo al carbono 5 de nucleótidos de citosina por acción de DNA metiltransferasas. Las citosinas metiladas son reconocidas por factores supresores de transcripción y se unen más difícilmente a los factores de transcripción, además de favorecer un mayor empaquetamiento y heterocromatinización del ADN; es decir, impiden la transcripción (5).
Por su parte, los RNAs no codificantes (ncRNAs)son moléculas de RNA que se transcriben de secuencias específicas de DNA pero no se traducen a proteínas y no intervienen de manera directa en su síntesis, a diferencia del mRNA, el rRNA o el tRNA. Hay varios tipos, aunque los más estudiados y relevantes son los miRNA, siRNA y lncRNA. Sus mecanismos de acción son variados, pudiendo destruir o silenciar moléculas de mRNA, favoreciendo la modificación de histonas, provocando cambios en el splicing, etc (4).
Finalmente, las modificaciones de la cromatina pueden deberse a cambios tanto en el DNA (por ejemplo, la metilación de citosinas) como en las histonas, aunque son mucho más habituales las modificaciones de este segundo tipo.
Estas modificaciones suelen tener como objetivo modificar la carga positiva que tienen las histonas, de modo que su relación con el DNA sea menos estrecha y que pueda acceder a él más fácilmente la maquinaria de transcripción, aunque hay algunos cambios que actúan a otros niveles.
Hay varios tipos de modificaciones, aunque los 3 más destacables son la metilación, la fosforilación y la acetilación de histonas, habitualmente de sus colas N-terminales.
Por su parte, la metilación de histonas ocurre por acción de metiltransferasas de histonas específicas y no tiene un efecto único en la transcripción: puede tanto favorecer como inhibir la expresión génica. Esto es así porque las metilaciones no modifican la carga de las histonas, afectando directamente la relación entre el DNA y ellas, sino que las histonas metiladas cambian su conformación espacial, dando lugar a estructuras secundarias de la cromatina que atraen dominios proteicos específicos pertenecientes a proteínas implicadas en la transcripción (6).
Por otro lado, la fosforilación de histonas se lleva a cabo mediante la intervención de kinasas. La adición de ácidos fosfóricos aporta cargas negativas a la cola de la histona, lo que reduce su afinidad por el DNA, también cargado negativamente, y favorece la accesibilidad a la secuencia de nucleótidos por parte de la maquinaria de transcripción (6).
La acetilación y desacetilación de histonas (concretamente la desacetilación) son los procesos epigenéticos que la romidepsina tiene como dianas, y que explicaremos más en profundidad. Este proceso está regulado por dos familias de enzimas: las acetiltransferasas de histonas (HATs) y las desacetilasas de histonas (HDACs). Las HATs toman el grupo acetilo de un acetil-CoA y lo transfieren a la cadena lateral de una lisina, anulando su carga positiva y reduciendo su afinidad por el DNA.
Figura 3. Reacciones de acetilación y desacetilación de lisinas llevadas a cabo por las enzimas HAT y HDAC, respectivamente. Creado con ChemDraw.
La desacetilación de histonas está a cargo de las HDACs, que toman el grupo acetilo que se había agregado a las lisinas y lo retiran, devolviéndole a la histona su carga positiva y permitiendo una unión más fuerte con el DNA.
En cuanto a la naturaleza de las HDAC, existen un total de 18 tipos diferentes en mamíferos, los cuales se agrupan en 4 clases: I, II, III y IV. Las dianas principales de la romidepsina son la HDAC1 y la HDAC2, ambas dentro de la clase I, y también tiene cierto efecto sobre las HDAC de clase II.
Figura 4. Modelo tridimensional de HDAC1 con un grupo acetato en su centro activo (PDB: 4BKX, Chain B). Estructura modificada con Chimera y creada con Sketchfab, clicar en la imagen para interactuar con el modelo.
El centro catalítico de las HDAC clase I lo forman un catión Zn2+, tres residuos de histidina, un residuo de tirosina y dos de aspartato. La tirosina forma un puente de hidrógeno con el oxígeno del grupo acetato a eliminar, mientras que una molécula de agua se une a las histidinas y ataca el enlace entre el grupo acetilo y el grupo amino de la cadena lateral de la lisina, formándose un intermediario de reacción estabilizado por el catión Zn2+ y, finalmente, rompiéndose el enlace amida para dar lugar a una lisina por un lado y a un grupo acetato libre por el otro (4, 6).
Figura 5. Modelo tridimensional de la superficie y del Zn pocketde la HDAC1(PDB: 4BKX, Chain B). Estructura modificada con Chimera y creada con Sketchfab, clicar en la imagen para interactuar con el modelo.
Figura 6.Mecanismo de acción de las HDAC, desacetilación del fármaco AHA por la HDAC8. Imagen tomada de Bonomi, R., Mukhopadhyay, U., Shavrin, A., Yeh, H. H., Majhi, A., Dewage, S. W., Najjar, A., Lu, X., Cisneros, G. A., Tong, W. P., Alauddin, M. M., Liu, R. S., Mangner, T. J., Turkman, N., & Gelovani, J. G. (2015). Novel Histone Deacetylase Class IIa Selective Substrate Radiotracers for PET Imaging of Epigenetic Regulation in the Brain. PloS one, 10(8), e0133512.
Cabe destacar que tan solo un 2-5% del genoma se encuentra regulado por la acetilación/desacetilación de histonas, dentro del cual encontramos genes muy importantes reguladores del ciclo celular o factores de apoptosis. Es por esto por lo que se convierte las HDAC en un target farmacológico muy interesante.
4. MECANISMO DE ACCIÓN
A rasgos generales, la romidepsina es un inhibidor de HDACs, lo que favorece que aumente el número de proteínas acetiladas, incluyendo proteínas histónicas y no histónicas. Al permanecer acetiladas las histonas, se mantiene una estructura de la cromatina más laxa y activa transcripcionalmente. Además, muchas proteínas citoplásmicas y nucleares también permanecen acetiladas, aunque se desconoce cómo afecta a la célula.
La romidepsina natural es un profármaco, y al entrar en las células el glutatión (GSH) reduce el puente disulfuro, dando un péptido monocíclico con dos grupos sulfhidrilo. El sulfhidrilo unido a la cadena de cuatro carbonos es capaz de introducirse en el sitio activo y quelar el Zn2+, lo que inhibe a la enzima de manera que ya no se puede unir el sustrato (acetato de Lys en colas de histonas) (7). La figura 7 es un estudio de modelado computacional que representa la interacción del fármaco con la enzima (8).
Figura 7.Interacciones entre romidepsina y HDAC1. Imagen tomada de Oda, A., Kato, K., Morino, M., Nakayoshi, T., Fukuyoshi, S., Saijo, K., Ishioka, C., & Kurimoto, E. (2018). Prediction of the three-dimensional structures of histone deacetylase 1 complexed with Romidepsin and FK-A5. Journal of Physics: Conference Series, 1136, 012019.
Como no hemos encontrado en el Protein Data Bank ninguna HDAC acomplejada con romidepsina, hemos seleccionado con fines didácticos una en la que se inhibe por Vorinostat o SAHA, otro inhibidor de HDAC que muestra un mecanismo parecido, y también aprobado frente al CTCL.
Figura 8. Centro activo de HDAC acomplejado con SAHA: en verde se muestran los aminoácidos del centro activo y en azul el inhibidor unido al Zn2+. Estructura modificada con Chimera (PDB:1ZZ1)
Figura 9. Modelo tridimensional de un homólogo bacteriano de HDAC inhibido con SAHA (PDB: 1ZZ1). Estructura modificada con Chimera y creada con Sketchfab, clicar en la imagen para interactuar con el modelo.
Inicialmente se pensó que el grupo sulfhidrilo se unía covalentemente a una cisteína del sitio activo (Cys151 en HDAC1), pero un experimento en el que se sustituyó esta por una serina puso de manifiesto que seguía existiendo inhibición, aunque era necesaria una concentración mayor. Por tanto, sumado al hecho de que la inhibición es reversible, se piensa que esta cisteína tiene un papel regulador en la afinidad por la romidepsina (9).
La efectividad del tratamiento epigenético se cree que se debe a la hipótesis de la “vulnerabilidad epigenética de las células cancerosas”, propuesta por Dawson y Korazides (10). Esta nos dice que mientras las células normales tienen múltiples mecanismos epigenéticos, las células tumorales dependen de unos pocos, que al fallar (inhibición de HDAC por ejemplo), llevan a catástrofe celular. Se basa en la observación de que las células normales permanecen inalteradas por estos inhibidores epigenéticos, al contrario que las cancerosas. Además, en el desarrollo de tumores, muchas veces las HDACs se encuentran sobreexpresadas, inhibiendo la expresión de genes reguladores del ciclo y supresores de tumores. Por ello, son actualmente un target farmacológico en investigación para el diseño de inhibidores (HDACi), dando lugar a este nuevo tipo de quimioterapia. Por tanto, lo fundamental es conseguir restablecer la expresión de proteínas antitumorales que llevan a la célula maligna a la muerte.
La romidepsina ejerce múltiples efectos encaminados a disminuir la población tumoral: relajación de cromatina, aumento de la transcripción, interferencia con la función de chaperonas, generación de ROS (especies reactivas de oxígeno), daños en el DNA, aumento de inhibidores endógenos del ciclo celular y promover apoptosis, tanto por vía extrínseca como intrínseca.
La romidepsina consigue la detención del ciclo celular y apoptosis principalmente, pero también inhibición de angiogénesis, inducción de autofagia y diferenciación. Los mecanismos que llevan a la célula cancerosa hacia la muerte son múltiples y variados, y dependen del tipo celular y la dosis. La mayoría de estos mecanismo se desconocen a día de hoy (7, 11).
Apoptosis: se ha visto que puede estar mediada por daños en el DNA, aumento de ROS, expresión de factores proapoptóticos, disminución de factores de supervivencia, aumento de la permeabilidad de la membrana mitocondrial y por interacciones con receptores de señales de muerte. Según el tipo de cáncer aparecen unas u otras vías implicadas. Por ejemplo, en líneas de células T malignas, se ha visto in vitro la apoptosis relacionada con ROS y daño en el DNA, activación de vías de señalización de estrés SAPK/JNK (pertenecen a superfamilia de MAPK) y UPR (respuesta a proteínas desplegadas) e inhibición de las vías PI3K-AKT-mTOR y Wnt/β-cateninas, ambas relacionadas con la progresión del ciclo (12).
Detención del ciclo celular: está relacionada con la inducción de p21 y p53. p21 o CDKN1A (cyclin dependent kinase inhibitor) inhibe las ciclinas dependientes de kinasa (CDK) y defosforila la proteína de retinoblastoma, lo que detiene el ciclo en G1. Su expresión está regulada por p53, que compite con las HDAC en el promotor de p21, induciendo su expresión. Este proceso se ve favorecido gracias a la inhibición de las HDAC. Además, la acetilación de p53 prolonga su vida media, estimulando aún más el proceso (11).
En cuanto a la generación de ROS parece que se debe a la reacción de los grupos sulfhidrilos de la romidepsina con oxígeno, dando lugar al mismo puente disulfuro y anión superóxido. La romidepsina se volvería a reducir por el GSH para pasar a su forma activa e inhibir a la HDAC o volver a reaccionar con oxígeno. Por tanto, la romidepsina es un fármaco inhibidor de HDAC, pero también productor de ROS y consumidor de GSH celular. Debido a esto último, la célula tumoral se vuelve más vulnerable a los fármacos y radicales libres, en especial, aquellas que presentan quimiorresistencias mediadas por GSH. Las ROS son inductores de daño en el ADN, desnaturalización de proteínas y permeabilización de la membrana mitocondrial, lo que supone el inicio de la vía intrínseca de la apoptosis (9, 13).
Figura 10. Relación de la romidepsinacon GSH y ROS. Estructura de la romidepsina obtenida de Drugbank y modificada con Chemdraw.
Figura 11. Efectos de la romidepsina. Se pueden observar algunos de los múltiples efectos del fármaco en las células tumorales. El conjunto de todas las vías activadas y desactivadas conducen a la célula hacia la detención del ciclo celular y apoptosis
5. USO CLÍNICO CONTRA LINFOMAS
La romidepsina se puede utilizar como tratamiento para dos tipos de cánceres, el linfoma cutáneo de células T (CTCL) y el linfoma periférico de células T (PTCL). Ambos son tipos de linfoma no Hodgkin (NHL), es decir, cánceres en los que proliferan y se malignizan linfocitos, en este caso linfocitos T (14).
Los CTCLs suponen un 4% de los NHLs y se dividen en varios tipos, dentro de los cuáles los principales son la micosis fungoide (más indolente) y el síndrome de Sézary (más agresivo), ambos caracterizados porque los linfocitos T malignos se concentran en la piel, aunque en el síndrome de Sézary también es habitual encontrarlos en la sangre (15, 22). Por su parte, los PTCLs son aproximadamente 10% de NHLs, son mucho más agresivos que los CTCLs y las células T cancerosas se encuentran principalmente en órganos linfoides secundarios, como los ganglios linfáticos o el bazo (16, 23).
Figura 12.Piel de un paciente de micosis fungoide. Imagen tomada de Ram-Wolff, C. (2014). Linfomas T Cutáneos de Tipo micosis fungoide/Síndrome de Sézary (incluida parapsoriasis). EMC – Dermatología, 48(2), 1–12. https://doi.org/10.1016/s1761-2896(14)67581-6
En 2009, la FDA (la Administración de Medicamentos y Alimentos de EEUU) aprobó el uso de la romidepsina como tratamiento para pacientes de CTCL y en 2011 para pacientes de PTCL que ya hayan recibido al menos una terapia previa. Sin embargo, hay estudios recientes que cuestionan su eficacia como tratamiento para los PCTLs, llevando a algunas compañías farmacéuticas a dejar de indicarla como tratamiento para estos cánceres (17).
Se administra de manera intravenosa, idealmente perfundiendo durante 4 horas una dosis total de 14mg/m2 los días 1, 8 y 15 de un ciclo de 28 días. Tras el final de cada ciclo se inicia uno nuevo, pudiendo modificarse la cantidad de dosis o los días de administración dependiendo de la respuesta del paciente al tratamiento. La romidepsina es transportada por la sangre unida a proteínas hasta llegar a sus tejidos diana, hasta que finalmente es metabolizada a nivel hepático por enzimas de la familia citocromo P450 (14, 24).
En cuanto a los efectos secundarios, puede producir algunos leves, como náuseas o vómitos, y otros algo más graves, como linfopenia, anemia o trombocitopenia, que pueden llegar a afectar hasta al 40% de los tratados (14, 24).
6. OTRAS APLICACIONES
Aparte de su eficacia contra los CTCLs y PTCLs, la romidepsina también parece tener cierta actividad antineoplásica en otros cánceres, aunque su uso clínico no está todavía aprobado. Algunos de los cánceres que parece que responderían a un tratamiento con romidepsina serían el cáncer de pulmón de células no pequeñas al combinarse con bortezomib (inhibidor del proteosoma 26S) (18), el cáncer de mama inflamatorio al combinarse con paclitaxel (inhibidor de polimerización de microtúbulos) (19) y ciertos tipos de cáncer de ovario al combinarse con inhibidores de las ciclooxigenasas, como la aspirina (20). Parece que las posibilidades de la romidepsina con otros fármacos son infinitas.
Por otro lado, también se tiene en cuenta a la romidepsina como un posible tratamiento contra el SIDA. Esto se debe a que la romidepsina es un agente reversor de la latencia, por lo que activa a los virus que estaban en estado latente, ocultos dentro de los linfocitos, y los expone a tratamientos antivirales o al sistema inmunitario, que debe haber sido previamente estimulado con una vacuna (21).
Para finalizar, nos gustaría lanzar una pregunta: ¿por qué la romidepsina es más eficaz en cánceres de sangre que en tumores sólidos? ¿Cuál es la diferencia fundamental para que uno sea mucho más susceptible que el otro a las modificaciones epigenéticas? La complejidad genética de los tumores sólidos o el medio en el que se encuentran expuestos al fármaco son algunas diferencias que podrían influir, pero aún se desconoce mucho de esta terapia. Por eso, “further research is needed!”.
BIBILIOGRAFÍA
Ueda, H., Nakajima, H., Hori, Y., Fujita, T., Nishimura, M., Goto, T., & Okuhara, M. (1994). FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. The Journal of antibiotics, 47(3), 301–310. https://doi.org/10.7164/antibiotics.47.301
Ueda, H., Manda, T., Matsumoto, S., Mukumoto, S., Nishigaki, F., Kawamura, I., & Shimomura, K. (1994). FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. III. Antitumor activities on experimental tumors in mice. The Journal of antibiotics, 47(3), 315–323. https://doi.org/10.7164/antibiotics.47.315
Shigematsu, N., Ueda, H., Takase, S., Tanaka, H., Yamamoto, K., & Tada, T. (1994). FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. II. Structure determination. The Journal of antibiotics, 47(3), 311–314. https://doi.org/10.7164/antibiotics.47.311
Al Aboud, N. M., Tupper, C., & Jialal, I. (2021). Genetics, Epigenetic Mechanism. In StatPearls. StatPearls Publishing.
Moore, L. D., Le, T., & Fan, G. (2013). DNA methylation and its basic function. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology, 38(1), 23–38. https://doi.org/10.1038/npp.2012.112
Bannister, A. J., & Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell research, 21(3), 381–395. https://doi.org/10.1038/cr.2011.22
VanderMolen, K. M., McCulloch, W., Pearce, C. J., & Oberlies, N. H. (2011). Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma. The Journal of antibiotics, 64(8), 525–531. https://doi.org/10.1038/ja.2011.35
Oda, A., Kato, K., Morino, M., Nakayoshi, T., Fukuyoshi, S., Saijo, K., Ishioka, C., & Kurimoto, E. (2018). Prediction of the three-dimensional structures of histone deacetylase 1 complexed with Romidepsin and FK-A5. Journal of Physics: Conference Series, 1136, 012019. https://doi.org/10.1088/1742-6596/1136/1/012019
Furumai, R., Matsuyama, A., Kobashi, N., Lee, K. H., Nishiyama, M., Nakajima, H., Tanaka, A., Komatsu, Y., Nishino, N., Yoshida, M., & Horinouchi, S. (2002). FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer research, 62(17), 4916–4921.
Dawson, M. A., & Kouzarides, T. (2012). Cancer epigenetics: from mechanism to therapy. Cell, 150(1), 12–27. https://doi.org/10.1016/j.cell.2012.06.013
Eckschlager, T., Plch, J., Stiborova, M., & Hrabeta, J. (2017). Histone Deacetylase Inhibitors as Anticancer Drugs. International journal of molecular sciences, 18(7), 1414. https://doi.org/10.3390/ijms18071414
Valdez, B. C., Brammer, J. E., Li, Y., Murray, D., Liu, Y., Hosing, C., Nieto, Y., Champlin, R. E., & Andersson, B. S. (2015). Romidepsin targets multiple survival signaling pathways in malignant T cells. Blood cancer journal, 5(10), e357. https://doi.org/10.1038/bcj.2015.83
Mizutani, H., Hiraku, Y., Tada-Oikawa, S., Murata, M., Ikemura, K., Iwamoto, T., Kagawa, Y., Okuda, M., & Kawanishi, S. (2010). Romidepsin (FK228), a potent histone deacetylase inhibitor, induces apoptosis through the generation of hydrogen peroxide. Cancer science, 101(10), 2214–2219. https://doi.org/10.1111/j.1349-7006.2010.01645.x
Yang L. P. (2011). Romidepsin: in the treatment of T-cell lymphoma. Drugs, 71(11), 1469–1480. https://doi.org/10.2165/11207170-000000000-00000
Bagherani, N., & Smoller, B. R. (2016). An overview of cutaneous T cell lymphomas. F1000Research, 5, F1000 Faculty Rev-1882. https://doi.org/10.12688/f1000research.8829.1
Xie, C., Li, X., Zeng, H., & Qian, W. (2020). Molecular insights into pathogenesis and targeted therapy of peripheral T cell lymphoma. Experimental hematology & oncology, 9(1), 30. https://doi.org/10.1186/s40164-020-00188-w
Bachy, E., Camus, V., Thieblemont, C., Sibon, D., Casasnovas, R. O., Ysebaert, L., Damaj, G., Guidez, S., Pica, G. M., Kim, W. S., Lim, S. T., André, M., García-Sancho, A. M., Penarrubia, M. J., Staber, P. B., Trotman, J., Hüttmann, A., Stefoni, V., Re, A., Gaulard, P., … Delarue, R. (2021). Romidepsin Plus CHOP Versus CHOP in Patients With Previously Untreated Peripheral T-Cell Lymphoma: Results of the Ro-CHOP Phase III Study (Conducted by LYSA). Journal of clinical oncology : official journal of the American Society of Clinical Oncology, JCO2101815. Advance online publication. https://doi.org/10.1200/JCO.21.01815
Schrump, D. S., Fischette, M. R., Nguyen, D. M., Zhao, M., Li, X., Kunst, T. F., Hancox, A., Hong, J. A., Chen, G. A., Kruchin, E., Wright, J. J., Rosing, D. R., Sparreboom, A., Figg, W. D., & Steinberg, S. M. (2008). Clinical and molecular responses in lung cancer patients receiving Romidepsin. Clinical cancer research : an official journal of the American Association for Cancer Research, 14(1), 188–198. https://doi.org/10.1158/1078-0432.CCR-07-0135
Robertson, F. M., Chu, K., Boley, K. M., Ye, Z., Liu, H., Wright, M. C., Moraes, R., Zhang, X., Green, T. L., Barsky, S. H., Heise, C., & Cristofanilli, M. (2013). The class I HDAC inhibitor Romidepsin targets inflammatory breast cancer tumor emboli and synergizes with paclitaxel to inhibit metastasis. Journal of experimental therapeutics & oncology, 10(3), 219–233.
Son, D. S., Wilson, A. J., Parl, A. K., & Khabele, D. (2010). The effects of the histone deacetylase inhibitor romidepsin (FK228) are enhanced by aspirin (ASA) in COX-1 positive ovarian cancer cells through augmentation of p21. Cancer biology & therapy, 9(11), 928–935. https://doi.org/10.4161/cbt.9.11.11873
Mothe, B., Rosás-Umbert, M., Coll, P., Manzardo, C., Puertas, M. C., Morón-López, S., Llano, A., Miranda, C., Cedeño, S., López, M., Alarcón-Soto, Y., Melis, G. G., Langohr, K., Barriocanal, A. M., Toro, J., Ruiz, I., Rovira, C., Carrillo, A., Meulbroek, M., Crook, A., … BCN02 Study Investigators (2020). HIVconsv Vaccines and Romidepsin in Early-Treated HIV-1-Infected Individuals: Safety, Immunogenicity and Effect on the Viral Reservoir (Study BCN02). Frontiers in immunology, 11, 823. https://doi.org/10.3389/fimmu.2020.00823
Ángela Aparicio Valencia e Inés Cristóbal Díaz; Universidad de Alcalá de Henares
La Plitidepsina es un principio activo con propiedades antitumorales, antivirales e inmunosupresoras de origen marino, aislada de la ascida Aplidium albicans, un invertebrado primitivo (1). Es comercializada por la empresa española PharmaMar S.A. bajo el nombre de Aplidin, el cual está siendo probado en varios ensayos clínicos. Es un compuesto de didemnina de segunda generación que pertenece a fármacos derivados de productos oceánicos naturales (2).
Figura 1. Aplidin. ¿Qué es la Plitidepsina? Antiviral de PharmaMar contra la Covid reduce casi al completo la carga viral. [cited 2022 Jan 6]. Available from: https://www.heraldo.es/noticias/salud/2021/01/26/un-antiviral-fabricado-en-espana-contra-la-covid-reduce-casi-al-completo-la-carga-viral-1416800.html
Se trata de un péptido citotóxico que aunque en la actualidad se sintetiza en el laboratorio, se aisló inicialmente de un organismo marino, de la ascidia Aplidium albicans, un pequeño animal filtrador que puede encontrarse en una de las bahías de las Islas Baleares. Este organismo puede localizarse en diferentes lugares del mundo, pero se piensa que tiene su origen en el mar Caribe y que pudo llegar al mar Mediterráneo pegado a los barcos que surcaban los mares de América a Europa.
La Plitidepsina ha surgido recientemente como nuevo candidato para el tratamiento terapéutico del SARS-CoV-2. Este fármaco ha mostrado resultados prometedores en pacientes hospitalizados con COVID-19 en cuanto a una reducción de la carga viral y una resolución clínica. Sus propiedades antivirales y antitumorales se deben a su actuación sobre el eEF1A, al cual inhibe y con ello impide la traducción de proteínas virales y tumorales (3).
Actualmente el fármaco se encuentra en fase de investigación para el tratamiento de tumores, ya que a día de hoy el uso de la Plitidepsina no está autorizado en Europa. Así la EMA y la FDA lo han categorizado como fármaco huérfano, por lo que la empresa está recurriendo a Bruselas para que sea cambiado de categoría.
Figura 2. Aplidium albicans: El animal marino que proporcionaría un fármaco (Plitidepsina) contra el coronavirus [Internet]. [cited 2022 Jan 6]. Available from:https://www.rafer.es/innovacion-laboratorio-clinico/aplidium-albicans-el-animal-marino-que-proporcionaria-un-farmaco-plitidepsina-contra-el-coronavirus/
ESTRUCTURA PLITIDEPSINA
La Plitidepsina químicamente es un depsipéptido cíclico, también conocida como dehidrodidemnina B y es comercializada por la empresa biotecnológica PharmaMar S.A. con el nombre de Aplidin. La dehidrodidemnina B es una clase de didemnina, un grupo de compuestos depsipeptídicos cíclicos aislados de tunicados del género Trididemnum. Así la Plitidepsina está formada por un péptido cíclico, en el que encontramos uno o varios enlaces éster en vez de enlaces peptídicos. Su estructura química es bastante similar a la de la didemnina B, salvo porque la Plitidepsina posee un piruvato en vez de un lactato en la posición N-terminal (1). Su fórmula molecular es C57H87N7O15 y tiene un peso molecular de 1.110,34 (2).
Figura 3. Estructura molecular de la Plitidepsina. Creado con Chimera.
MECANISMO DE ACCIÓN
En este apartado vamos a describir diferentes mecanismos de acción de la Plitidepsina. Estos mecanismos hacen que el fármaco sea activo frente a la infección por el SARS-CoV-2, y suponen un nuevo tratamiento anticancerígeno.
PLITIDEPSINA Y SARS-CoV-2
Los coronavirus pertenecen a la familia Coronaviridae dentro de la cual encontramos cuatro géneros distintos: Alphacoronavirus, Betacoronavirus, Gammacoronavirus y Deltacoronavirus. El SARS-CoV-2 pertenece al género Betacoronavirus, este es un virus esférico con envoltura que contiene un RNA monocatenario de entre 26-32 kB de polaridad positiva. Este virus infecta aquellas células que presentan en su membrana el receptor ACE-2, el cual se encuentra principalmente en las células pulmonares, por lo que en términos generales ocasiona una infección respiratoria. (4)
Para entender los efectos de la Plitidepsina sobre el SARS-CoV-2 es fundamental conocer los elementos que conforman el genoma de este virus. Su genoma está formado por diferentes marcos abiertos de lectura (ORF), los de mayor tamaño son el ORF1A y ORF1B que codifican para dos poliproteínas denominadas respectivamente como pp1a y pp1b, estos constituyen dos tercios del genoma del virus. Estas poliproteínas son posteriormente escindidas generando 16proteínas no estructurales (nsps) que forman el complejo de replicación y transcripción viral, entre las enzimas que pertenecen a este complejo encontramos la RNA polimerasa RNA dependiente. A partir del tercio restante del genoma se sintetizan distintos RNA subgenómicos que codifican para proteínas estructurales como la proteína S (Spike), la proteína E (envoltura), la proteína M (membrana) y la proteína N (nucleocápside). Estas proteínas estructurales son esenciales para la entrada del virus en las células y para el ensamblaje de las nuevas partículas víricas. (5)
Figura 4. Genoma SARS-CoV-2. Centro de Coordinación de Alertas y Emergencias Sanitarias. Aportaciones de esta actualización INFORMACIÓN CIENTÍFICA-TÉCNICA Información microbiológica acerca de SARS-CoV-2. 2021.
MECANISMO DE REPLICACIÓN E INVASIÓN
El ciclo replicativo del SARS-CoV-2 se inicia cuando la proteína S se une al receptor de sus células diana (ACE-2, principalmente las de los pulmones), así la envoltura del virus y la membrana de la célula se fusionan lo que permite la entrada en la célula del RNA monocatenario del SARS-CoV-2. Una vez dentro, se produce la traducción de los dos grandes marcos de lectura del virus, ORF1A y ORF1B que originan las poliproteínas pp1a y pp1b. Estas a su vez se escinden en distintas proteínas no estructurales gracias a la acción de proteasas virales. Estas proteínas tienen la capacidad de formar vesículas de doble membrana a partir del RER donde tiene lugar la replicación viral. Aquí la RNA pol RNA dependiente a partir del RNA del virus (+) origina copias de RNA de polaridad negativa, las cuales son utilizadas para producir los RNA (+) que formarán parte de los nuevos virus. Además gracias a las moléculas de RNA (-) se produce la transcripción de los RNA subgenómicos, estos darán lugar a las proteínas estructurales. Una vez sintetizadas, las vesículas con las diferentes proteínas y los genomas virales pasan al golgi donde se empaquetan para formar los nuevos virus. Por último, las vesículas son liberadas fuera de la célula por un proceso de exocitosis. (6)
Cabe destacar que los coronavirus poseen una exonucleasa (actividad proofreading) que les permite evitar la acumulación de mutaciones, por lo que poseen genomas más grandes que otros virus de RNA.
ACCIÓN ANTIVIRAL DE LA PLITIDEPSINA
El papel antiviral de la Plitidepsina reside en que es un potente inhibidor del factor de elongación 1α (eEF1α) de las células eucariotas. Este factor proteico participa en la traducción de las células eucariotas, más específicamente en la fase de elongación de la cadena polipeptídica. Su función es transferir los aminoacil-tRNAs al sitio A del ribosoma para así incorporar los aminoácidos correspondientes a la cadena polipeptídica. De esta manera los compuestos que inhiben al eEF1α son capaces de suprimir la síntesis de nuevas proteínas.
El eEF1α es utilizado por el SARS-CoV-2 para la traducción del RNA viral de cadena positiva y así poder sintetizar las distintas poliproteínas, además de que también es fundamental para la síntesis de las proteínas estructurales. En consecuencia la Plitidepsina inhibe la traducción de los ORF1A y ORF1B, lo que conduce a una disminución en la producción de poliproteínas y por lo tanto una menor producción de proteínas replicativas no estructurales como la RNA pol RNA dep, esencial para la síntesis de los genomas de los nuevos virus. Por este mecanismo la Plitidepsina también inhibe la traducción de los RNA subgenómicos y por lo tanto la síntesis de las proteínas estructurales, por lo que impide el ensamblaje de los nuevos virus y la replicación de estos. (1) (7)
Así sabemos que este nuevo fármaco no actuaría directamente sobre el virus sino que le impediría completar su ciclo replicativo y en consecuencia la síntesis de nuevas partículas víricas.
Figura 5. Mecanismo de acción de la Plitidepsina contra el SARS-CoV-2. Creado en BioRender.
PLITIDEPSINA Y CÁNCER
La Plitidepsina constituye un agente antitumoral que actualmente se encuentra en fase de estudio. Este fármaco posee numerosas dianas que pueden tener efectos anticancerígenos diversos, en este trabajo vamos a comentar algunos de ellos.
RUTA JNK
La ruta JNK o C-Jun NH2-terminal kinasas son un subgrupo de proteínas activadas por mitógenos (MAP Kinasas). Estas proteínas participan en vías de transducción de la señal que regulan la proliferación celular mediante la transcripción y regulación de distintos genes, además de que también desempeñan un papel fundamental en las vías apoptóticas. Una de las principales dianas de esta vía es el factor de transcripción c-Jun, de manera que JNK fosforila a c-Jun en dos residuos de serina activándolo y aumentando su actividad transcripcional. Este factor de transcripción controla la proliferación y apoptosis gracias a su capacidad de regular la expresión y función de moléculas reguladoras del ciclo celular como las ciclinas o p53. (8)
La ruta JNK se ve activada por múltiples estímulos como el estrés físico o las citoquinas a través de las MAP kinasas. Estudios recientes han demostrado que JNK también participa en algunas formas de muerte celular como la necrosis, estimulada por el Factor de Necrosis Tumoral (TNF) y promoviendo la producción de especies reactivas de oxígeno. (8)
Así la Plitidepsina es capaz de detener el ciclo celular e inducir la apoptosis gracias a la activación sostenida de la ruta JNK, junto con la inducción de estrés oxidativo por la alteración de la homeostasis del glutatión y la activación de la GTPasa Rac1 (oncogén). Todo ello finalmente conduce a la apoptosis dependiente de caspasas. En cuanto a esto, diferentes estudios han demostrado que la Plitidepsina induce la apoptosis en células de tumores sólidos por la activación sostenida de la ruta JNK. (9) (10)
PROPIEDADES ANTIANGIOGÉNICAS
Otro aspecto importante de la Plitidepsina con respecto a su papel antitumoral es que posee propiedades antiangiogénicas, puesto que se encarga de inhibir la expresión de genes que estimulan la angiogénesis, como el Factor de Crecimiento Endotelial Vascular (VEGF). Por otro lado, la Plitidepsina también es capaz de inhibir la respuesta de las células endoteliales frente a estímulos angiogénicos, por lo que se inhibe la síntesis y proliferación de nuevos vasos sanguíneos, haciendo que los tumores tengan menor aporte de oxígeno y nutrientes. (11) (12) (2)
MICROAMBIENTE TUMORAL Y CICLO CELULAR
La Plitidepsina es capaz de modificar el microambiente tumoral de manera que inhibe la proliferación del tumor y por lo tanto hace que disminuya la división de las células cancerígenas. (9)
En cuanto a su efecto antiproliferativo debemos comentar que este fármaco provoca la detención del ciclo celular en G1 y en G2/M, este fenómeno se ha visualizado principalmente en células leucémicas. Se ha comprobado que a concentraciones bajas la Plitidepsina inhibe la proliferación al detener el ciclo celular en G1 y G2/M y a concentraciones altas induce la apoptosis. (11)
También cabe destacar que la Plitidepsina puede unirse al factor de elongación 1α (eEF1α) de células eucariotas inhibiéndolo, por lo que también sería capaz de alterar la síntesis proteica en células tumorales.
Figura 6. Esquema del mecanismo de acción de la Plitidepsina frente al cáncer. Creado con GitMind.
ACTUALIDAD
Hemos explicado los diferentes mecanismos de acción de la Plitidepsina frente al SARS-CoV-2 y el cáncer, uno de los más importantes se basa en la inhibición de eEF1α. Este factor de elongación pertenece a la maquinaria de traducción de las células eucariotas, por lo que es fundamental preguntarse qué efecto podría tener su inhibición en las células sanas del paciente. Muchos estudios han analizado los efectos adversos de la Plitidepsina sobre las células eucariotas, ya que casi todos los inhibidores de la traducción en estas células suelen ser citotóxicos en los ensayos realizados con células de mamíferos.
Los estudios de la citotoxicidad de la Plitidepsina demuestran un impacto citostático más que citotóxico en la proliferación celular, además de que también se ha observado que el SARS-CoV-2 se muestra más susceptible a la acción del fármaco que la célula huésped que lo contiene (1). Para confirmar estos resultados será necesario realizar más estudios y perfeccionar el fármaco para evaluar que este es seguro.
En cuanto a la acción de la Plitidepsina contra el SARS-CoV-2 se ha demostrado que su efecto antiviral es mucho más potente que el del fármaco Remdesivir. Así se ha observado que el fármaco reduce significativamente la carga viral después de 24 horas desde su aplicación, y es capaz de disminuir la expresión de los RNAs subgenómicos tan solo 4 horas después. (1)
Actualmente este fármaco se encuentra en periodo de estudio, pero en Australia sí está autorizado para pacientes con mieloma múltiple refractario (7). La Plitidepsina supone un medicamento muy prometedor contra el SARS-CoV-2 en pacientes graves, pero para ello es necesario realizar más ensayos y conocer su eficacia contra las distintas variantes del virus, así como los efectos secundarios sobre el paciente.
BIBLIOGRAFÍA
1. Reina J. Plitidepsin, an inhibitor of the cell elongation factor eEF1a, and molnupiravir an analogue of the ribonucleoside cytidine, two new chemical compounds with intense activity against SARS-CoV-2. Rev Española Quimioter. 2021;34(5):402–7.
2. Candel FJ, Peñuelas M. Delafloxacin: Design, development and potential place in therapy. Drug Des Devel Ther. 2017;11:881–91.
3. White KM, Rosales R, Yildiz S, Kehrer T, Miorin L, Moreno E, et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science (80- ). 2021;371(6532):926–31.
4. Centro de Coordinación de Alertas y Emergencias Sanitarias. Aportaciones de esta actualización INFORMACIÓN CIENTÍFICA-TÉCNICA Información microbiológica acerca de SARS-CoV-2. 2021; Available from: https://www.mscbs.gob.es/profesionales/saludPublica/ccayes/alertasActual/nCov/documentos/20210621_MICROBIOLOGIA.pdf
5. Ezpeleta D, García Azorín D. Manual COVID-19 para el neurólogo general [Internet]. Sociedad Española de Neurología. 2020. 12–16 p. Available from: https://www.mendeley.com/catalogue/3d265ec7-03ae-325c-83d1-
6. Fernández-Camargo DA, Morales-Buenrostro Luis Eduardo. Biología del SARS-CoV-2. Rev Mex Traspl. 2020;9(S2):139–48.
7. Papapanou M, Papoutsi E, Giannakas T, Katsaounou P. Plitidepsin: Mechanisms and clinical profile of a promising antiviral agent against covid-19. J Pers Med. 2021;11(7).
8. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19(2):142–9.
9. Cáncer TDEL. Bioactivos marinos en el tratamiento del cáncer. Rev electron. 2015;40(7).
10. González-Santiago L, Suárez Y, Zarich N, Muñoz-Alonso MJ, Cuadrado A, Martínez T, et al. Aplidin® induces JNK-dependent apoptosis in human breast cancer cells via alteration of glutathione homeostasis, Rac1 GTPase activation, and MKP-1 phosphatase downregulation. Cell Death Differ. 2006;13(11):1968–81.
11. Muñoz-Alonso MJ, González-Santiago L, Zarich N, Martínez T, Alvarez E, Rojas JM, et al. Plitidepsin has a dual effect inhibiting cell cycle and inducing apoptosis via Rac1/c-Jun NH2-terminal kinase activation in human melanoma cells. J Pharmacol Exp Ther. 2008;324(3):1093–101.
12. Nalda-Molina R, Valenzuela B, Ramon-Lopez A, Miguel-Lillo B, Soto-Matos A, Perez-Ruixo JJ. Population pharmacokinetics meta-analysis of plitidepsin (Aplidin ®) in cancer subjects. Cancer Chemother Pharmacol. 2009;64(1):97–108.
Tomar café acorta nuestra vida… ¿o no?
escrito por grupo3_B_9 | 10 enero, 2022
por Lucía Dueñas Prieto & Edurne Gómez Maroto, estudiantes de 3º Biología Sanitaria (Universidad de Alcalá, UAH)
Sabemos que nuestro estilo de vida y hábitos influyen en la salud y en el funcionamiento de nuestro organismo. Durante años, el consumo de café ha estado ligado a efectos negativos sobre la salud y al envejecimiento debido, principalmente, al acortamiento de los telómeros, pero… ¿Cómo afecta realmente el consumo de café a nivel molecular? ¿Es esta idea totalmente cierta?
Este artículo se centrará en el estudio del café y en aclarar su modo de acción sobre los telómeros en relación al envejecimiento molecular.
1. IMPORTANCIA DEL TELÓMERO:
La longitud de los telómeros es un biomarcador tanto del pasado replicativo como del potencial replicativo de las células. Cada vez hay más pruebas que apoyan la idea de que los telómeros desempeñan un papel importante en la senescencia, puesto que se ha demostrado que aquellos individuos con telómeros más cortos tienen un mayor riesgo de muerte prematura en comparación con aquellos con telómeros más largos. La longitud de los telómeros es predictiva de los años de vida sana.
1.1. ¿QUÉ SON LOS TELÓMEROS?
Los telómeros son los extremos de los cromosomas, los cuales van a resultar fundamentales para que los distintos cromosomas no se unan entre sí. Se caracterizan por ser regiones de DNA no codificante y altamente repetitivas. Son estructuras que van a dar estabilidad estructural a los cromosomas, y en la división celular. Las repeticiones teloméricas permiten que no se pierda esta información.
Imagen 1. Situación de los telómeros y repeticiones teloméricas. Imagen realizada en Biorender. (https://app.biorender.com/).
Los eucariotas presentan una característica diferente respecto a los procariotas, y es que tienen un final abierto, por lo que hay un problema: el acortamiento telomérico. Este consiste en que cada replicación se va acortando la secuencia del final, lo que nos lleva a la secuencia de Hayflick, que es el número limitado de replicaciones que puede tener la línea de una célula; en humanos es de hasta 40-60 replicaciones.
1.2. ESTRUCTURA DE LOS TELÓMEROS: G-CUADRUPLEXOS.
Los G-cuadruplexos son un tipo estructural de DNA. Su estructura se basa en 3 bases (XXX) + 3 G (GGG) + 3 bases (XXX). Estas estructuras rompen su estructura de DNA B, la molécula se abre y se unen las guaninas entre ellas formando pares de Hoogsteen, que es la estructura más estable entre guaninas. Se trata de 3 planos de una tétrada de guaninas que se estabilizan con un catéter metálico (normalmente es K+ y a veces Na+). Hablamos de G-tétradas, debido a que se produce una asociación de cuatro guaninas emparejadas a través de enlaces de hidrógeno de Hoogsteen, que se apilan verticalmente.
Los G-cuadruplexos son fundamentales en el funcionamiento del telómero, pues protegen la terminación de los genes de la acción de nucleasas.
Vamos a explicar brevemente la replicación de los telómeros, para entender el papel de algunas proteínas que se mencionarán en el artículo. Hay dos proteínas en los telómeros:
La shelterina, la cual se une en la secuencia telomérica y fomentan el plegamiento de esta secuencia (T-loop), anudan el extremo del telómero y protege la secuencia de la acción de las nucleasas.
La telomerasa, enzima polimerasa reversa, pues tiene un complejo de RNA y proteína, es decir, tiene su propia secuencia de RNA. Sintetiza DNA a partir de un molde de RNA, lo que forma el complejo de TERT. La telomerasa es una enzima altamente regulada para que la longitud de los telómeros se mantenga más o menos constante.
Para que comience la elongación de los telómeros es necesario abrir los G-cuadruplexos para que actúe la maquinaria enzimática de la replicación. Una vez que están abiertos, la telomerasa se encarga de sintetizar DNA. Cuando se termina de sintetizar todo el telómero, cesa su función y se retira. A continuación, el complejo CST recluta la DNA pol α y primasa, que sintetiza el cebador, y continúa la DNA pol δ. Cuando termina, actúa una ligasa para poder unir . De esta manera, se alarga el telómero.
Otras enzimas importantes que participan en el proceso de replicación del telómero son:
RTLE1: es una proteína helicasa que abre los G-cuadruplexos, es fundamental para el mantenimiento de la estructura del telómero, pues tiene una actividad antirrecombinasa. Cuando el DNA tiene un extremo libre, con un overhang (como ocurriría con el telómero si no estuviese cerrado), es el momento en el que se activan las señales de reparación del DNA: recombinación homóloga, no homóloga y por medio de transposones (o retro).
Mec1 y Tel1. Tel1 es fundamental para reclutar la telomerasa, con la ayuda de Mec1. Además, se encargan de coordinar la respuesta al daño en el DNA mediante la fosforilación de proteínas implicadas en la reparación del DNA y vías de control. Así estas dos proteínas pueden actuar cuando el DNA esta expuesto a agentes oxidantes. (1)
2. CONEXIÓN ENTRE ESTRÉS OXIDATIVO Y ACORTAMIENTO TELOMÉRICO:
2.1 CONCEPTOS EN RELACIÓN AL DAÑO OXIDATIVO.
A continuación vamos a definir algunos conceptos en relación al daño oxidativo:
Radicales libres. Es una especie química que va a ser altamente reactiva, con capacidad oxidativa, debido a que presentan uno o más electrones desapareados, y tienden a captar un electrón de moléculas estables. Los radicales libres se forman como productos intermedios en reacciones químicas, por lo que como estas reacciones tienen lugar constantemente en el cuerpo, va a haber unas especies químicas para protegernos de los radicales libres, esta es la función de los antioxidantes.
Antioxidante. Los antioxidantes son nutrientes que “retardan o previenen la oxidación de otras moléculas”. El modo de acción de los antioxidantes es “romper” y terminar la reacción de oxidación-reducción, eliminando intermediarios del radical libre o inhibiendo otras reacciones de oxidación. De esta manera, son capaces de disminuir el efecto perjudicial que originan los radicales libres. Estas moléculas, de diferente origen y estructura, se pueden encontrar en una gran variedad de alimentos como vegetales, frutas, vino tinto, chocolate, aceites, y café.
Estrés oxidativo. El estrés oxidativo es una patología celular debida al aumento de la actividad oxidativa en el interior celular, como consecuencia de que las sustancias antioxidantes no son suficientes para combatir la cantidad de radicales libres en sangre. Origina cambios estructurales y funcionales en estas, provocando así envejecimiento celular y con ello una futura apoptosis; por lo tanto causa deterioro tisular y desarrollo de patologías. También, como veremos en este artículo, provoca daños en el DNA, afectando a los telómeros.
Especies reactivas del oxígeno (ROS). Estas especies van a ser formadas de forma exógena y endógena, desde propios hábitos (fumar, inhalar humo del tabaco, consumo de alcohol y otras drogas, y consumir pocos antioxidantes, o gastarlos muy deprisa debido al metabolismo), que podríamos evitar, hasta el propio el ambiente en el que nos encontramos (radiación, luz solar y radiación UV, y contaminación del aire).
Estar expuestos a alguno de los elementos mencionados anteriormente, van a dar lugar especies reactivas del oxígeno (ROS), moléculas altamente reactivas debido a la presencia de una capa de electrones de valencia desapareada. Son moléculas inestables que contienen oxígeno y que reaccionan fácilmente con otras moléculas en la célula. Los ROS incluyen anión superóxido, peróxido de hidrógeno, radical hidroxilo y especies reactivas del nitrógeno, los cuales tienen que ser combatidos por antioxidantes. Estas especies se producen en la mitocondria durante procesos oxidativos del metabolismo.
Si alcanzamos una situación de estrés oxidativo, y hay sobreproducción de ROS, esto va a conllevar a un deterioro de los componentes celulares (ácidos nucleicos, proteínas y lípidos). Centrándonos en los ácidos nucleicos, pueden causar lesiones de bases, roturas en el DNA, entrecruzamientos entre cadenas…
2.2. ACORTAMIENTO DEL TELÓMERO EN RELACIÓN AL ESTRÉS OXIDATIVO.
Sabemos que los telómeros están en los extremos de los cromosomas, y son ricos en guanina, así que adopta estructuras del tipo G-cuadruplexo, dificultando la actuación de la telomerasa, debido a que hay que abrirlas para que se pueda llevar a cabo la elongación de los telómeros. (2)
Sin embargo, la guanina es la nucleobase más propensa a la oxidación (ya podemos ir deduciendo cúal es el problema). Como los G-cuadruplexos son estructuras ricas en guanina esta será una estructura que responde al estrés oxidativo, porque las guaninas provocarán daño oxidativo en el DNA, dando lugar a lesiones que provocan mutaciones y problemas en la replicación, traducción y transcripción del DNA. (2)
El por qué de la guanina es la nucleobase más propensa a la oxidación, se debe a que tiene un bajo potencial redox. Como resultado de la oxidación se forma 8-oxo-7,8-dihidroguanina, y el problema reside en que ahora esta guanina oxidada aparea con una adenina, en lugar de con una citosina como ocurre en situaciones normales, lo que conlleva a una mutación si no es reparada por los sistemas moleculares. Así, las ROS pueden ocasionar modificaciones en las guaninas, las cuales pueden afectar a la estructura de los G-cuadruplexos al reducir la estabilidad térmica de sus motivos, afectando a la unión de proteínas a la estructura. (2)
Vemos a continuación la estructura de la guanina y de la guanina oxidada:
Vemos que el enlace entre la guanina – citosina, es un triple enlace, mucho más fuerte y estable que el que se forma entre la 8-oxo-guanina – adenina, el cual es un doble enlace, así entre G-C hay un emparejamiento de bases de Watson y Crick, y en el caso de 8-oxo-G – A hay un emparejamiento de bases de Hoogsteen. Esto supone que la ausencia de un tercer enlace de hidrógeno en el emparejamiento de Hoogsteen indica menor estabilidad, como ya hemos comentado, lo que conduce a la obstrucción de la formación de la tétrada de los G-cuadruplexos. (2)
Vemos cómo sería la tétrada de G-cuadruplexos con guanina:
Varios grupos de investigación han estudiado este suceso, observando que se llega a oxidar hasta un 50% de la guanina en los G-cuadruplexos, pues es una estructura susceptible al estrés oxidativo. (2)
Lo que ocurre es que esta estructura de G-cuadruplexos se forma en los extremos de los cromosomas para protegerlos de la acción de las nucleasas, porque son extremos libres. Si en presencia de ROS los G-cuadruplexos se despliegan, y no es reparado, estos extremos van a estar expuestos a las nucleasas. Un nivel elevado de 8-oxo-guanina dificulta la actividad de la telomerasa, lo que va a producir un acortamiento de los telómeros, la función y su mantenimiento. (2)
El acortamiento de los telómeros va a producir senescencia prematura. Estudios han demostrado que la pérdida de los telómeros afecta a muchos procesos celulares, produciendo apoptosis, envejecimiento, carcinogénesis e inestabilidad cromosómica. Si esto no se repara, se producen roturas en el DNA y aparición de mutaciones puesto que ha habido un apareamiento 8-oxo-G – A que supone ante una posible futura replicación la aparición de un apareamiento A – T, cambiando completamente la base inicial. (2)
Hay estudios con fibroblastos humanos normales en los que por la incorporación de ROS, se aceleró el acortamiento de los telómeros en la replicación, lo que supuso roturas teloméricas de una sola hebra debido a los radicales libres. (2)
Roturas de una hebra en los telómeros supone la activación de vías de reparación, homóloga o no homóloga, pudiéndose dar lugar, en el caso de la reparación no homóloga, a los círculos teloméricos que llevarían a una situación anómala, de muerte o malignificación de la célula (2).
Vemos a continuación una imagen aportada por dicho estudio, se trata de “Múltiples fragmentos de ADN telomérico extra cromosómico en una célula en metafase A-T de un cultivo expuesto a una dosis alta de peróxido de hidrógeno (algunos de los fragmentos se indican con flechas) (…)”.
A día de hoy, el café sigue siendo una de las bebidas más consumidas a nivel mundial debido, en gran parte, a su capacidad de mantener a los individuos en estado de alerta, a parte de por su buen aroma y sabor.
Generalmente, el consumo de café está relacionado con efectos negativos sobre la salud (existen creencias a nivel médico acerca de sus potenciales efectos adversos) y con estilos de vida poco saludables (relacionado con el tabaquismo, menos horas de sueño…), lo cual le otorga en ocasiones una imagen perjudicial.
Sin embargo, algunos estudios recientes demuestran que estaría asociado a un menor riesgo de padecer ciertas enfermedades o retrasar el envejecimiento, lo que se podría relacionar con el hecho de que contiene una gran concentración de antioxidantes.
3.1. GENERALIDADES DEL CAFÉ. ORIGEN Y COMPONENTES.
Se denomina café a la bebida preparada a partir de las semillas del fruto de los cafetos (arbusto tropical del género Coffea spp. ). Comprende muchas especies, sin embargo, sólo se cultivan Arábica y Robusta. Produce frutos carnosos rojos con dos núcleos que contienen cada uno un grano o semilla (3).
Granos de café.
Fruto de la planta del café.
Planta del café (Coffea spp. ), también conocida como cafeto.
El café está compuesto por una gran cantidad de sustancias de diferente naturaleza química (se estiman alrededor de 1000). La mayoría han sido identificadas y están relacionadas con su aroma y sabor. La concentración de estas sustancias en el café es diferente en cada una de las variedades de café y el grano de tostado. (Tabla 1) (3).
En este caso, nos centraremos en dos de ellas: la cafeína y los ácidos clorogénicos, los cuales son muy abundantes, y además, poseen propiedades antioxidantes.
3.1.1. Cafeína:
Tiene otros nombres como mateína o teína. Se trata de una molécula pequeña, que se conoce como 1,3,7-trimetilxantina. En su estructura contiene bases púricas no canónicas como la xantina (4). Es una de las tres metilxantinas que se encuentran en el café (3).
Imagen 9. Estructura molecular de la cafeína (1,3,7-trimetilxantina). Ilustración obtenida de https://molview.org/?cid=2519 (aquí también puedes ver la imagen en 3D).
Se encuentra de forma natural en el té (Camellia sisensis), cacao (Theobroma cacao) y obviamente, en el café. Además, se añade en bebidas de consumo habitual como son las bebidas energizantes y cola (3).
Sus funciones se basan principalmente en que actúa como antagonista del receptor de adenosina (del tipo A2a) los cuales son receptores inhibitorios de la señal sináptica. La cafeína estimula al Sistema Nervioso Central (SNC) permitiendo que la transmisión sináptica permanezca activa y de esta manera, se origina un estado de alerta y disminución de la somnolencia. También tiene efectos sobre el sistema cardiovascular, es estimulante de la respiración y se le atribuye una ligera acción diurética (4).
Actualmente se están realizando ensayos clínicos para poder estudiar su posible efecto en la prevención de enfermedades respiratorias pulmonares en prematuros, así también como en tratamientos contra la ansiedad, la diabetes de tipo II o la arteriosclerosis, junto con otras enfermedades cardiovasculares (3).
3.1.2. Ácidos clorogénicos:
Químicamente son ésteres fenólicos (polifenoles). Derivan de la unión éster entre el ácido cafeico y el ácido quínico. En el café se han identificado un total de 11 ácidos clorogénicos, pero generalmente se refiere a aquel que se encuentra en mayor cantidad, que es el 5-O-cafeolquínico (3).
Imagen 10. Estructura del ácido 5-O-cafeolquínico. Ilustración obtenida de https://molview.org/?cid=5280633(Aquí también puedes ver la imagen en 3D)
Se encuentra de manera natural en el té negro (Camellia sinensis) y en el café (3).
Entre algunas de sus funciones se encuentran: inhibe las metaloproteínas de la matriz, regula el metabolismo de la glucosa y de los ácidos grasos, favorece la secreción biliar y tiene cierta acción hipertensiva, favoreciendo la vasodilatación (3)(5).
Gran parte del ácido clorogénico es metabolizado en el colon por la microbiota, disminuyendo su actividad antioxidante pero favoreciendo su biodisponibilidad (3).
Ha sido utilizado en ensayos sobre el tratamiento del cáncer en estado avanzado y tratamientos para la tolerancia a la glucosa (puede ser de ayuda para las personas que sufren de diabetes de tipo II) y contra la obesidad (3) (5).
3.2. ¿CONSUMIR CAFÉ ES BENEFICIOSO O PERJUDICIAL?. EFECTOS SOBRE LOS TELÓMEROS. VENTAJAS Y DESVENTAJAS.
Debido a las importantes propiedades del café, existe un considerable interés sobre sus efectos por parte de Salud pública, sobre todo en los últimos años, donde se tiene en cuenta cada vez más cómo nuestra forma de vida afecta a nuestra salud.
Por ello, a pesar de que existen varios estudios que investigan cómo afecta el consumo de café al desarrollo de ciertas enfermedades, o en este caso, a la longitud de los telómeros, es un campo que está poco desarrollado en general porque los estudios son muy recientes. Se necesita más investigación, resultados más concluyentes y mecanismos que los puedan explicar con más precisión.
Una de las primeras investigaciones, publicada en The Journal of Nutritionrealizó un estudio a más de 4700 enfermeras para descubrir si los niveles variables de consumo de café o cafeína estaban asociados con la longitud de los telómeros (en este caso, leucocitarios). Para ello, la información acerca del consumo de café se obtuvo a partir de cuestionarios de frecuencia alimenticia, con diferentes variables, y se midió la longitud relativa de los telómeros en leucocitos mediante técnicas de biología molecular (6).
Tras realizar el experimento, se encontraron asociaciones lineales significativas con telómeros más largos para un mayor consumo de café total con cafeína. Más concretamente, en comparación con las no bebedoras de café, las probabilidades de tener una longitud de los telómero por encima de la mediana fueron alrededor del 29% en aquellas enfermeras que bebían de 2 a 3 tazas de café al día, y un 36% para aquellas que bebían 3 o más tazas al día. Sin embargo, no se obtuvieron resultados significativos para el consumo de café descafeinado (6).
Los resultados del experimento nos indicarían que la capacidad antioxidante del café sería mayor debido a la cafeína. Sin embargo, después de realizar un ajuste adicional por el consumo total de café (como un ajuste indirecto de los posibles factores de confusión de los otros antioxidantes en el café), desapareció la correlación. Esto sugiere que los otros compuestos del café podrían ser los responsables de la asociación entre el café y la longitud de los telómeros, sin descartar por completo a la cafeína (6).
Esta idea además, podría verse reforzada por el hecho de que, durante el proceso de descafeinización, no solo se reduce la concentración de cafeína, sino también se puede reducir las concentraciones de otros antioxidantes como los ácidos clorogénicos (6).
En conclusión, los hallazgos de este primer estudio sugirieron que el consumo de café (especialmente con cafeína) se asociaba con telómeros más largos, pero se necesitaban estudios adicionales para poder aclarar esta idea y que explicaran como los compuestos de café estarían involucrados en en el mantenimiento de los telómeros (6).
En otro estudio posterior, se decidió observar los cambios en la longitud del telómero frente a diferentes estímulos externos, como la temperatura, el alcohol o la cafeína. Para realizar el experimento, se utilizó un cultivo de levaduras (Saccharomyces cerevisiae), a las que se les sometió a un total de 13 estímulos estresantes durante 400 generaciones para estudiar los mecanismos responsables de las alteraciones de la longitud de los telómeros en varias condiciones de estrés (7).
Mutaciones en al menos un 6% de los genes TLM (mantenimiento de la longitud de los telómeros) conducen a la alteración del tamaño de los telómeros. La homeostasis precisa de la longitud de los telómeros depende de una gran red genética que incluye alrededor de 400 genes (conservados en gran medida desde el punto de vista evolutivo). Esta red puede verse afectada precisamente, por varias señales ambientales y diferentes mecanismos de regulación (7).
En el caso concreto de la cafeína, se identificó a las proteínas Tel1 y Mec1 como proteínas directamente afectadas. Es decir, por primera vez se identificó que estas proteínas medían el estrés por la cafeína (7).
La cafeína es un inhibidor de las quinasas relacionadas con la fosfatidil inositol-3 quinasas (quinasas similares a PI3K) como la ATR humana y la ATM y sus contrapartes de levadura, Tel1 y Mec1. Por lo tanto, se estudió si las mutaciones en estos genes diana abolirían el acortamiento de los telómeros causado por la cafeína (7).
Durante el experimento se llegó a la conclusión de que la supresión de tanto Tel1 o Mec1 individual no frena la respuesta a la cafeína (acortamiento de los telómeros). Sin embargo, un doble mutante tel1Δ- mec1Δ es completamente insensible al efecto telomérico de la cafeína, en consonancia con la función conocida que desempeñan estas dos quinasas en la biología de los telómeros (7).
Imagen 12. Células de tipo salvaje (sin modificar genéticamente), así como las dos colonias independientes en donde se eliminaron los genes codificantes para MEC1 y TEL1 mostraron acortamiento por el efecto de la cafeína. Sin embargo, la cepa de dobles mutantes tel1Δ- mec1Δ no mostró acortamiento telomérico por el efecto de la cafeína. (7)
Por tanto, gracias a este estudio se pudo llegar a la conclusión de que, realmente, la cafeína provoca el acortamiento de los telómeros al inhibir las quinasas reguladoras de tipo ATM / ATR (7).
Imagen 13. Se muestra como afecta la cafeína a la longitud de los telómeros en cepas que mostraban deleciones u otras mutaciones. El eje X muestra la longitud inicial de cada mutante y el eje Y muestra el acortamiento tras 100 generaciones. (7)
Por último, se necesitaba un estudio en el que se pudiera comparar el efecto de la cafeína y el café de manera conjunta.
Los hallazgos del estudio realizado por Larry Tucker (Universidad Brigham Young, en EE.UU), basado en la encuesta nacional NHANES, sugieren que cuanta más cafeína consumían los participantes, más cortos eran los telómeros. Por cada 100 mg de cafeína consumida, los telómeros eran 35,4 pares de bases más cortos tras eliminar el efecto de la edad y de otros factores (8).
Sin embargo, el consumo de café demostró un efecto opuesto sobre la longitud de los telómeros: cuanto más café bebían, más largos eran sus telómeros, de forma independiente a las covariables (8).
Entonces, propone que el café en sí tiene propiedades beneficiosas para la longitud de los telómeros, pero se debe a los otros compuestos y no a la cafeína (8).
Por lo tanto, significa que el consumo de cafeína procedente de otras fuentes distintas del café, como bebidas energéticas, suplementos y refrescos de cola, supone telómeros más cortos y es tan poco saludable como lo es para los que no beben café (8).
El objetivo de otros estudios más recientes se ha basado en encontrar una posible relación entre el consumo de café y el desarrollo de diferentes patologías, observando la variación del tamaño de los telómeros en las mismas. En un estudio realizado en el año 2020 por Ferruchi (Universidad de Yale, EE.UU) se evaluó la asociación transversal entre la ingesta de café y la longitud de los telómeros en los controles de cuatro estudios previos realizados para la detección de varios tipos de cáncer (9).
La conclusión general fue, otra vez, que los bebedores moderados y los bebedores en exceso (más de 3 tazas de café al día) tienen entre 2 y 3 veces más probabilidad de tener una longitud de telómeros por encima de la mediana, a pesar de que fuera poco probable que el consumo de café desempeñara un papel en las posibles asociaciones con la enfermedad (9).
2.2.1. Aspectos beneficiosos del consumo de café.
– Aspecto antioxidante (3):
La actividad antioxidante del café se debe tanto por los ácidos clorogénicos (concretamente del 5-O-cafeoilquínico) como a la presencia de cafeína y otros compuestos derivados del tostado.
Los ácidos clorogénicos son reconocidos como grandes antioxidantes. La capacidad antiradical hidroxilo (OH.) del café depende del ácido 5-O-cafeoilquínico. Actúa como captador de radicales libres superóxido.
Por otra parte, el proceso de tostado del café induce la formación de compuestos (como las melanoidinas) que también poseen actividad antioxidante. Como gran parte de los ácidos clorogénicos se pierden durante el tostado, el origen de nuevas moléculas con capacidad antioxidante compensa este hecho.
La cafeína tiene la capacidad de inhibir los efectos del estrés oxidativo provocado por radicales hidroxilos (OH.), peróxidos (ROO.) y oxígeno singlete. A pesar de que la cafeína se considera un gran antioxidante, los resultados de los estudios comentados sugieren que la cafeína no sería el componente con más propiedades beneficiosas, porque también puede actuar acortando los telómeros, tal y como hemos comentado anteriormente.
Los estudios han demostrado que el café y sus componentes, menos la cafeína, pueden proteger contra el daño oxidativo del DNA porque constituye un alimento con alta capacidad antioxidante al disminuir los niveles de los radicales ROS. De esta manera, se previene el daño provocado en la secuencia o estructura del DNA, y más concretamente, del telómero.
– Expresión de la TERT de la telomerasa (10)*.
Sin embargo, hace poco se ha descubierto que el consumo exclusivo de cafeína tiene, sorprendentemente, aspectos positivos sobre la longitud del telómero, contradiciendo los hallazgos de los estudios anteriores.
Los resultados de un estudio realizado recientemente por la Escuela de Biotecnología, Universidad de Ciencia y Tecnología de Tianjin han revelado que la cafeína promueve la expresión de la transcriptasa inversa de la telomerasa (TERT), esto ocurre tanto a niveles de ARNm como de proteínas. Como consecuencia, permite una mayor tasa de extensión de la longitud de los telómeros y previene la senescencia celular.
Este estudio se basó en un experimento realizado sobre ratones, a los cuales se les trató con cafeína durante ocho meses. Se observó la extensión de la longitud de los telómeros en el bazo y timo de los ratones, además de un cambio estructural histológico del timo, bazo e hígado de los ratones y la reducción de los niveles de beta-galactosidasa (un biomarcador de la senescencia) en las células.
Imagen 15. Posibles efectos de la cafeína sobre los telómeros: aumento de la expresión de TERT y disminución de la senescencia celular. (10)
Estos resultados sugieren que la cafeína podría promover la expresión de TERT para retrasar la senescencia celular y el envejecimiento.
3. CONCLUSIÓN:
La ingesta de café es generalizada en gran parte del mundo. Está relacionada con una serie de consecuencias beneficiosas pero también perjudiciales para la salud.
La longitud de los telómeros es un biomarcador de la senescencia de las células, y por tanto, del envejecimiento. Podemos concluir según los estudios realizados, que a medida que aumenta la ingesta de café, los telómeros tienden a ser más largos; sin embargo, un mayor consumo de cafeína supone el acortamiento de los mismos. Por lo tanto, el consumo de café moderado podría ser positivo para retrasar el envejecimiento.
Estos estudios no suponen la última palabra sobre los beneficios para la salud del café en cuanto a la longitud de los telómeros, dado que hallazgos recientes se contradicen con otros resultados anteriores; sino un comienzo y una llamada de atención para realizar más investigaciones sobre una posible vía para mejorar la salud y la calidad de vida de las personas.
4. BIBLIOGRAFÍA:
Singh, A., Kukreti, R., Saso, L., & Kukreti, S. (2019). Oxidative stress: Role and response of short guanine tracts at genomic locations. International Journal of Molecular Sciences, 20(17), 4258. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6747389/
Tchirkov, A., & Lansdorp, P. M. (2003). Role of oxidative stress in telomere shortening in cultured fibroblasts from normal individuals and patients with ataxia-telangiectasia. Human Molecular Genetics, 12(3), 227–232. https://doi.org/10.1093/hmg/ddg023
Gotteland, M., & de Pablo, S., V. (2007). Algunas verdades sobre El café. Revista Chilena de Nutricion: Organo Oficial de La Sociedad Chilena de Nutricion, Bromatologia y Toxicologia, 34(2), 105–115. https://doi.org/10.4067/s0717-75182007000200002
Liu, J. J., Crous-Bou, M., Giovannucci, E., & De Vivo, I. (2016). Coffee consumption is positively associated with longer leukocyte telomere length in the nurses’ Health Study. The Journal of Nutrition, 146(7), 1373–1378. https://doi.org/10.3945/jn.116.230490
Romano, G. H., Harari, Y., Yehuda, T., Podhorzer, A., Rubinstein, L., Shamir, R., Gottlieb, A., Silberberg, Y., Pe’er, D., Ruppin, E., Sharan, R., & Kupiec, M. (2013). Environmental stresses disrupt telomere length homeostasis. PLoS Genetics, 9(9), e1003721. https://doi.org/10.1371/journal.pgen.1003721
Tucker, L. A. (2017). Caffeine consumption and telomere length in men and women of the National Health and Nutrition Examination Survey (NHANES). Nutrition & Metabolism, 14(1), 10. https://doi.org/10.1186/s12986-017-0162-x
Steiner, B., Ferrucci, L. M., Mirabello, L., Lan, Q., Hu, W., Liao, L. M., Savage, S. A., De Vivo, I., Hayes, R. B., Rajaraman, P., Huang, W.-Y., Freedman, N. D., & Loftfield, E. (2020). Association between coffee drinking and telomere length in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. PloS One, 15(1), e0226972. https://doi.org/10.1371/journal.pone.0226972
Tao, L., Zhang, W., Zhang, Y., Zhang, M., Zhang, Y., Niu, X., Zhao, Q., Liu, Z., Li, Y., & Diao, A. (2021). Caffeine promotes the expression of telomerase reverse transcriptase to regulate cellular senescence and aging. Food & Function, 12(7), 2914–2924. https://doi.org/10.1039/d0fo03246h
ENFERMEDAD DE HUNTINGTON
escrito por 3-TeresaNatalia | 10 enero, 2022
María Teresa Larriba González y Natalia Guío Marugán
Grado en biología sanitaria, Universidad de Alcalá de Henares
INTRODUCCIÓN
La enfermedad de Huntington es una enfermedad neurodegenerativa, progresiva y mortal, causada por la repetición ininterrumpida de las bases CAG en el gen HTT, lo que provoca un alargamiento de poliglutamina anormal en la proteína Huntingtina.
Esta enfermedad ha servido como base para estudiar otros trastornos neurodegenerativos, que tienen en común el agregado anormal de proteínas, efectos tóxicos en las células y neuroinflamación.
La neurodegeneración de los individuos aparece antes de la aparición de los síntomas y se caracteriza por cambios cognitivos, motores y psicológicos.
Actualmente, a partir de la patogenicidad, se están estudiando posibles tratamientos tanto para frenar la enfermedad como para retrasarla o prevenirla, ya que todavía no existe ninguna cura.
GEN HTT
La enfermedad de Huntington es una enfermedad hereditaria originada por la mutación de un gen situado en el cromosoma 4 que está presente en todas las células del cuerpo. Este gen será el que codifique una proteína llamada Huntingtina (HTT). Por lo tanto, el Huntington es una enfermedad neurodegenerativa causada por una mutación en el gen que codifica la proteína huntingtina.
Esta mutación consiste en una repetición ininterrumpida de las bases CAG, que se traduce en el mal plegamiento de la proteína Huntingtina, originando así la enfermedad de Huntington.
En las personas con esta enfermedad, el codón CAG, que es el responsable de formar el aminoácido glutamina, está repetido de 36-120 veces.
Fotografía 1. El exceso de repeticiones del segmento CAG conduce a la producción de una versión anormalmente larga de la proteína Huntingtina. Roche Farma, S.A. (2021). Enfermedad de Huntington- Información básica [Fotografía]. Roche Pacientes. Disponible en https://rochepacientes.es/enfermedad-huntington/causas.html.
PROTEÍNA
Como consecuencia de las repeticiones del trinucleótido CAG en el gen HTT, se produce un estiramiento de poliglutamina en la proteína huntingtina. Esta proteína tiene muchas secuencias consenso llamadas HEAT que son importantes para la interacción con otras proteínas. Estos motivos HEAT provocan que se forme una súper hélice hidrofóbica, la cual protege a la proteína de posibles escisiones.
Están presentes en proteínas encargadas del transporte intracelular y son posibles responsables de provocar el apilamiento de la huntingtina en la formación de complejos proteicos. Además de intervenir en el tráfico intracelular, es necesaria para la formación de sinapsis excitatorias.
La Huntingtina se expresa en altos niveles en las neuronas del SNC donde parece localizarse en el citoplasma y estar asociadas a membranas vesiculares.
Fotografía 2. Estructura de la proteína. Swaminathan, J. Personal del MSD del Instituto Europeo de Bioinformática. (23 de marzo de 2010). Estructura de la proteína 3D en cristalografía [Fotografía]. Protein Data Bank in Europe. Disponible en http://www.ebi.ac.uk/pdbe-srv/view/entry/3io4/summary.
ENFERMEDAD
Esta enfermedad es del tipo hereditaria autosómica dominante y depende de la edad, y de la repetición de los trinucleótidos CAG.
Cuanto más larga sea esta repetición, mayor es la posibilidad de padecer esta enfermedad. De hecho, una repetición de 40 o más veces de los trinucleótidos CAG provoca la enfermedad en personas mayores de 65 años. Esta longitud está afectada por las mutaciones y el ambiente.
La prevalencia es de 4 a 10 personas por cada 100.000 habitantes en los países occidentales.
La edad media es de 40 años, con una mortalidad a los 15-20 años desde la aparición de la enfermedad.
Fotografía 3. Más de 40 repeticiones CAG en la proteína huntingtina mutada. European Huntington’s Disease Network. (2021). About Huntington’s Disease [Fotografía]. EHDN 2016. Disponible en http://www.ehdn.org/es/about-hd/#inheritance.
PATOGÉNESIS
Las repeticiones del trinucleótido CAG llevan a que las hebras de poliglutamina formen una lámina beta que se mantiene unida mediante enlaces de hidrógeno, formando de esta manera los agregados anormales que se acumulan en el cerebro. Estos agregados se pueden transmitir de una célula a otra, como los priones. La pérdida de funcionalidad de proteínas que forman parte de estos agregados producen un efecto deletéreo, que lleva a la neurodegeneración.
Estos agregados se producen en el núcleo neuronal, pero también pueden aparecer en el citoplasma, dendritas y en la terminal axónica. Las células gliales también contribuyen a la enfermedad de Huntington ya que muchos de estos agregados también actúan sobre ellas.
La huntingtina se puede dividir en fragmentos tóxicos y la acumulación de estos fragmentos es característica también de esta enfermedad.
Otro argumento es que el número de poliglutaminas está correlacionado con la tasa de agregación y con la aparición de la enfermedad. Esto sugiere que haya un vínculo directo entre la agregación y la toxicidad celular. Las poliglutaminas expandidas pueden interferir con regiones ricas en glutamina presentes en los dominios de muchos factores de transcripción. De hecho, la huntingtina mutante interactúa con reguladores de la transcripción lo que lleva a su interrupción.
Las neuronas se ven comprometidas debido a la disminución de la transcripción de los genes esenciales en la neurotransmisión y por los defectos en la entrega de proteínas y orgánulos a lo largo de sus axones.
Fotografía 4. Formación progresiva de los agregados de la proteína Huntingtina. Li H, Li SH, Cheng AL, Mangiarini L, Bates GP, Li XJ (julio de 1999). Ultrastructural localization and progressive formation of neuropil aggregates in Huntington’s disease transgenic mice [photograpg]. Hum Mol Genet. https://pubmed.ncbi.nlm.nih.gov/10369868/
Debido a la pérdida de la función de la proteína salvaje, los agregados de proteínas motoras y agregados que bloquean los axones, la huntingtina inhibe el transporte axonal rápido de orgánulos. Este transporte se necesita para la correcta entrega a las membranas nucleares durante la transmisión sináptica y su reciclaje. Así, un fallo en la entrega de GABA o AMPA, inhibe la excitabilidad sináptica en la enfermedad de Huntington.
La Huntingtina altera la función mitocondrial y como consecuencia causa la producción de especies reactivas de oxígeno (ROS), que a su vez dañan a las mitocondrias. Además, la Huntingtina se expresa en células inmunitarias que secretan citoquinas proinflamatorias que causan la neuroinflamación. Hay informes que han descrito un deterioro del proteosoma debido a la expresión expandida de poliglutamina en la huntingtina.
Los primeros síntomas muestran cambios de personalidad, cognición y control motor sutiles. Los movimientos de coordinación gruesa, como la marcha y la postura, se deterioran más tarde que los movimientos finos. Los individuos se pueden volver irritables, la multitarea se vuelve difícil y aumenta la ansiedad y los olvidos.
Después de este primer periodo sintomatológico, los enfermos comienzan a mostrar signos de corea, pérdida de coordinación, lentitud, insuficiencia motora y movimientos oculares sacádicos lentos. Sin embargo, la corea no es un buen indicador de la gravedad de la enfermedad, ya que en ocasiones puede aparecer temprano o de forma transitoria.
Las funciones ejecutivas como la organización, la planificación y la adquisición de nuevas habilidades motoras empeoran con el tiempo. El habla se deteriora más rápidamente que la comprensión. Los síntomas psiquiátricos y conductuales aparecen con cierta frecuencia, pero no muestran una progresión con la gravedad de la enfermedad. La depresión es típica y se estima que el suicidio es entre 5 y 10 veces más frecuente que en el resto de la población.
Los enfermos de Huntington son incapaces de mantener una contracción voluntaria constante. Tampoco son capaces de mantener una presión constante de la mano, lo que se llama agarre de la lechera.
La inconstancia motora es independiente de la corea y sí que avanza progresivamente, siendo un indicador de la gravedad de la enfermedad. A medida que los déficits motores y cognitivos se agravan, los pacientes acaban muriendo, normalmente por complicaciones de las caídas, inanición, disfagia o aspiración.
DIAGNÓSTICO
Los individuos con riesgo de heredar estas repeticiones se pueden identificar mediante análisis genéticos, ya que las personas con esta enfermedad presentan repeticiones CAG en su genoma. Además, se realizan test de imagen cerebrales, para mostrar los posibles cambios sufridos en la estructura encefálica.
El diagnóstico formal se hace basándose en signos como movimientos involuntarios, distonía, bradiquinesia e incoordinación, es decir, en la sintomatología neurológica.
TRATAMIENTO
No existe ninguna cura, pero hay medicamentos que pueden reducir algunos de sus síntomas, como antidepresivos, medicamentos para reducir los cambios de humor e irritabilidad, tetrabenazina, que reduce los movimientos involuntarios de la corea al agotar la dopamina, y antipsicóticos, que bloquean la dopamina. Sin embargo, la mayoría de estos medicamentos causan efectos secundarios problemáticos.
Fotografía 7. Uso de tetrabenazina para reducir los movimientos involuntarios de la corea de Huntington. Gramón Bagó de Uruguay S.A. (2016). Productos- Tetrazol [Fotografía]. Gramón Bagó. Disponible en https://www.gramonbago.com.uy/contenido/Tetrazol-25-mg-5343.
Además, las intervenciones sociales son a menudo igual de efectivas que el tratamiento con medicamentos en pacientes con alteraciones en la conducta. Los grupos de apoyo son buenas fuentes de información y conocimiento que pueden ayudar a los pacientes a superar las dificultades de la enfermedad. También se proporcionan ayudas en las tareas diarias (terapia ocupacional), alimentación, comunicación (logopedia), movilidad y equilibrio (fisioterapia).
No obstante, se siguen realizando estudios para descubrir nuevos métodos tanto para tratar la enfermedad, como para predecirla. Se ha avanzado en la identificación de posibles formas de “desactivar» el gen defectuoso que la causa. Además, se están llevando a cabo ensayos clínicos para encontrar agentes modificadores de la enfermedad Huntington que ralentice o revierta.
Fotografía 8. Grupo de apoyo psicológico. Centro de Terapia y Bienestar Emocional. (28 de junio de 2016). Grupos de apoyo psicológico [Fotografía]. A la Sombra del Árbol. Disponible en http://centrodepsicoterapia.mx/el-grupo-de-apoyo-psicologico/.
CONCLUSIÓN
En conclusión, la enfermedad de Huntington es un trastorno neurodegenerativo que está incrementando en los últimos años y para el que se requieren muchos estudios.
Además, esta investigación podrá servir de base en la intervención de otros trastornos neurodegenerativos más frecuentes con los que comparte muchas características, como Alzheimer o Parkinson.
BIBLIOGRAFÍA
Walker FO. (20 de junio de 2007). Huntington’s disease. Lancet.
Ross CA, Tabrizi SJ. (enero de 2011). Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol.
Jimenez-Sanchez M, Licitra F, Underwood BR, Rubinsztein DC. (5 de julio de 2017) Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb Perspect Med.
McColgan P, Tabrizi SJ. (enero de 2018). Huntington’s disease: a clinical review. Eur J Neurol.
SÍNDROME DE BLOOM
escrito por galen14 | 10 enero, 2022
Belén Segovia y Gabriela Zapata
El Síndrome de Bloom (BS) es una enfermedad hereditaria autosómica recesiva, rara causada por una mutación en el gen BLM, que deriva en dos características principales:
Predisposición a desarrollar todo tipo de cánceres a una edad temprana.
Inestabilidad genética caracterizada por anomalías citogenéticas que incluyen numerosas roturas cromosómicas y un alto número de intercambios de cromátidas hermanas.
Se caracteriza por que los individuos que lo padecen tienen una estatura más baja de lo normal acompañada de facciones características de la enfermedad como son una cara larga y estrecha, nariz y orejas prominentes, con manifestaciones dermatológicas como es una erupción facial desencadenada por la exposición al sol (fotosensibilidad). Todo esto acompañado por una voz aguda, mayor susceptibilidad de padecer infecciones (inmunodeficiencia), y en ellos aumenta el riesgo de padecer cáncer. También padecen de problemas de fertilidad debido al hipogadismo, dificultades de aprendizaje, y retraso en el crecimiento y desarrollo.
Este es un síndrome de inestabilidad cromosómica, y las mutaciones somáticas que se producen como consecuencia de esa inestabilidad, son responsables del mayor riesgo de padecer cáncer. Entre todas las complicaciones, quizá las neoplasias malignas sean las más prominentes y potencialmente mortales. De hecho, los pacientes con BS tienen entre 150 y 300 veces más probabilidades de desarrollar una neoplasia maligna que la población general. Muchos de estos pacientes desarrollan algún tipo de cáncer a lo largo de su vida, y entre ellos destacan la leucemia, linfoma o cáncer colorrectal, de laringe, de mama y de piel.
Además de los síntomas anteriores, los pacientes con BS presentan niveles de inmunoglobulinas reducidas, lo que conlleva a una mayor susceptibilidad a neumonía, bronquiectasias y enfermedad pulmonar crónica. Aquellos que lo padecen también pueden presentar anomalías endocrinas particularmente anomalías del metabolismo de los carbohidratos, resistencias a la insulina y susceptibilidad a diabetes tipo II, dislipidemia e hipotiroidismo.
Este síndrome prevalece en la población judía Ashkenazi del este de Europa e Israel, lo que explica aproximadamente un tercio de los casos del síndrome de Bloom. De los judíos ashkenazis, 1 de cada 48000 personas lo presentan, y un 1% son portadores de este síndrome. En Estados Unidos se han notificado 170 casos, y raramente se ha notificado en otros países.
ENZIMA (GENÉTICA, FUNCIÓN NORMAL, MECANISMO DE ACCIÓN Y FORMAS INTERMEDIAS)
El Síndrome de Bloom surge por mutaciones en ambas copias del gen BLM, localizado en el brazo largo del cromosoma 15 (15q26.1), por lo tanto la mutación que causa el síndrome es autosómica y, en este caso, recesiva. En esta región del cromosoma se han encontrado diversas patologías normalmente relacionadas con retrasos en el crecimiento y en el desarrollo mental por deleciones o duplicaciones.
El gen situado en ese loci corresponde a una proteína de la familia de las helicasas RecQ, la RecQL3. Esta familia está formada por un solo miembro de origen procariota representado por la proteína RecQ de Escherichia coli, dos miembros de origen eucariota inferior representados por las proteínas Sgs1 y Rqh1, y cinco miembros de origen humano, las proteínas WRN (RecQL2), BLM (RecQL3), RecQ4 (RecQL4), RecQL (RecQL1) y RecQ5 (RecQL5).
Las helicasas son un grupo diverso de enzimas que se unen al DNA y desenrollan las dos hebras en espiral de la molécula del DNA. Este desenrollado es necesario en procesos en el núcleo de la célula, incluyendo la copia del DNA en preparación para la división celular y reparación del DNA dañado.
Cuando una célula se prepara para dividirse, el DNA que compone los cromosomas se copia de forma que cada nueva célula tendrá dos copias de cada cromosoma, una de cada progenitor. El DNA está dispuesto en las cromátidas hermanas, que están unidas una a otra durante las primeras etapas de la división celular. Las cromátidas hermanas de vez en cuando intercambian pequeñas secciones de DNA durante este tiempo, un proceso denominado intercambio de cromátidas hermanas. Se cree que estos intercambios pueden ser una respuesta al daño de DNA durante el proceso de copia.
BLM es una helicasa de ADN RecQ dependiente de ATP 3′-5 ′, uno de los estabilizadores del genoma más esenciales involucrados en la regulación de la replicación del ADN, recombinación y vías homólogas y no homólogas de reparación de rotura de doble cadena. Ayuda a evitar el exceso de intercambios de las cromátidas hermanas y también participa en otros procesos que ayudan a mantener la estabilidad del DNA durante el proceso de copia.
Se han identificado 151 mutaciones diferentes del gen BLM en las personas con síndrome de Bloom con diferentes orígenes étnicos. Principalmente las mutaciones dan lugar a un codón de paro prematuro que da lugar a una proteína truncada (80% de los casos), o se crea una proteína sin su actividad helicasa (20% de los casos).
Existe una mutación genética particular que elimina seis nucleótidos y los reemplaza con otros siete en la posición 2281 del DNA. Es la mutación blmAsh, en población judía Ashkenazi, que da lugar a la síntesis de una versión anormalmente corta, no funcional de la proteína BLM. Aparte de esta mutación, hay otras mutaciones del gen BLM que cambian aminoácidos en la secuencia de la proteína o crean una señal de parada precoz en su codificación. Como consecuencia de la ausencia o deficiencia de proteína BLM funcional, la frecuencia de intercambio de las cromátidas de las cromátidas hermanas es de alrededor de 10 veces más alta que el promedio.
Debido a la mutación de esta proteína, puede ocurrir la rotura de los cromosomas por la zona de telómeros, que se produce con mayor frecuencia en los individuos afectados. Estas roturas, que ocurrirían por una mala interacción de la BLM con los G-cuadruplexos, alteran las actividades normales de las células y provocan los problemas de salud asociados con este síndrome. Sin la proteína BLM, la célula es menos capaz de reparar el daño del ADN provocado por la luz UV, lo que deriva en una mayor sensibilidad a la luz solar.
Estos cambios genéticos permiten a las células dividirse de forma incontrolada dando lugar a una predisposición en el desarrollo de cánceres.Por otro lado, además de las mutaciones el gen de la BLM sufre otros tipos de alteraciones incluyendo un aumento en el número de copias transcripcionales, aumentando el número de proteínas en muchos cánceres.
De esta manera, se ha visto que las mutaciones en la BLM inducen al desarrollo de tumores y déficits en el desarrollo debido a la ausencia de su acción, lo que provoca intercambios entre las cromátidas hermanas y cromosomas homólogos que acaba provocando graves mutaciones en los individuos. Estas mutaciones se han visto en la línea hematopoyética, pero esto abriría nuevas preguntas para investigar cómo: ¿en qué células pueden darse estas modificaciones?, ¿en qué estado del desarrollo ocurre la recombinación? o ¿están estas mutaciones restringidas al precursor hematopoyético o podrían ocurrir en cualquier estado de desarrollo?
Imagen 2. Esquema resumen.
2. ANOMALÍA EN EL SÍNDROME A NIVEL BIOMOLECULAR
La BLM es una proteína que se compone de un conjunto de dominios que median sus funciones. Es una helicasa que utiliza la energía de la hidrólisis del ATP para moverse por una cadena simple de DNA y para poder romper los puentes de hidrógeno. Las helicasas están implicadas en la apertura del dúplex, lo cual se incluye los procesos de replicación, transcripción, recombinación homóloga, y otras formas de reparación del DNA, y todas ellas participan en el mantenimiento de la integridad del genoma.
Como se predice por su homología con las helicasas RecQ, la función de la BLM es hidrolizar el ATP para desenrollar el ADN moviéndose en una sola hebra en la dirección 3´-5´, y prefiere los sustratos que incluyen moléculas que simulan la replicación del DNA e intermediarios de recombinación (incluyendo horquillas de replicación, uniones de Holiday, bucles de desplazamiento y G-cuadruplexos). Es importante para la estabilidad de la horquilla de reparación, incluso en procesos de daño del DNA. Para ello está implicada en las siguientes procesos:
Participa en la apertura de las dos cadenas para crean cadenas simples que son un sustrato de la RAD51, una enzima muy importante en la reparación del DNA.
Implicada en reclutamiento de proteínas para estabilizar la apertura de la horquilla como las ssDNA-binding protein (RPA y RAD51). Estas son recombinasas que previenen que se formen apareamientos incorrectos por recombinación homóloga.
Se encarga de desplazar las uniones de Holliday y de disolverlas sin sobrecruzamiento.
Tiene funciones relacionadas con la interacción con estructuras como los G-cuadruplexos, encargados de mantener la estabilidad del genoma en regiones como los telómeros o los rRNA.
La BLM en conjunto con la topoisomerasa IIIα desenrolla las moléculas de DNA con dos uniones de Holliday y las disuelve sin crear productos de recombinación. La topoisomerasa I es una enzima que se une a la doble hebra de DNA y permite el relajamiento del superenrollamiento que se crea en la hebra de DNA. La topoisomerasa IIIα es un tipo de topoisomerasa I, por lo que tiene actividad de corte y empalme que se da en una sola hebra en ssDNA.
La BLM forma un complejo con la topoisomerasa IIIα y otros componentes. La región de la BLM que interactúa con la topoisomerasa se encuentra localizada en el extremo N-terminal de los 200 últimos aminoácidos de la proteína. De esta manera las proteínas BLM que presentan deleciones en esta región no son capaces de interactuar con la topoisomerasa IIIα.
Una vez analizados los procesos en los que media la proteína, podemos analizar sus funciones celulares más detalladamente:
A. REPLICACIÓN
La helicasa BLM juega un papel esencial en varios eventos que tienen lugar durante la replicación del ADN. Cuando se produce un daño en la replicación, el ADN se vuelve más propenso a romperse y cualquier estrés replicativo podría ser una fuente de una transformación tumoral temprana
Progresión y reparación de la horquilla de replicación: el daño inducido durante la replicación puede ser inducido por estructuras de ADN atípicas que pueden bloquear la progresión de la horquilla, como G-quadruplex, uniones de Holliday, complejos específicos de proteína-ADN, híbridos de ARN, etc. La enzima BLM es necesaria para la progresión y la estabilidad de la horquilla de replicación en condiciones fisiológicas normales y también es fundamental, después del daño en la replicación, para la estabilización y progreso de las horquillas bloqueadas al desenrollar estructuras de ADN inusuales, evitar la recombinación inapropiada y la supresión de la activación de un nuevo origen.
DNA telomérico: las secuencias teloméricas repetidas forman estructuras muy ordenadas, como T-Loops, horquillas y G-quadruplexos, los sustratos preferidos para BLM, que deben disolverse para permitir la replicación adecuada del ADN telomérico.La BLM también participa en el mantenimiento de la integridad de los telómeros por la vía de alargamiento alternativo de los telómeros (ALT), la cual es independiente de la vía de la telomerasa.
DNA ribosomal: las secuencias repetidas del DNA ribosomal se replican de manera única. La alta actividad de transcripción de estas regiones del genoma induce la formación de un R-Loop que si no se resuelve provoca rupturas del DNAr. BLM podría estar implicada en el mantenimiento de la integridad de este DNA, seguramente desenrollando las estructuras complejas que se formen.
B. RECOMBINACIÓN HOMÓLOGA
BLM interacciona con varios componentes de la maquinaria de recombinación homóloga (HR). La vía de la recombinación homóloga utiliza una secuencia homóloga como plantilla para reparar el ADN dañado o reiniciar las horquillas de replicación bloqueadas. BLM es una de esas proteínas necesarias que actúan en la replicación y recombinación desempeñando funciones tanto pro como anti-recombinogénicas.
Reanuda el bloqueo de las horquillas de replicación cuando disuelve las uniones de Holliday.
Reparación de la ruptura de la doble hebra: la ruptura de doble hebra es una de las formas más letales de daño en el DNA. En el primer paso, BLM actúa estimulando la actividad nucleasa de la exonucleasa 1 (EXO1) y la helicasa / endoexonucleasa DNA2 para asegurar la resección extensa 5′ de los extremos del ADN generados por la ruptura de doble hebra. Los overhangs creados se recubren rápidamente con la proteína RPA para estabilizarlas y protegerlas de la degradación (BLM tiene actividad recombinasa en este paso).
El overhang invade la hebra opuesta para buscar un fragmento homólogo a partir del cual pueda darse la reparación. Esta invasión de la hebra es seguida por la síntesis de ADN en el extremo invasor y conduce al desplazamiento de la hebra paralela, formando un D-Loop.
Si la homología encontrada es suficiente, BLM permitirá que continúe la reparación por la vía de recombinación homóloga. Las etapas finales de esta reparación se llevarán a cabo mediante la resolución de las uniones de Holliday (HJs), ya sea por el complejo disolvasoma BLM sin recombinación (actividad anti recombinasa de BLM), o por resolución, lo cual generará productos de recombinación.
El complejo BLM disolvasoma está compuesto por diversas enzimas: la BLM, topoisomerasa IIIα y y RMI1 y RMI 2, para procesar las uniones dobles de Holliday (dHJ) generadas durante el paso de invasión de la cadena de la vía de reparación por disolución.
Vía alternativa de alargamiento de telómeros (ALT): en algunos cánceres se utiliza esta vía, en la que BLM interacciona con los telómeros y otras proteínas de esta vía.
BLM desenrolla los dúplex híbridos de ARN-ADN, conocidos como R-Loop, y G4, en sitios transcripcionalmente activos, específicamente después del daño del ADN. La resolución de estas estructuras es esencial para permitir el acceso de la maquinaria de reparación a las lesiones inducidas, asegurando así una reanudación normal de la transcripción.
El gen que codifica para la proteína BLM es un gen de 1417 aminoácidos ubicado en el cromosoma 15q26.1, que posee una actividad helicasa dependiente de ATP 3′ → 5 ‘. Su expresión está muy regulada en el ciclo celular, expresándose niveles más altos en las fases S y G2.
La proteína BLM, como ya hemos mencionado, es una helicasa de tipo RecQ, cuya función se basa en desenrollar el DNA, una actividad que utiliza ATP para moverse a lo largo de una hebra simple y para romper los enlaces de hidrógeno que mantienen unidas las dos cadenas de DNA. Su actividad dependiente de ATP es muy relevante en procesos tales como la replicación, transcripción, recombinación y reparación del ADN, por lo que estas proteínas presentan un sitio de unión a ATP. Las RecQ pertenecen a la superfamilia de las helicasas SF2 y juegan un papel crucial en el metabolismo del ADN, asegurando así el mantenimiento de la integridad y la estructura del genoma.
Sin embargo, el dominio helicasa no es suficiente para la actividad de la BLM. Asociado a este dominio helicasa está el dominio RQC, y ambos forman el núcleo catalítico de la BLM. El dominio helicasa es el más conservado en la familia de las RecQ, ya que es común a las proteínas de la familia SF2.
Este RQC se compone de un motivo de unión Zn 2+ y un dominio de “alas hélice de ADN” (winged helix domain) que conjuntamente actúan como el lugar de anclaje a la doble hebra del DNA. En este motivo se encuentra también una pinza-β que actúa insertando el complejo en el DNA y causando la separación del dúplex. El extremo C-terminal del RQC es el dominio “helicasa y ribonucleasa D C-terminal (HRDC)” que no se une directamente al DNA pero puede ayudar en las actividades helicasa y ATPasa. Es necesario para las disoluciones de las uniones de Holiday.
El extremo N- terminal de la proteína presenta la región de reconocimiento (la BLM forma un complejo con numerosas enzimas como son la topoisomerasa IIIα, RM1 y RM2, RPA…). La combinación de la actividad de desenrollamiento y reconocimiento cataliza la función del intercambio entre cadenas, que requiere promover previamente la disolución de las uniones holiday o el retroceso de las horquillas de replicación estancadas. No se han encontrado mutaciones que causen el síndrome de Bloom fuera de los dominios de RQC y helicasa.
Los miembros de la familia RecQ tienen siete motivos helicasa conservados, insertados entre dominios N-terminales y C-terminales únicos de tamaño variable. Existen cinco RecQ en humanos, las cuales tienen un dominio de helicasa estructuralmente conservado que contiene cajas Walker A y B y una caja DEAH que funciona en el desenrollamiento, siendo este dependiente de ATP y Mg2 +. Otros dominios como el RQC (RecQ C-terminal) y HDRC también son comunes a las proteínas de esta familia.
Estudios recientes basados en diversas bases de datos hacen ver que existe un pequeño dominio de unión a ácido nucleico en la región C-terminal de todos los miembros de la familia RecQ, lo que sugiere características comunes de reconocimiento de ADN en las helicasas.
Las helicasas RecQ se pueden clasificar en 2 grupos: el grupo que contiene las proteínas WRN, BLM, RecQ4, Sgs1 y Rqh1 cuyos tamaños van desde 1208 a 1447 aminoácidos ácidos, y el grupo que contiene las proteínas RecQL y RecQ5 con 410 a 649 aminoácidos y que se componen prácticamente del dominio helicasa central. Cabe destacar que, como podemos visualizar en la imagen, las proteínas BLM, WRN, RecQL y RecQ5, presentan una señal de localización nuclear (NLS) en la región C-terminal.
Además se sabe que hasta hoy solo RecQ, RecQL, BLM, WRN y Sgs1 tienen una actividad ADN helicasa dependiente de ATP 3′-5 ’. La helicasa WRN tiene una actividad enzimática exonucleasa 3′-5 ′ adicional dentro de su región N-terminal.
Imagen 8. Se representan las proteínas con las que interactúa la proteína BLM, la cual se compone de los dominios de núcleo de ATPasa / helicasa, RQC y HRDC. Podemos ver las proteínas clave involucradas en diferentes vías de reparación e interactuando con BLM junto con sus posiciones de unión a BLM. Extraída de: (https://www.frontiersin.org/files/Articles/634789/fgene-12-634789-HTML/image_m/fgene-12-634789-g001.jpg).
4. PATOLOGÍA Y CÁNCER
El cáncer es la complicación médica más frecuente y grave que aparece en pacientes con Síndrome de Bloom, y suele llevar a la muerte. En este síndrome se dan una amplia variedad de tipos de cáncer y ubicaciones anatómicas en personas con SB. Las células con mutaciones en BLM conducen a la acumulación de DNA con daños con alta sensibilidad hacia varios fármacos quimioterapéuticos.
La distribución de los cánceres es parecida a la que sucede en la población general, pero ocurren a edades más tempranas y una sola persona puede experimentar múltiples cánceres. En esta enfermedad los más frecuentes son la leucemia y el linfoma, y dentro de los tumores sólidos los más comunes son los cánceres del tracto digestivo, en particular el adenocarcinoma del tracto intestinal superior e inferior. Los carcinomas de células de cabeza y cuello también se han diagnosticado con frecuencia, especialmente en la base de la lengua, la epiglotis y el esófago.
Como dato curioso cabe destacar que la población judía Ashkenazi tiene alta prevalencia de cáncer colorrectal precisamente por la mutación en este gen. De acuerdo con su función de cuidar el genoma, se han visto mutaciones en el gen BLM asociadas con el riesgo de cáncer colorrectal. Aproximadamente el 0,11% de pacientes presentan la mutación heterocigótica BLM que confiere un riesgo de penetrancia de bajo a moderado para desarrollar cáncer colorrectal., y entre estos pacientes se encuentran personas con ascendencia judía asquenazí, portadores de alta frecuencia de la mutación heterocigótica BLMAsh.
Múltiples estudios muestran que la proteína BLM tiene una implicación directa en la regulación de varios oncogenes y genes supresores de tumores . BLM potencia la degradación y alteración de la función del factor de transcripción oncogénico c-Jun, moderando así el efecto de este último en la transformación neoplásica.
Es más, el gen c-Myc, que regula la transcripción de muchos genes fundamentales y se sobreexpresa en varios tipos de cánceres, también es un objetivo de BLM. De hecho, BLM disminuye los niveles de c-Myc y retrasa la iniciación del tumor en múltiples líneas de células de ratón. Es por esto que muchos autores denominan a la proteína como una “cuidadora de la supresión de tumores”
El efecto directo y específico de la función de BLM en la prevención del cáncer ya que BLM trabaja en conjunto con p53 para efectuar la apoptosis; por lo tanto, previene la transformación tumoral. Se han realizado múltiples estudios en células con síndrome de Bloom en los que se identifican anomalías tanto en la acumulación como en la activación de p53 inducida por daños en el ADN.
Además de la predisposición al cáncer que tienen los individuos que presentan mutaciones en la BLM, también existe esta predisposición en pacientes con sobreexpresión de la BLM. En estos últimos casos, hay una eficiencia muy alta de reparación así como de recombinación por la vía homóloga, lo que se traduce en una acumulación del daño en el DNA e hiper recombinación.
5. SISTEMA INMUNE
Se afirma también que la proteína BLM está involucrada en el desarrollo y mantenimiento del sistema inmune. En pacientes con el síndrome de Bloom se observan niveles de inmunoglobulinas IgM e IgA anormales y de IgG reducidos. Con los niveles disminuidos de BLM, las células precursoras linfoides de la médula ósea y las células B maduras del bazo y la cavidad peritoneal disminuyen significativamente, e incluso se ven fallos en el desarrollo de las células T.
Debido a la acumulación de ADN dañado en células deficientes en BLM, también se ha observado una mayor expresión del gen inflamatorio estimulado por interferón y niveles aumentados en sangre periférica. BLM juega un papel indispensable en el desarrollo, proliferación, mantenimiento, estabilidad y función de las células inmunes y contribuye a la inmunodeficiencia en pacientes afectados por BS.
6. MÉTODOS DIAGNÓSTICOS Y TRATAMIENTO
Debido a que este síndrome es raro, no existen pautas de tratamiento basadas en evidencias científicas. Entre las principales preocupaciones destacan las anomalías de la piel, los problemas de crecimiento y nutrición, las anomalías endocrinológicas y el riesgo de neoplasias malignas, que son la mayor causa de muerte.
Como remedio para los problemas de crecimiento se emplean preparados y alimentos hipercalóricos. Además el tratamiento con hormona de crecimiento podría mejorar el crecimiento lineal, aunque su uso es controvertido por algunos estudios que identifican un mayor riesgo para el desarrollo de cáncer en niños.
En algunos casos en los que hay una disminución de los niveles de inmunoglobulinas y se sufren infecciones, se pueden aplicar antibióticos y tratamiento con inmunoglobulinas intravenosas o subcutáneas.
Resulta fundamental el cuidado de la piel con cremas y protectores y su vigilancia en cuanto a la aparición de posibles cánceres.
La enfermedad sigue un patrón de herencia autosómico recesivo y es por eso que se ofrece consejo genético a las parejas en riesgo cuando ambos son portadores de la mutación, informándoles de un riesgo del 25% de tener un hijo afectado en cada embarazo.
El diagnóstico se intuye clínicamente en base a la identificación de síntomas propios del síndrome de Bloom y se confirma mediante la identificación de variantes patogénicas bialélicas del gen BLM en las pruebas moleculares. También puede confirmarse mediante un análisis citogenético que identifica un mayor número de intercambios entre cromátidas hermanas (esta prueba se puede realizar en el embarazo) o mediante técnicas de secuenciación y deleción/duplicación de BLM.
BIBLIOGRAFÍA
– Ababou M. (2021) Bloom syndrome and the underlying causes of genetic instability. Mol Genet Metab. 2021 May;133(1):35-48. doi: 10.1016/j.ymgme.2021.03.003. Epub 2021 Mar 10. PMID: 33736941.
– Kaur E, Agrawal R and Sengupta S (2021) Functions of BLM Helicase in Cells: Is It Acting Like a Double-Edged Sword? Front. Genet. 12:634789. doi: 10.3389/fgene.2021.634789
Científicos e investigadores. Una reflexión contra la excelencia.
escrito por C. Menor-Salvan | 10 enero, 2022
Alguna vez me han preguntado por qué siempre distingo entre investigador y científico, y cual es la diferencia.
Hay una respuesta corta y rápida: Ser científico es una condición, una forma de vida. El científico lo es por cómo piensa, qué se pregunta y cómo interroga a la realidad. La Ciencia es una forma de gobernar el pensamiento, junto con una gran cantidad de conocimiento. Investigador hace referencia a la aplicación, a la parte práctica. A la acción de investigar. El investigador no tiene por qué ser un científico.
Entonces yo hago la siguiente reflexión, que espero sirva a los nuevos científicos que llegan a mi clase y comienzan en este mundo: Actualmente, a lo largo de la carrera nos educan en la competición y nos inundan con el mito del talento, la excelencia y con la «orientación al logro» (digo actualmente, porque cuando yo estudié no había nada de eso: los profesores, salvo alguna excepción, eran meros autómatas que transmitían una información desde unos papeles amarillos por el uso curso tras curso y con quienes teníamos mínima interacción).
Constantemente estamos sometidos a lo que J. Krishnamurti llamaba la «violencia de la comparación«: el fracaso del otro como medida de mi propio logro. Hay que sacar mejor nota que el otro, hay que conseguir la beca antes que el otro, hay que publicar más artículos que el otro, hay que conseguir el contrato compitiendo con el otro; ser el primero en la clase, la oposición, el primero en publicar algo, etc.
Todos los seres humanos desean poder, riqueza, posición social. El deseo de poder se expresa de muchos modos: está en el profesor, la pareja, en un estudiante respecto de otro. Este deseo de posición dominante es una de las formas de agresividad del hombre. La agresividad y el sometimiento a ella pervierten toda relación a lo largo de la vida. El hombre ha aceptado esto como natural, con todos los conflictos y desdichas que conlleva. Básicamente, en ello se encuentra involucrada la medida -el más, el menos, lo mayor y lo menor- que en esencia implican comparación […]. Ello comienza casi al nacer y continúa a lo largo de la vida-este constante medir el poder, prestigio, riqueza. Esto se fomenta en las escuelas y universidades. Todo su sistema de calificar consiste en una evaluación comparativa. Cuando A es comparado con B, que es brillante, agresivo, inteligente, esa comparación misma destruye a A. Esta destrucción toma la forma de la competencia, la imitación y la conformación a los patrones establecidos por B. Ello engendra antagonismo, celos, ansiedad, miedo, y termina por volverse la condición en la que A vive el resto de su vida, siempre midiendo, siempre comparándose. Esta comparación es uno de los muchos aspectos de la violencia.
– Jiddu Krishnamurti. Cartas a las Escuelas.
El concepto de «excelencia» es distinto en el ámbito científico-académico a como entendemos la palabra en el lenguaje normal. Alguien «excelente» mostraba un conjunto de características mas o menos relativas. Cuando decimos «es una excelente persona», es obvio que es algo relativo al resto de personas. Si decimos que alguien es un «excelente fontanero», claramente, en su profesión, muestra un conjunto de capacidades que nos hace preferirle a otros fontaneros. Pero, pensemos en alguien que es una «excelente persona». ¿cómo cuantificamos eso? ¿haríamos un ranking? ¿cual es la persona más excelente que conoces?. ¿Cómo cuantificamos la excelencia de un fontanero? ¿por el número de obras al año? ¿por las buenas referencias? ¿por la relación calidad-precio?. Seguramente sea difícil medir esa excelencia, incluso para algo aparentemente objetivo, que es cómo trabaja un profesional. Al final es un conjunto de factores muchas veces subjetivos o relativos. Si esto es difícil, ¿vamos un paso más allá y, después de implementar una medida de la excelencia, impedimos que los fontaneros que no la alcancen puedan trabajar?
Nos educan no sólo para cuantificar y valorar el «logro» o el «éxito», la «consecución de objetivos y la excelencia» y en «ser el primero» por encima de todo, sino en la aceptación del sacrificio como algo deseable. Vivimos en una especie de culto al sufrimiento. Se asocia el sufrimiento con ese logro y el disfrute con culpabilidad o pérdida de tiempo. Los investigadores aceptan el sacrificio como parte del trabajo y lo imponen a los más jóvenes. «tienes que ir al extranjero (no por que desees aprender allí, sino como imposición de carrera o para ser mejor que el otro)», «es muy difícil ser investigador y tener familia» «tienes que sacrificar relaciones personales» «tienes que dedicar todo tu tiempo a tu trabajo sin descanso» «si eres investigador, prepárate para pasar penurias económicas y precariedad» «no tengas hijos hasta los 45» «como yo lo pasé mal/trabajé gratis/tuve que exiliarme, tu también debes pasarlo mal/etc.». Hemos llegado a la aberrante asociación «ser investigador» = «sufrir» que aceptamos como algo normal, como parte de la profesión.
Como quien va al gimnasio y asocia el dolor, las agujetas y el cansancio con el avance en sus objetivos. «Cómo me he machacado» dicen. En todo momento se usa una terminología bélica: «hay que luchar por…». Muchas veces pienso en éste perverso pensamiento y recuerdo a mi maestro de Aikido, que no toleraba en el Dojo ninguna expresión de agresividad, dominio, superioridad y ego. Disciplinas como el Aikido o el Yoga nos muestran que la asociación tóxica de dolor y sufrimiento con logro debe terminar: a través de ellas fortalecemos el cuerpo y la mente, mantenemos un cuerpo saludable y mejoramos en el estudio, sin necesidad de competir, ni de sufrir, y llevando una práctica vibrante y agradable con, esencialmente, buenas sensaciones.
En el momento en que te intereses por cuales son «buenos» y cuales «malos» entre tus compañeros, estas abriendo una apertura en tu corazón por donde entrará la malicia. Poner a prueba, competir y criticar a otros te debilita y finalmente te derrota
O-Sensei Morihei Ueshiba
Nos intentan hacer creer que vivimos en una confrontación constante con nuestros compañeros y con nosotros mismos, como seres imperfectos en constante test y prueba, intentando mejorarse y ser mejores que otros. Sea lo que sea que signifique eso. Nos presionan para ser «primero», para ser «antes que», en definitiva, para valorar nuestro éxito en la medida del fracaso que perciben los que están en la lista o el ranking tras nosotros. Durante todo el tiempo nos lavan el cerebro con términos como «resiliencia», «talento» y «excelencia». Estas tres palabras, indicadores claros de que estamos ante un idiota postmoderno, deberían hacernos saltar las alarmas: «resiliencia» es la capacidad para recuperarse de una agresión o perturbación. ¿debemos prepararnos entonces para ser maltratados?. Si alguien os pide «resiliencia» como cualidad para un trabajo, tened por seguro que os van a tratar mal.
Pero vamos a revelar una parte importante del engaño: «talento» y «excelencia» son los términos usados por burócratas y políticos para justificar la explotación, la competición, la falta de oportunidades y financiación y la falta de derechos. «Sólo concedemos X contratos o proyectos, porque estamos fomentando la excelencia y el talento», cuando en realidad quieren decir «Sólo concedemos X contratos porque no queremos invertir más en Ciencia, os haremos creer que nos basamos en la excelencia, sea lo que sea eso, pero solo vamos a financiar lo que queremos/creemos conveniente/a quien hemos financiado ya previamente y ha tenido resultados (el efecto Mateo)». Cuando en una convocatoria de proyectos o contratos incluyen la palabra «excelencia», sólo os están diciendo que hay un pedazo de pastel muy pequeño y esperan que los comensales peleen como perros rabiosos por él. Son un engaño, como toda la cultura del talento. Palabrería que crea un ambiente tóxico, lleva a la infelicidad, la frustración y la decepción. Y la frustración o la aceptación del propio fracaso lleva a la desmotivación y a acumular más fracaso según los parámetros de quienes nos están aplicando sus medidas de excelencia. En el momento en el que aceptamos el fracaso, dejamos de esforzarnos. «¿para qué?» es una pregunta habitual, «si no voy a conseguir esa plaza o proyecto».
Estos sentimientos negativos abundan en los jóvenes científicos y son contrarios a los que deben tenerse al hacer Ciencia: el ambiente en un laboratorio debe ser relajado y motivador. La exploración de la Naturaleza es y debe ser gozosa, no debe ser una fuente de sufrimiento o estrés. Un científico no está interesado en competir, en lograr, en el «talento». Hacemos ciencia porque con ella aprendemos cosas, tenemos experiencias interesantes, vemos la Naturaleza bajo otros ojos y, en definitiva, tenemos una vivencia. No importa lo inteligente que seas, ni tu talento, sea lo que sea eso. La Ciencia es una forma de manejar nuestro pensamiento y enfrentarnos a la realidad. La Ciencia es un modo de relacionarnos con el mundo. Es exploración, aprender y enseñar. Descubrir y transmitir. Parafraseando a Karl Popper,la Ciencia nos interesa porque queremos saber algo del enigma del mundo en que vivimos y del otro enigma del conocimiento humano de este mundo.
Un científico intenta dominar el arte de aprender y, los que tienen la suerte de ser al mismo tiempo científicos y profesores de ciencias, tienen la doble tarea de practicar el arte de aprender y el arte de enseñar.
Pero daré a conocer lo poco que he aprendido para que alguien mejor que yo pueda atisbar la verdad y, en su obra, pueda probar y criticar mi error. Así, me regocijaré a pesar de todo de haber sido un medio a través del cual salga a la luz la verdad.
Alberto Durero
Un investigador os hablará de cuantos «papers» publica y sus cuartiles, cuantos fondos y proyectos consigue, de sus logros, de su talento, de líderes y liderar (otra palabreja postmoderna de moda), de la cantidad de charlas que da y de los sitios y colegas prestigiosos con los que se codea. Vivirá observando baremos, índices de impacto, métricas y rankings. Un investigador no desea el error, porque, durante toda nuestra enseñanza, se nos enseña a temer y odiar el error. A ridiculizar o castigar a quien se equivoca. Un investigador buscará destacar sobre el otro, y aprovechará cualquier posibilidad de promocionarse. Es a lo que nos han enseñado y forma parte del trabajo del investigador científico actual.
Durante la celebración del sexagesimo aniversario de Max Planck, en 1918, Einstein dijo que en el templo de la ciencia hay tres tipos de personas. Muchas se dedican a la ciencia en razón del goce de su poder intelectual superior; para ellos, la investigación es una especie de deporte que satisface su ambición personal. Una segunda clase de investigadores se dedica a la ciencia para conseguir fines exclusivamente utilitarios. Pero, en lo que respecta a la tercera: si el ángel del Señor viniera y sacara del templo a todas las personas que pertenecen a esas dos categorías, quedarían unas pocas personas, entre ellas Planck, y ésta es la razón por la que lo queremos.
Edward O. Wilson. Consiliencia o la unidad del conocimiento
Un científico mas bien os hablará de las cosas que ha visto y aprendido; cuando habla de publicaciones, explica lo que ha publicado, no de dónde lo ha hecho o cuanto ha publicado, o de su impacto; habla de lo que ha experimentado, lo que observa, sus ideas, lo que le ha fascinado en su exploración de la vida y la naturaleza. No le interesará dónde has publicado o con quién trabajas, o si has estado en un centro extranjero de nombre sonoro, sino qué te apasiona, qué preguntas e ideas tienes y qué opinas. Un científico abraza el error, porque el error, que puede saber amargo, madura en el delicioso fruto de la Ciencia y el aprendizaje.
La Ciencia se ha construido sobre la refutación de ideas previas. En el momento en el que el investigador trabaja en la Ciencia en un ambiente de violencia y confrontación, como ocurre en cualquier batalla, un error puede ser fatal. Así, es fácil ver que la construcción y el trabajo de la Ciencia no es compatible con los sentimientos de estrés, competitividad, frustración y miedo al fracaso a los que se someten a los científicos. En el momento en que eres científico (no investigador), no existe el fracaso; existen, mas bien, resultados negativos, refutación de tus ideas previas, resultados que no puedes interpretar y limitaciones, falacias y paradojas cognitivas. Todos estos factores son tan importantes o más para la Ciencia que la verificación de una conjetura o teoría. Es importante por ello, que el científico deje de lado el orgullo y acepte con humildad su verdadera posición de observador y transmisor de conocimiento. Naturalmente, «científico» e «investigador» no son dos categorías absolutas y separadas. Podríamos decir, si acaso, que «investigador» es el trabajo y «científico» una condición, una forma de vida. Todos navegamos entre las dos aguas, salvo excepciones (investigadores que son meros trabajadores y tienen poco de científico y, mas raramente, científicos idealistas empeñados en alejarse de la realidad). A mi me parece, a veces, complicado encontrar el equilibrio.
Quienes os digan que hay que competir, que todo esto que digo es incompatible con tener objetivos, con el esfuerzo, con tener resultados y eso que llaman «éxito», sólo os están engañando o manipulando. Se puede ser un científico y tener disciplina, marcarse objetivos o realizar un gran esfuerzo. La diferencia es que éste esfuerzo no estará motivado por la competitividad, por el miedo al fracaso o por la búsqueda del logro, sea cual se este (ya sea salvar a la humanidad, publicar con alto índice de impacto o satisfacer el placer de recibir elogios), sino motivado por la búsqueda del aprendizaje, por comprender un poquito más un pequeño aspecto de la realidad, por responder y/o plantear preguntas; y también, por qué no, realiza un gran esfuerzo por el pundonor del trabajo bien hecho, por el arte, digamos. Para el científico no hay ciencia pequeña o irrelevante. El investigador desea el impacto, porque le aportará fama y prestigio. Para el científico, toda la ciencia es necesaria e importante. Un dato que aportas puede que no cambie ningún paradigma, pero dentro de 5, 10 o 20 años puede ser el factor clave de un gran descubrimiento. Hay muchos ejemplos de ello, y los profesores deberían hacer hincapié en esa «ciencia pequeña» tan importante. La calidad de la ciencia no se mide con su impacto o según dónde se publica, sino según el modo en que ha sido realizada, según su técnica y discusión y según la veracidad y honestidad de su resultado, da igual si es un «pequeño resultado» o no. Es la velocidad y el simplismo de la sociedad moderna lo que lleva a juzgar la calidad de un resultado científico por el título de la revista en que se publica o según cuanto se cite en los medios.
Eso se aplica a cualquier Ciencia: saber un poco más del mundo en que hemos vivido y el universo del que formamos parte.
Según todos estos factores, un investigador quizá pueda ser mejor que otro. No lo se. Quizá podamos inventar métricas y rankings para decidir artificialmente quien es absolutamente mejor o peor investigador en base a una métrica relativa. Igual que las notas de clase: ¿será mejor biólogo molecular quien haya sacado matrícula en la asignatura de biología molecular?. Igual, simplemente, en esa métrica particular obtuvo un valor mas alto, pero, ¿es mejor o peor? ¿en qué y en base a qué? Igual fue pura casualidad. Igual es un desastre en el laboratorio. O igual no. Tratar de decidir quién es mejor o peor científico es tan ridículo como observar un parque en el que juegan grupos de niños y tratar de hacer un ranking de los que mejor juegan. «Juanito ha subido al tobogán un 20% más que Carlitos». «Anita sube más arriba en el columpio que el 75% restante de los niños». ¿no os suena ridículo?.
Determina cual de estos niños juega «mejor» y luego establece un ranking de «excelencia». A quien esté el último, no le dejes jugar en el parque. ¿podríamos hacer algo así? ¿cómo?. Con la Ciencia ocurre algo similar.
Podemos intentarlo, claro, pero ellos estarán ajenos a ello, y, si intentamos que entren en nuestro juego de rankings (dando de merendar o un juguete, o permitiendo jugar en el parque sólo al primer cuartil en el número de veces que suban a lo más alto de determinado columpio), solo lograremos arruinar su tiempo de juegos, desmoralizarles y convertir el parque en un campo de batalla. He visto, a lo largo de mi carrera, personas moralmente deshechas por su trabajo como investigadores. Personas inteligentes y con muchas ideas y pasión abandonando la Ciencia. He visto agresiones e intentos de asesinato. Suicidios. Yo mismo he sufrido estrés y depresión causada por mi trabajo. Esto es lo que provocan aquellos que hablan de «talento», «excelencia», «resiliencia», «competitividad», etc. ¿es esto lo que queremos para nosotros, nuestros alumnos y compañeros? ¿donde queda, en éste ambiente de «competitividad por la excelencia» la amabilidad y la bondad?
La bondad solo florece en libertad. No puede hacerlo en el suelo de la coacción bajo ninguna de sus formas, ni bajo compulsión, ni es el resultado de la búsqueda de recompensas. No puede existir cuando hay temor.
Jiddu Krishnamurti
Alguien podría decirnos, «es el sistema». Que no os mientan: el sistema lo hacen personas como vosotros y yo. En nuestra mano está cómo queremos que sea. Podéis enfocar vuestra carrera de dos formas: como un investigador que quiere ser el primero o como un científico que quiere descubrir. En ambos casos haréis básicamente lo mismo (mucho trabajo, burocracia, participar en concursos por plazas y proyectos, intentar publicar papers lo mejor posible…etc.) La diferencia es que, en el primer caso, es muy probable que viváis una vida de frustración, de estar pendientes de rankings y baremos, de lo que hacen otros, de alimentar el ego, de sufrir y hacer infelices a otros. En el segundo caso es posible que lo paséis mal a veces, que sea difícil, que os disgustéis. Pero siempre encontraréis gozo, motivaciones y alegrías. En el primer caso viviréis pensando en el fracaso, en el segundo caso no hay fracaso ni éxito, simplemente viviréis una vida gozosa como científicos, independientemente de dónde estéis en el ranking. Y, recordad algo muy importante: en el mundo de la Ciencia casi todo el mundo es inteligente, y «excelente». Si queréis marcar la diferencia, sed amables, humildes y generosos. Sed científicos, no investigadores.
Los actores se ponen maquillaje y representan la belleza y la fealdad; pero cuando se termina la obra, ¿Dónde están la belleza y fealdad?. Los jugadores de ajedrez compiten por ser los primeros e intentan aventajarse unos a otros en sus movimientos; pero cuando se acaba el juego y se retiran las piezas, ¿Dónde queda entonces la competición?
Control de calidad de proteínas en el retículo endoplasmático
escrito por Grupo 19 | 10 enero, 2022
Por Beatriz Naranjo Martínez, Mª del Carmen Nogales Valenciano y Sara Ortiz Planchuelo. Grado en biología sanitaria – Universidad de Alcalá de Henares
El retículo endoplasmático (RE) es un orgánulo de la célula que regula la síntesis, plegamiento, maduración, estabilización, tráfico y degradación de aproximadamente un tercio de las proteínas totales de la célula. El destino de estas proteínas es ser secretadas por la célula, así como residir en la membrana plasmática, el aparato de Golgi, los lisosomas y el propio RE.
Estas proteínas sufren modificaciones postraduccionales, plegamiento y maduración hasta alcanzar su estado funcional terciario o cuaternario.
El plegamiento ocurre gracias a la intervención de chaperonas y otras enzimas, que reconocen las proteínas desplegadas o mal plegadas y las pliegan para que logren una conformación estable funcionalmente activa. No obstante, este proceso es el punto donde más errores se producen en todo el proceso de síntesis de proteínas. Por ello, el RE posee un sistema de control de calidad del plegamiento, que consiste en exportar únicamente aquellas proteínas plegadas correctamente y dirigir a la degradación las que no lo están, evitando así la acumulación de proteínas aberrantes que puede conducir a la apoptosis.
Un fallo en este control de calidad puede generar múltiples enfermedades relacionadas con el mal plegamiento de proteínas, como diabetes mellitus, hígado graso, neurodegeneración, inflamación y cáncer.
Síntesis de proteínas en el retículo endoplasmático
Las proteínas destinadas al RE comienzan su síntesis en los ribosomas libres en el citosol, donde se traduce, en primer lugar, un péptido señal de 16 a 30 aminoácidos, que contiene un núcleo de aminoácidos hidrofóbicos, que es esencial para su función (Figura 1, paso 1).
Una ribonucleoproteína citosólica, la partícula de reconocimiento de señal (SRP), se une a la subunidad 60S y al péptido señal en cuanto éste emerge del ribosoma (Figura 1, paso 2), de manera que detiene la traducción para que el polipéptido naciente pueda ser translocado al RE.
Este complejo se dirige a la membrana del RE, donde el receptor de la SRP reconoce el extremo de SRP no unido al ribosoma (Figura 1, paso 3). La unión del complejo ribosoma-péptido señal-SRP al receptor de SRP conduce a la apertura del complejo Sec61 o translocón en la membrana del RE, que actúa como un poro acuoso. Esta unión al translocón permite la liberación de SRP por la hidrólisis de GTP, la reanudación de la traducción y la entrada del polipéptido al RE a medida que se va sintetizando (Figura 1, paso 4). Todo este proceso requiere ATP. [1,2,3]
Dependiendo del destino de la proteína naciente, la síntesis continua de forma distinta:
Si la proteína se va a secretar, a medida que la cadena polipeptídica se va alargando, pasa a través del canal del translocón hacia la luz del RE, donde el péptido señal es cortado inmediatamente por la peptidasa señal y posteriormente degradado (Figura 1, paso 5). La cadena continúa sintetizándose (Figura 1, paso 6) hasta la terminación de la traducción del mRNA, de manera que se libera el ribosoma (Figura 1, paso 7) y se cierra el translocón (Figura 1, paso 8).
Si la proteína va a formar parte de una membrana biológica, poseerá un péptido de señal-anclaje, que posee una región hidrofóbica que le sirve para anclarse a la membrana del RE y no ser escindida. [4]
Figura 1. Translocación cotraduccional de proteínas de secreción al RE. Idea tomada de Biología Celular y Molecular de Harvey Lodish | Editorial Médica Panamericana. Available at: https://www.medicapanamericana.com/es/libro/biologia-celular-y-molecular. Accessed Feb 15, 2021. Creada con BioRender.com.
Plegamiento de proteínas
Una vez el polipéptido emerge del translocón, ha de plegarse para adquirir su conformación funcionalmente activa (estado nativo), la cual es la más estable [5, 6]. Para hacer posible la espontaneidad del proceso, se “ocultan” los residuos hidrófobos de aminoácidos no polares en el núcleo de la proteína y se exponen las cadenas laterales hidrófilas al entorno acuoso.
Primero se produce el plegamiento co-traduccional temprano, en el cual se forman estructuras secundarias. Una vez la proteína se libera del translocón, se produce el plegamiento postraduccional, que predomina sobre el primero y da lugar a las proteínas funcionales.
El proceso de plegamiento ha de llevarse a cabo en un tiempo biológicamente aceptable. Para ello, se limitan el número de conformaciones que pueden adoptar los polipéptidos, mediante la formación de interacciones hidrofílicas como puentes salinos y enlaces disulfuro [5].
La formación de estas interacciones co‐ y postraduccionales es catalizada por una maquinaria de modificación y plegamiento de proteínas residente en RE, que comprende una red de chaperonas, enzimas glucosilantes, oxidorreductasas e isomerasas que actúan simultánea o secuencialmente [5, 6].
A menudo las proteínas pequeñas con un solo dominio adoptan su estado nativo de manera espontánea, sin necesidad de la intervención de esta maquinaria, al contrario de lo que ocurre en el caso de proteínas con estructuras más complejas que se plegaran más lentamente y, por tanto, requieren de diferentes enzimas para alcanzar su conformación activa en un tiempo aceptable biológicamente [7].
Chaperonas
Las chaperonas moleculares se definen como «proteínas que interactúan, estabilizan o ayudan a una proteína no nativa a adquirir su conformación nativa, pero no están presentes en la estructura funcional final» [8]. Fueron identificadas por su mayor abundancia después de su exposición al choque térmico, por lo que son conocidas como Hsp, por sus siglas en inglés Heat Shock Proteins [5].
Estos catalizadores moleculares se encuentran en todos los orgánulos y compartimentos en las células encargadas de la síntesis y modificación post-traduccional, dado que se han conservado a lo largo de la evolución [9]. En el RE destacan la Hsp70 BiP (Grp78), la Hsp90 Grp94 (gp96) y las chaperonas de lectina calnexina (CNX) y calreticulina (CRT), que son exclusivas del RE [5]. La mayoría de los factores de plegamiento del RE se unen a Ca2+ y dependen de él para funcionar.
Las chaperonas actúan como enzimas acelerando las etapas limitantes de la reacción de plegamiento, es decir, disminuyen la barrera energética entre el estado nativo y no nativo de la proteína [6]. Para ello, reconocen dominios hidrofóbicos expuestos al medio acuoso, los cuales señalizan que la proteína está mal plegada, es un intermediario de plegamiento o forma parte de un polímero no ensamblado. Una vez reconocidos estos dominios hidrofóbicos, los ocultan y forman interacciones no covalentes con ellos para dar lugar al estado nativo, que es estable contra la agregación multimérica irreversible.
Si la proteína ha alcanzado su conformación nativa, no podrá volver a unirse a las chaperonas, pero si este plegamiento se ha producido de manera incompleta, habrá residuos hidrofóbicos expuestos que permitirán una nueva oportunidad para llevar a cabo el plegamiento correctamente [4].
Así, las chaperonas pueden diferenciar específicamente las etapas de plegamiento para una amplia gama de proteínas y, por tanto, actúan como supervisoras de la calidad del plegamiento proteico [9].
Control de calidad del RE
A pesar de la intervención de las maquinarias de plegamiento proteico, el plegamiento es el punto más propenso a errores desde la transcripción hasta lograr la proteína funcional.
El RE posee una concentración de calcio y un potencial redox mayor que el citosol, lo cual facilita el funcionamiento adecuado de las maquinarias de plegamiento. Esto le otorga la capacidad de realizar un control de calidad del plegamiento, mediante el monitoreo de la cantidad de proteínas mal plegadas en el RE, y un control de cantidad, al eliminar copias excesivas de ciertas proteínas para coordinar su plegamiento con las demandas fisiológicas y patológicas de la célula [9].
Este control consiste en detectar aquellas proteínas que tienen defectos de plegamiento o se encuentran en exceso para dirigirlas a vías de degradación, pasando únicamente a la vía secretora aquellas proteínas correctamente plegadas [10].
En RE de mamíferos existen dos mecanismos distintos de control de calidad: la vía de plegamiento general y la vía específica de glucoproteínas [5].
En ausencia de N-glucanos dentro de los primeros 50 residuos de aminoácidos, la proteína naciente se dirige hacia la vía de plegamiento general, de manera que se une primero a la chaperona BiP (Grp78). En presencia de dichos N-glucanos, la proteína se dirige hacia la vía de plegamiento específica de glucoproteínas, de manera que interacciona con las chaperonas calnexina-calreticulina (CNX / CRT) para sufrir procesos de modificación en el oligosacárido hasta alcanzar el plegamiento adecuado. No obstante, debido a la importancia de BiP en la translocación, ésta puede interaccionar primero de manera transitoria con la glucoproteína [11]. Ambos mecanismos actúan de forma coordinada en la maduración proteica para que únicamente sean exportadas las proteínas funcionales.
Cuando las proteínas alcanzan una conformación estable funcionalmente activa, pueden ser exportadas mediante vesículas al aparato de Golgi.
Las proteínas que permanecen mal plegadas tras varios intentos de plegamiento son finalmente retrotraslocadas al citosol para ser degradadas en el proteasoma mediante la vía de degradación asociada al RE (ERAD). En caso de que se formen cuerpos de inclusión proteicos debido a la agregación, éstos son degradados por autofagia mediante la vía de degradación en lisosomas asociada al RE (ERLAD).
Ante condiciones de estrés reticular, la acumulación de proteínas mal plegadas puede superar un umbral crítico, lo cual induce la activación de la respuesta de proteína desplegada (UPR). Este mecanismo trata de promover la supervivencia celular mediante el restablecimiento de la homeostasis, por un lado, controlando la síntesis y degradación de estas proteínas, y por otro, regulando la transcripción de los factores que intervienen en esta proteostasis.
Cuando el estrés del RE es demasiado alto, la UPR puede no ser suficiente para recuperar la homeostasis, de manera que los niveles de proteínas mal plegadas siguen siendo altos y se activan vías pro-apoptóticas para evitar la supervivencia de células aberrantes [7].
Vía de plegamiento general
Como ya hemos mencionado, en la vía general de plegamiento intervienen una chaperona de la familia Hsp70, BiP, y enzimas de plegamiento entre las que destacan las proteínas disulfuro isomerasas (PDI) (PDIA1), y peptidil prolil cis‐trans isomerasas (PPI) [12].
La proteína de unión a inmunoglobulina (BiP) es la chaperona más abundante y versátil de la célula, considerándose el regulador maestro del plegamiento de proteínas en el RE. Entre sus funciones destacan:
Interviene en la translocación de cadenas nacientes al lumen reticular.
Participa en el plegamiento y la oligomerización de proteínas.
Interviene en la preparación de las proteínas no nativas para ser translocadas al citosol y degradas en el proteasoma.
Regula la respuesta a proteína desplegada (UPR).
Ayuda a mantener la homeostasis del Ca2+, lo cual es fundamental para el plegamiento de proteínas, pues la mayoría de los factores son dependientes de Ca2+ [4, 6].
La actividad ATPasa de BiP está regulada por las proteínas J (ERdj1–7) y factores de intercambio de nucleótidos (Grp170 y Sil1). Los ERdjs actúan reclutando a BiP para realizar diferentes procesos como la traslocación (ERdj1 y ERdj2) (Figura 3, paso 1), el plegamiento (con ERdj3 y ERdj6) (Figura 3, paso 2) y la degradación (ERdj4 y ERdj5) (Figura 3, paso 3a) [8].
Cuando se une ATP al dominio de unión de nucleótidos de BiP, el dominio de unión a sustrato alcanza su conformación abierta, en la cual se une de manera transitoria a las regiones hidrofóbicas que están expuestas en la proteína plegada incompletamente (Figura 2, paso 1). Al hidrolizarse este ATP, se desacoplan los dominios, y este último adquiere su conformación cerrada, uniéndose de forma más estrecha a su proteína sustrato, de manera que facilita su plegamiento (Figura 2, paso 2). Las proteínas ERdj intervienen en ambos pasos, en primer lugar, transfiriendo las proteínas desplegadas y, en segundo lugar, produciendo la hidrólisis de ATP.
El ADP resultante de la hidrólisis es sustituido por ATP gracias a la intervención de factores de intercambio de nucleótidos (Figura 2, paso 3). Esto produce un cambio conformacional que libera la proteína diana, permitiendo que la chaperona sea reutilizada tras la intervención de las proteínas ERdj (Figura 2, paso 4).
Figura 2. Ciclo de la ATPasa BiP. Creada con BioRender.com.
Grp94 es otra chaperona de la familia Hsp90, sin actividad ATPasa, que se une a los sustratos después de BiP (Figura 3, paso 4) y es esencial en muchos procesos en el RE, incluido el plegamiento de proteínas, el control de calidad del RE, la respuesta al estrés y la amortiguación de Ca2+ [5, 6].
Además de estas chaperonas, en el plegamiento intervienen enzimas, destacando las peptidil prolil cis‐trans isomerasas (PPI) y las disulfuro isomerasas (PDI).
Las peptidil prolil cis‐trans isomerasas (PPI) catalizan la isomerización del enlace peptídico que precede a los residuos de prolina desde la conformación trans a la cis y viceversa. Esto es importante porque, durante el plegamiento, pueden ser necesarias múltiples isomerizaciones cis-trans hasta alcanzar la estructura proteica correcta. Este cambio conformacional es extremadamente lento, por lo que se considera un paso limitante en el plegamiento de proteínas [5].
Las disulfuro isomerasas (PDI) catalizan la formación, reducción e isomerización de puentes disulfuro, que estabilizan la estructura de la proteína nativa y los complejos oligoméricos. Esta reacción también es un paso limitante de la velocidad en el plegado [12].
Cuando las proteínas están bien plegadas, salen del RE y viajan al Golgi (Figura 3, paso 3b), mientras que las proteínas mal plegadas se dislocan al citosol para su degradación [5].
Figura 3. Vía de plegamiento general. Creada con BioRender.com.
Vía de plegamiento específica de glucoproteínas
La mayoría de las proteínas solubles y de membrana que se dirigen a la vía secretora reciben glucanos ligados a N a medida que se traducen y translocan al RE (Figura 4, paso 1). Esto juega un papel crucial en el plegamiento de proteínas en el RE, principalmente a través de su interacción con calnexina (CNX) y calreticulina (CRT) [12, 13].
La N-glucosilación comienza cuando la oligosacariltransferasa (OST), una enzima anclada al translocón, transfiere un complejo de 14 azúcares (2 moléculas de N-acetil-glucosamina, 9 moléculas de manosa y 3 moléculas de glucosa) de un dolicol pirofosfato de la membrana del RE al residuo N de asparagina (Asn) de una secuencia aceptora (N-glicosilación) (Figura 4, paso 2) [5, 7, 12].
Las chaperonas calnexina y calreticulina (CNX / CRT) únicamente se unen a intermediarios proteicos monoglucosilados. Estos intermediarios se logran gracias a la previa escisión secuencial de las dos primeras glucosas de las glucoproteínas por las glucosidasas I y II (Figura 4, paso 3).
La calnexina es una proteína integral de la membrana del RE y la calreticulina es su parálogo soluble en el lumen de éste. Ambas proteínas poseen un dominio de unión a glucanos similar a lectina y un brazo flexible, el dominio P, que recluta a otras chaperonas. Además, se estabilizan cuando se unen a calcio [4].
A través de sus dominios P, se unen a chaperonas de función específica como ERp57, que es una disulfuro isomerasa (PDI) [14]; ciclofilina B, que es una peptidil prolil cis‐trans isomerasa (PPIasas); y ERp29, que tiene función de chaperona general (Figura 4, paso 4) [7, 12]
Una vez realizada la función de plegamiento, la glucosidasa II recorta la glucosa restante, liberando la proteína del complejo de chaperonas (Figura 4, paso 5).
La glucoproteína glucosiltransferasa (UGGT1) reconoce imperfecciones estructurales en las glucoproteínas que no han conseguido alcanzar su estado nativo tras el plegamiento y monoglucosila de nuevo sus cadenas laterales a partir del donador UDP-glucosa, de manera que pueden unirse otra vez a la calnexina-calreticulina para intentos de plegamiento adicionales (Figura 4, paso 5) [12].
Además, esta proteína monoglucosilada también actúa como sustrato de la glucosidasa II, que compite con UGGT1 con efectos opuestos. La situación en el RE va a condicionar la disponibilidad de estas dos enzimas y, por tanto, el desplazamiento de este equilibrio; por ejemplo, cuando se produce estrés en el retículo aumenta la expresión de UGGT1, por lo que este equilibrio se desplaza hacia la monoglucosilación para que se incremente la tasa de plegamiento.
Por tanto, UGGT1 actúa como un sensor de reconocimiento de proteínas mal plegadas, pues determina si una proteína sale del RE o se retiene para una mayor intervención. [7, 15]
Una proteína puede recorrer el ciclo CNX/CRT numerosas veces hasta que alcanza su conformación nativa. Para salir de este ciclo, el residuo de manosa más externo del brazo que contiene glucosa es eliminado por una manosidasa (Figura 4, paso 6 y 7), evitando que la UGGT1 vuelva a glucosilar.
Si las proteínas consiguen un plegamiento correcto, tras esta escisión, pueden ser exportadas fuera del RE [7]. Sin embargo, si las proteínas no consiguen plegarse adecuadamente tras varios ciclos, se dirigen a la vía de degradación de proteínas asociada a RE (ERAD).
Figura 4. Vía de plegamiento específica de glucoproteínas. Creada con BioRender.com.
ERAD
Cuando las proteínas no adquieren su forma nativa tras varios intentos de plegamiento, las células activan la vía de degradación asociada al RE (ERAD) para evitar que las proteínas se acumulen dando lugar al estrés del RE, el cual pondría en peligro la supervivencia celular. En este mecanismo las proteínas mal plegadas son retrotranslocadas al citosol para su posterior ubiquitinación y degradación por el proteosoma 26S, pues el RE no posee mecanismos de degradación [1].
Vía ERAD de proteínas glucosiladas
Salida del ciclo de calnexina/calreticulina
Se cree que la salida de las glucoproteínas mal plegadas del ciclo de CXN/CRT ocurre cuando la α1,2-manosidasa I del RE (ERManI) corta el residuo de manosa más externo del brazo que contiene glucosa, dando lugar a un residuo de 8 manosas en vez de 9 como el glucopéptido original, de manera que la UGGT1 no puede volver a monoglucosilar. [17, 18, 19].
No obstante, esta desmanosidación también ocurre en las proteínas bien plegadas, por lo que una proteína cuyo N-glicano se recorta de 9 a 8 manosas puede ser sustrato tanto para el transporte a Golgi como para ERAD [20].
Recientemente se ha demostrado que, si la proteína se pliega adecuadamente, se incorpora a vesículas COPII que se dirigen al Golgi. Sin embargo, si no se pliega, las proteínas similares a la α-manosidasa I (EDEM1-3) reconocen el glucano recortado y producen un recorte adicional de manosa que conducirá a los siguientes pasos de la vía de degradación (Figura 5) [18, 20].
A partir de aquí, ERAD ocurre en un proceso de múltiples pasos que comprende el reconocimiento, translocación y ubiquitinación de proteínas ER para la degradación proteasomal citosólica.
Reconocimiento
La acción secuencial de ERManI y EDEM1 en glicoproteínas mal plegadas da como resultado la exposición de un resto de manosa unido a α1,6, que es reconocido por el dominio receptor de manosa-6-fosfato de las lectinas: osteosarcoma amplificado 9 (OS-9) y proteína transactivada XTP3 (XTP3-B). Estas lectinas junto con los sustratos ERAD son reclutados Sel1L, una proteína asociada con el complejo de retrotranslocación [17, 18].
Translocación y ubiquitinación
Antes de la translocación, las proteínas deben abrir su estructura parcialmente plegada, mediante la ruptura de los puentes disulfuro por disulfuro isomerasas (PDI) y BiP.
En el complejo de retrotranslocación de la membrana del RE destaca la proteína Derlin-1, que forma canales de retrotranslocación en la membrana del RE [20].
Como se ha mencionado, OS-9 y XTP3-B unidos a la proteína desplegada son reclutados por la SEL1L, que se encuentra asociada con Hrd1, una ubiquitina ligasa transmembrana que a su vez está unida al complejo de retrotranslocación.
La ubiquitinación de Hdr1 produce un cambio conformacional en éste, que permite la inserción del polipéptido por el canal Hdr1. A medida que la proteína emerge en el citosol, será poli-ubiquitinado por Hrd1 y las enzimas E2 de conjugación de ubiquitina asociadas. La AAA-ATPasa p97, se recluta en la membrana del RE a través de su asociación con VIMP y proporciona la energía necesaria para extraer las proteínas de la membrana del RE.
Finalmente, la cadena polipeptídica es dirigida hacia este proteasoma por Dsk2 y Rad23, una vez la N-glicanasa ha recortado el glicano restante de la cadena polipétidica para que esta última pueda entrar por el poro del proteasoma dónde finalmente serán degradadas. [20, 21].
Vía ERAD de proteínas no glucosiladas
La vía ERAD de las proteínas no glicosiladas está menos estudiada, pero fundamentalmente difiere del proceso de las glucosiladas en el reconocimiento de los sustratos desplegados (Figura 5). La decisión de dirigir las proteínas a la vía de degradación implica la unión de la proteína a las co-chaperonas pro-degradación: ERdj4, que está asociado con Derlin1, y ERdj5, que actúa como disulfuro isomerasa, induciendo un mayor despliegue de las proteínas para dirigirlas a ERAD, mientras que ERdj4 está asociado con Derlin1 [20].
Figura 5. Vía ERAD de proteínas glucosiladas y no glucosiladas. Creada con BioRender.com.
ERLAD
Las proteínas mal plegadas que no pueden ser reconocidas y degradadas por ERAD, son autofagocitadas para su degradación en lisosomas asociada al ER (ERLAD) [16, 22].
Estrés del RE
La eficiencia del plegamiento de proteínas en el RE se puede ver alterada por un amplio grupo de alteraciones celulares que conducirán a la acumulación de proteínas mal plegadas dentro del orgánulo que, si supera un umbral crítico, provocará estrés en el RE. Las condiciones que desencadenan este estrés incluyen: falta de nutrientes, hipoxia, mutaciones puntuales en proteínas secretadas que intervienen en el plegamiento o causan agregación y pérdida de la homeostasis del calcio. Así, las células han desarrollado un sofisticado sistema para detectar y responder frente al estrés antes de que peligre su supervivencia [23].
UPR determina el destino celular bajo estrés en el RE
Ante el estrés del RE, las células activan una compleja red de vías de señalización intracelular interconectadas, la respuesta de proteína desplegada (UPR). Esta vía es iniciada por tres transductores ubicados en la membrana del RE, PERK, IRE1 (α y β) y ATF6, que contienen un dominio luminal RE capaz de detectar directa o indirectamente la acumulación crítica de proteínas mal plegadas, de manera que transmiten esta información al citoplasma y al núcleo [23, 24].
La UPR se activa para restaurar la homeostasis del plegamiento de proteínas del RE, pero si el estrés ER no se mitiga y amenaza la supervivencia celular, la UPR desencadena la apoptosis [25]. Esta vía se activa para restaurar la homeostasis del plegamiento de proteínas del RE.
UPR adaptativa: hacia la supervivencia celular
Ante el estrés agudo del RE, la UPR se activa para restaurar la homeostasis del plegamiento de proteínas del RE. Esto implica, en primer lugar, la expansión de la membrana del RE y un aumento en el plegamiento y transporte de proteínas en el RE, y, posteriormente, la atenuación transitoria de la síntesis de proteínas y un aumento en la degradación de proteínas asociadas al RE (Figura 6).
IRE1α
IRE1α (proteína 1α que requiere inositol) es una glicoproteína transmembrana del RE, que posee un dominio citoplasmático con actividad quinasa y RNasa, y un dominio luminal que detecta a las proteínas mal plegadas [4].
Esta proteína mantiene en un estado reprimido en condiciones sin estrés a través de una asociación con BIP (26). Durante el estrés del retículo endoplásmico, BIP se disocia para unirse a las proteínas mal plegadas. Esto conduce a la fosforilación y dimerización parcial de IRE1α, que estimula su actividad RNasa que realizar el corte y empalme de la proteína de unión a caja X (XBP1).
XBP1s controla la transcripción de genes que codifican proteínas implicadas en el plegamiento de proteínas, la degradación asociada a RE (ERAD), el control de calidad de proteínas y la síntesis de fosfolípidos, esta última para dar como resultado una expansión del RE.
IRE1α también media la descomposición del ARNm para reducir la carga de plegamiento de proteínas en el RE, lo que se denomina descomposición dependiente de IRE1 regulada (RIDD). Por otro lado, induce ‘vías de estrés de alarma’, incluidas las impulsadas por la quinasa N-terminal JUN (JNK) y el factor nuclear κB (NF-κB), mediante la unión a proteínas adaptadoras [25].
PERK
PERK (quinasa del RE pancreático)es una proteína transmembrana, que reprime la traducción de proteínas en éste. Esta proteína fosforila la serina 51 de eIF2α (subunidad α del factor de inicio de la traducción 2 en eucariotas) en respuesta al estrés.
La fosforilación de eIF2α, por un lado, reduce la formación de complejos de iniciación de la traducción y, por tanto, lleva a una disminución de la traducción general. Este control permite reducir el estrés del RE mediante la reducción del número de proteínas mal plegadas [9]. Por otro lado, la fosforilación de eIF2α permite la transcripción de ATF4, un factor de transcripción que actúa sobre genes implicados en el metabolismo de aminoácidos, las respuestas frente a estrés oxidativo, la autofagia y la apoptosis [25].
ATF6
ATF6 (factor de transcripción activador 6) es una proteína transmembrana del RE que se localiza en células no estresadas. En las células sometidas a estrés RE, ATF6 es transportado al aparato de Golgi donde es escindido por las proteasas S1P y S2P para producir un fragmento citosólico soluble (ATF6f), que ingresa al núcleo para inducir la expresión de genes diana relacionados con ERAD y el plegamiento de proteínas como los genes que codifican para las chaperonas BiP. (9) [25].
Estas ramas de señalización de la UPR no se activan de forma simultánea, sino que la activación de ATF6α e IRE1α ocurre de inmediato y disminuye con el tiempo, mientras que la activación de PERK sigue a la de ATF6α e IRE1α y permanece durante el estrés crónico del RE.
En condiciones normales, BiP interactúa con los dominios luminales de estos transductores, actuando como regulador negativo de su activación. Ante el estrés del RE, BiP se une a las proteínas mal plegadas, lo que permite su liberación de los transductores. La liberación de BiP de IRE1 y PERK permite su homodimerización y activación, mientras que su liberación de ATF6 permite el transporte de éste al compartimento de Golgi para la proteólisis intramembrana regulada. Esta activación regulada por BiP proporciona un mecanismo directo para detectar la capacidad de plegamiento del RE. Además, las proteínas mal plegadas pueden actuar como ligandos activadores para estos sensores de estrés a través de la unión directa al dominio luminal de IRE1α y PERK.
Estos vías de transducción actúan como bucles de retroalimentación homeostática para atenuar el estrés del ER, de manera que, si tiene éxito en reducir la cantidad de proteína mal plegada, la señalización de la UPR se atenúa y la célula sobrevive.
Figura 6. UPR. Las proteínas transmembrana localizadas en el retículo endoplásmico IRE1, PERK y ATF6 detectan la carga de proteína desplegada en la luz del orgánulo, actuando como receptores de estrés. Transducen la señal del RE al núcleo para regular la transcripción y la síntesis de proteínas. En conjunto, producen la respuesta de la proteína desplegada (UPR). Creada con BioRender.com.
Algunos factores ambientales, el envejecimiento y mutaciones genéticas puede dar lugar a un mal funcionamiento de la UPR, lo cual se ha demostrado que induce enfermedades relacionadas con el plegamiento anómalo de proteínas como diabetes, ateroesclerosis, neurodegeneración, inflamación y cáncer.
UPR terminal: hacia la muerte celular
Ante el estrés del RE prolongado, las respuestas adaptativas pueden resultar insuficientes para restaurar la homeostasis del plegamiento de proteínas, de manera que la UPR activa una vía de señalización alternativa proapoptótica, que induce la muerte celular para evitar la supervivencia de células aberrantes.
Aun no se conocen los detalles moleculares de esta vía, pero hay evidencias de que las dos quinasas UPR, PERK e IRE1α, involucran un conjunto de salidas pro-apoptóticas que conducen a la degeneración celular si el estrés del RE no se puede resolver (Figura 7) [10, 27].
La hiperactivación de PERK conduce a la pausa prolongada de la traducción de proteínas, debido a la fosforilación de eIF2α, lo cual es incompatible con la supervivencia. Además, PERK induce la transcripción de ATF4, que activa a su vez la transcripción de CHOP (proteína homóloga C/EBP), un factor de transcripción que media la muerte celular mediante la regulación de la expresión de varias proteínas:
Inhibe la expresión de las proteínas anti-apoptóticas de la familia BCL-2, lo que conduce a la activación de proteínas BH3. Éstas desactivan las proteínas protectoras mitocondriales y las proteínas pro-apoptóticas BAX y BAK para que permeabilicen la membrana mitocondrial externa.
Activa la expresión de ERO1α (ER oxidorreductina 1), que crea un entorno hiperoxidante en el RE, perjudicando el plegamiento de proteínas y, por tanto, aumenta la señal a favor de la muerte.
Activa la expresión de GADD34 (proteína inducible por daño del ADN), que media la desfosforilación de eIF2α, de manera que se reanuda la síntesis de proteínas, aumentando la carga de proteínas en el RE. Por tanto, se forma un bucle de retroalimentación al amplificar la señal tóxica a favor de la muerte [28].
La hiperactivación de IRE1α fosforilada conduce a la formación de oligómeros en vez de homodímeros, lo cual tiene los siguientes efectos:
Su dominio RNasa tiene afinidad por los sustratos RIDD (desintegración de ARNm dependiente de IRE1), actuando como una endonucleasa de cientos de ARNm localizados en ER, de manera que agota la carga de proteínas en el RE y los componentes de la maquinaria de plegamiento, empeorando aún más el estrés.
La actividad RNasa reduce los niveles de microARN que normalmente reprimen dianas pro-apoptóticas, como la proteína pro-oxidante TXNIP (proteína que interactúa con tiorredoxina), cuyo aumento activa el inflamasoma y una vía pro-apoptótica dependiente de Caspasa-1.
Recluta TRAF2 (receptor 2 asociado al receptor del factor de necrosis tumoral), que activa ASK1 (quinasa 1 reguladora de la señal de apoptosis). Ésta activa a su vez JNK (quinasa terminal c-Jun NH2) y MAPK (proteína quinasa activada por mitógenos p38) que, mediante fosforilación, activan la BH3 pro-apoptótica e inhibe la BCL-2 anti-apoptótica [10, 27].
Figura 7. UPR terminal. IRE1α y PERK activan factores pro-apoptóticos e inhiben factores anti-apoptóticos ante el estrés agudo en el RE. Creada con BioRender.com.
Bibliografía
(1) Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci 2016 -01;73(1):79-94.
(2) Brodsky JL. Translocation of proteins across the endoplasmic reticulum membrane. Int Rev Cytol 1998;178:277-328.
(3) Zapun A, Jakob CA, Thomas DY, Bergeron JJ. Protein folding in a specialized compartment: the endoplasmic reticulum. Structure 1999 -08-15;7(8):173.
(4) Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell 2006 -05-05;125(3):443-451.
(5) Ellgaard L, McCaul N, Chatsisvili A, Braakman I. Co- and Post-Translational Protein Folding in the ER. Traffic 2016 -06;17(6):615-638.
(6) Redondo Juárez P. Fallos en el control de calidad en la síntesis de proteínas: origen de enfermedades raras. Mistakes in quality control of protein synthesis: the origin of rare diseases 2016.
(7) Lamriben L, Graham JB, Adams BM, Hebert DN. N-Glycan-based ER Molecular Chaperone and Protein Quality Control System: The Calnexin Binding Cycle. Traffic 2016 -04;17(4):308-326.
(8) Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol 2009 -06;16(6):574-581.
(9) Fu XL, Gao DS. Endoplasmic reticulum proteins quality control and the unfolded protein response: the regulative mechanism of organisms against stress injuries. Biofactors 2014 Nov-Dec;40(6):569-585.
(10) Hetz C, Papa FR. The Unfolded Protein Response and Cell Fate Control. Molecular cell 2018 Jan 18,;69(2):169-181.
(11) Molinari M, Helenius A. Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science 2000 -04-14;288(5464):331-333.
(12) Kozlov G, Gehring K. Calnexin cycle – structural features of the ER chaperone system. FEBS J 2020 -10;287(20):4322-4340.
(13) Price JL, Culyba EK, Chen W, Murray AN, Hanson SR, Wong C, et al. N-glycosylation of enhanced aromatic sequons to increase glycoprotein stability. Biopolymers 2012;98(3):195-211.
(14) Oliver JD, Roderick HL, Llewellyn DH, High S. ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol Biol Cell 1999 -08;10(8):2573-2582.
(15) Soldà T, Galli C, Kaufman RJ, Molinari M. Substrate-specific requirements for UGT1-dependent release from calnexin. Mol Cell 2007 -07-20;27(2):238-249.
(16) Fregno I, Molinari M. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Critical Reviews in Biochemistry and Molecular Biology 2019 March 4,;54(2):153-163.
(17) Wang Q, Groenendyk J, Michalak M. Glycoprotein Quality Control and Endoplasmic Reticulum Stress. Molecules 2015 -07-28;20(8):13689-13704.
(18) Roth J, Zuber C. Quality control of glycoprotein folding and ERAD: the role of N-glycan handling, EDEM1 and OS-9. Histochem Cell Biol 2017 -02;147(2):269-284.
(19) Caramelo JJ, Parodi AJ. Getting in and out from calnexin/calreticulin cycles. J Biol Chem 2008 -04-18;283(16):10221-10225.
(20) Oikonomou C, Hendershot LM. Disposing of misfolded ER proteins: A troubled substrate’s way out of the ER. Mol Cell Endocrinol 2020 -01-15;500:110630.
(21) Hwang J, Qi L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem Sci 2018 -08;43(8):593-605.
(22) Grumati P, Dikic I, Stolz A. ER-phagy at a glance. J Cell Sci 2018 -09-03;131(17).
(23) Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011 -11-25;334(6059):1081-1086.
(24) Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016 -01-21;529(7586):326-335.
(25) Liu CY, Kaufman RJ. The unfolded protein response. J Cell Sci 2003 -05-15;116(Pt 10):1861-1862.
(26) Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012 -01-18;13(2):89-102.
(27) Oakes SA, Papa FR. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu Rev Pathol 2015;10:173-194.
(28) Nam SM, Jeon YJ. Proteostasis in the Endoplasmic Reticulum: Road to Cure. Cancers (Basel) 2019 -11-14;11(11).
El sistema de ubiquitinas y la eliminación de proteínas
escrito por Grupo26 | 10 enero, 2022
Ignacio Moratilla Rivera y Miryam Muñoz Martín
3º Biología Sanitaria
Introducción:
Si pensamos en degradación de materiales en la célula en general, siempre nos viene a la cabeza los lisosomas como máximo exponente de esta función. No vamos a negar su imprescindible papel en las células, pero tampoco es el único responsable de realizar estas funciones.
Existe otro sistema que es el responsable de la degradación de proteínas citosólicas contribuyendo a la proteostasis celular. Este sistema está constituido por unas pequeñas proteínas denominadas ubiquitinas y unos complejos proteicos conocidos como proteosomas. Ambos junto a otras enzimas constituyen un proceso en cadena en el que finalmente las proteínas serán degradas.
Este mecanismo no solo se va a limitar a la eliminación de proteínas mal plegadas o aberrantes, sino que también va a desempeñar un papel crucial en la regulación del ciclo celular y fallos en él pueden llevar a cataclismos fisiológicos serios como el Parkinson, Alzheimer o enfermedades cardiovasculares.
Ubiquitinas: las proteínas omnipresentes.
Las ubiquitinas son unas proteínas monoméricas que se encuentra presenten en todos los organismos eucariotas (de ahí su nombre, ya que son ubicuas). No se han encontrado en procariotas, pero tienen proteínas con motivos estructurales similares, ThiS y MoaD que son sus posibles antecesores. Una evidencia de ello son las dos glicinas del extremo C-terminal, que están presentes en estas proteínas y en las ubiquitinas.
Figura 1: Imagen que representa la estructura tridimensional de Ubq (PDB: 5J8P), ThiS (PDB: 1ZUD) y MoaD (PDB: 1JW9). Apréciese la similitud estructural entre ellas, además de su coincidencia en los aminoácidos C-terminales. Imagen realizada con BioRender y Chimera
Lo que más llama la atención es su alto grado de conservación, lo que pone en evidencia su importante papel dentro de la célula. Presentan 76 residuos de aminoácidos, y el ser humano solo se diferencia en 3 de estos con las levaduras.
Estas pequeñas proteínas pueden ser vistas como “chivatas” ya que marcan por unión covalente las proteínas que deben ser degradas en proteosomas.
El residuo que produce la unión es una Gly situada en el extremo C-terminal y que es común en todas las ubiquitinas. La glicina se une a la proteína diana por un enlace isopeptídico, que se diferencia del enlace peptídico en que uno de los grupos (sea NH2 o COOH) forma parte de un grupo R. En este caso ocurre entre en el grupo COOH de la Gly terminal con el grupo NH2 de la cadena lateral de una Lys de la proteína diana en gran número de ocasiones, pero también se ha visto que pueden unirse con grupos tiol de la Cys y grupos hidroxilo de Ser y Thr.
Figura 2: Representación tridimensional de la disposición de la Gly y Lys formando el enlace isopeptídico. Esta unión permitirá el engarzamiento con la proteína diana. Imagen realizada con Molecular Constructor y BioRender
Las ubiquitinas no actúan solas en los procesos de marcaje de las proteínas a degradar, sino que estás se unen unas con otras formando cadenas. A este proceso se le conoce como poliubiquitinación y es clave para el reconocimiento de la proteína en el proteosoma. En la formación de la cadena cada eslabón de unión está formado por una Lys de la ubiquitina proximal y la Gly C-terminal de la distal.
A parte de las ubiquitinas, dentro de la célula podemos encontrar unas proteínas muy similares denominadas modificadores similares a ubiquitinas o Ubl (ubiquitin-like modifer proteins). Estas a parte de tener mecanismos comunes de unión con Lys, las Gly C-terminal y producir modificaciones en las proteínas, tienen un plegamiento casi idéntico al de la ubiquitina.
Proceso en cadena: E1, E2 y E3.
Hasta que la ubiquitina llega a marcar la proteína a degradar, tienen que actuar una serie de enzimas que la activen y unan.
E1: ubiquitina activasa
Esta proteína es la responsable de activar la ubiquitina mediante la unión a su extremo C-terminal de un residuo de cisteína (enlace tioéster). Para la formación de este enlace se requiere de ATP, que forma un intermediario con la ubiquitina antes de unirse a la enzima. La reacción de adenilación está muy conservada ya que se ha visto también en la proteína MoaD de bacterias.
Figura 3: Representación 3D del intermediario Ubq-AMP en el centro activo de la enzima E1 (PDB: 4NNJ) . Los lazos de colores pastel hacen referencia a la estructura de E1. Imagen realizada con Chimera y BioRender.
El intermediario Ub-AMP se sitúa en el centro activo de la E1, cerca de donde se encuentra la cisteína de unión. La estructura del nucleótido se estabiliza por puentes de hidrógeno con la enzima E1, reduciendo la entropía y favorecer el proceso de formación del enlace tioéster.
E2: enzima conjugadora
Las E2 unen las ubiquitinas que se habían unida a E1 también con un enlace en un residuo de cisteína.
Figura 4: Representación tridimensional de la transferencia de la Ubq desde E1 hasta E2 (PDB: 5KNL). La unión de E2 a E1 permite el paso de la ubiquitina por la formación de un nuevo enlace tioéster con un residuo de Cys de E2. Imagen realizada con Chimera y BioRender.
No es una única enzima E2, sino que son varias cada una con diferentes funciones. Estas después serán reconocidas por diferentes. El papel que puede tener E2 se discute, ya que podría ser más sencillo que E3 ligue directamente la ubiquitina desde E1 hasta la proteína diana.
Como es habitual, los eucariotas preferimos darle un vuelta de tuerca a todos los procesos moleculares. La presencia de E2 constituye un punto de regulación importante en la ubiquitinación y en la especificidad de los sustratos que se etiquetan.
La cisteína de E2 se encuentra en una hendidura de la estructura en la que debe entrar el complejo E1-Ubq para que se produzca la transferencia de una a otra.
E3: ubiquitina ligasa
La E3 tiene la habilidad de tomar la ubiquitina de la E2 y transferirla a la proteína diana. ¿Quiénes pueden ser estas proteínas dianas? Pues una gran diversidad de ellas. Es posible gracias a la existencia de varias enzimas E3 capaces de unir la ubiqutina a diferentes sustratos.
Figura 5: El modelo tridimensional pretende mostrar que la enzima E3 presenta un motivo de unión a E2-Ubq (PDB: 5TTE), y después rompe la unión entre ambas para realizar la transferencia de la Ubq hasta una proteína diana del tipo que sea mediante la catálisis del enlace isopeptídico. Imagen realizada con Chimera y BioRender
Dicha unión ocurre entre el C-terminal de la ubiquitina y una lisina de la proteína a degradar. Pero tienen la función también de enlazar unas ubiquitinas con otras para formar cadenas de poliubiquitinas.
Las tres clases de ubiquitina ligasa existentes se diferencian en función de su dominio activo: RING finger, HECT y U-box. Dentro de cada clase a su vez existen subclases, esto permite incrementar la especificidad del reconocimiento de las proteínas que queremos ubiquitinar. Para ello cada tipo de E3 cuanta con dominios de reconocimiento hacia determinadas proteínas y hacia diferentes tipos de E2.
RESUMEN DEL PROCESO ENCADENADO
Figura 6: Resumen del proceso de encadenado. Imagen realizada con BioRender.
Proteosomas: toneles de degradación
El sistema del proteosoma de ubiquitina (UPS) es un complejo multiproteico muy conservado que se encarga de la degradación enzimática de las proteínas. Está formado por una partícula o core central 20S. Este núcleo presenta dos anillos exteriores, con siete subunidades alfa, y dos anillos interiores con siete subunidades beta. Tiene forma de barril, en cuyo interior hay seis centros activos con actividad proteasa. El extremo amino-terminal de las subunidades alfa pueden abrirse para favorecer el paso de la proteína al interior del proteosoma.
Figura 7: Imagen ilustrativa tridimensional de un proteosoma con cada una de las estructuras que lo componen. Imagen realizada con BioRender
Además de la partícula central 20S, presenta partículas reguladoras 19S, compuestas por una tapa y una base. Esta última presenta seis moléculas de ATPasa, cuyos extremos carboxi-terminales encajan entre las subunidades alfa, provocando en ellas el cambio conformacional que abre la compuerta.
La ubiquitinación de las diferentes lisinas de las proteínas desencadena diferentes funciones, desde degradación de proteínas, hasta señales para reparar el DNA, activación de factores de transcripción, endocitosis…
En el proceso de liberación de aminoácidos de la proteína se distinguen dos partes, según la utilización de energía metabólica: hay una parte del proceso que es dependiente de ATP, mientras que otra parte es independiente de ATP.
Las proteínas son desplegadas por reguladores mediante cambios conformacionales provocados gracias a la hidrólisis de ATP. Después, las proteínas desplegadas son traslocadas a los centros activos con actividad proteasa del núcleo central, dando lugar a pequeños péptidos de tres a quince aminoácidos. Por último, estos péptidos son degradados de forma independiente de ATP por endopeptidasas, carboxipeptidasas y aminopeptidasas.
Figura 8: Proceso llevado a cabo durante la degradación de las proteínas ubiquitinadas. Imagen realizada con BioRender.
Comprender la estructura y el funcionamiento del proteosoma ha permitido conocer su asociación con enfermedades cardiovasculares, diabetes, enfermedades neurológicas y cáncer. El gen que codifica el proteosoma se localiza en el cromosoma 6, cerca de la región donde se codifican las moléculas del MHC-II.
En la actualidad, se han descrito cuatro tipos de proteosomas:
Proteosoma constitutivo, clásico o proteosoma 26S.
Es una estructura grande, macromolecular y multiproteica que tiene carácter enzimático. Se localiza principalmente en el citosol, aunque también en el núcleo de todos los eucariotas, por lo que es el principal complejo proteolítico celular, además, es la misma vía que la célula usa para presentar los péptidos en la superficie en el contexto de MHC-I.
Reconoce los sustratos de modo dependiente de ubiquitina, de modo mediado por un adaptador y de manera independiente de ubiquitina, en las tres formas requiere de ATP, siendo la vía dependiente de ubiquitina la más usada por microorganismos eucariotas.
Inmunoproteosoma
Fue descubierto en células WEHI-3 al ser tratadas con IFN-gamma. Se localiza en las células dendríticas de la médula del timo y en las células epiteliales tímicas medulares.
Se observó que presentan baja actividad tipo caspasas y una actividad alta de tipo tripsina en comparación con el proteosoma 26S, por lo que podría producir diferentes tipos de péptidos, debido a su tendencia a producir escisión detrás de aminoácidos hidrofóbicos o básicos. Además, optimiza la respuesta efectora frente a determinados antígenos mediante la activación selectiva de clones de linfocitos CD8+.
Su estudio nos permite comprender mejor los mecanismos de aparición de determinados tipos de cánceres y enfermedades autoinmunes.
Proteosomas intermedios
Es un proteosoma intermedio entre el proteosoma clásico y el inmunoproteosoma. Se ha observado en tumores del hígado, colon, intestino delgado y células dendríticas. El hallazgo de estos proteosomas intermedios en células dendríticas podría servir como herramienta para la inmunoterapia.
Timoproteosoma
Son los proteosomas que se encuentran en el timo. Son necesarios para que pueda llevarse a cabo de manera correcta el mecanismo de selección positiva de los linfocitos CD8+, aunque no se sabe muy bien cómo lo hace. Genera de forma constitutiva ligandos de péptidos alterados durante el mecanismo de selección positiva, lo cual jugará un rol importante en el proceso de la tolerancia inmune.
Importancia del sistema: ciclo celular, enfermedades neurodegenerativas y cáncer.
Esta función que presentan los proteosomas los hace especialmente importantes a la hora de controlar el ciclo celular. Las kinasas dependientes de ciclinas (CDK) controlan la progresión del ciclo celular tras ser activadas por ciclinas. Cuando ya han llevado a cabo su función es necesario que sean eliminadas. Un mal funcionamiento de estas CDK puede producir un proceso tumoral.
La ciclina será poliubiquitinizada y, posteriormente, degradada por el proteosoma, para que pueda continuar el ciclo celular. En concreto, para que la célula finalice la mitosis, requiere que el componente regulador ciclina B se disocie. En las células de vertebrados esta disociación será dependiente de los proteosomas.
En el cáncer, la mutación de ciertos genes provoca que las células sean inmortales. En estas células (sea por mutación o activación) habrá una sobreexpresión del proteosoma, el cual estará degradando la ciclina en exceso. Esto lo que provoca es que la célula nunca pare de dividirse.
Teniendo esto en cuenta, muchos fármacos y quimioterapias tienen como diana la alteración o inhibición de los proteosomas, haciendo que las células entren en apoptosis y el cáncer se frenaría.
Hay muchos tipos de ciclinas que se unen a CDK específicos, por ello, los CDK-inhibidores (p21 y p27) efectúan su acción cuando hay algún daño celular o cromosómico. Estos CDK-inhibidores inhiben el ciclo celular en estos casos siempre y cuando el proteosoma esté inhibido o regulado, ya que en células tumorales se encontraría sobreexpresado y degradando CDK-inhibidores, impidiendo la regulación de este punto de control.
Otro punto de control del ciclo celular que da lugar a procesos cancerígenos es llevado a cabo por factores de supervivencia celular (ikB y NFKB). A este nivel, la inhibición del proteosoma también provocaría la apoptosis celular al no haber señales de supervivencia.
El inmunoproteosoma participa en la regulación del desarrollo de tumores. Se ha observado en pacientes humanos con leimoisarcoma uterino, que carecen de una de las subunidades del inmunoproteosoma. También se ha demostrado su sobreexpresión en leucemias agudas. Por lo que se sugiere que, dependiendo del tipo de cáncer, el inmunoproteosoma puede ser una consecuencia de la enfermedad o puede actuar como un factor de desarrollo. Por ello, se ha convertido en un objetivo importante para luchar contra el cáncer, ya que las células cancerígenas son más subceptibles a los inhibidores de proteosomas. Por ejemplo, en el Mieloma Múltiple, que es un trastorno del plasma sanguíneo debido a la proliferación de células tumorales en la médula ósea, se utilizan inhibidores del proteosoma, estimulando así señales apoptóticas.
Las enfermedades neurodegenativas se caracterizan por la acumulación de proteínas aberrantes que tienden a unirse entre sí. El origen de muchas enfermedades neurodegenerativas se debe a un mal funcionamiento del inmunoproteosoma.
En la enfermedad del Parkinson, las neuronas dopaminérgicas dañadas producen un exceso de proteínas anómalas que interaccionan entre sí formando “placas amiloideas”. La célula trata de eliminarlas mediante la vía ubiquitina-proteosoma. Llegada a una edad, estos mecanismos no funcionan bien, provocando la apoptosis neuronal generando la enfermedad.
La Enfermedad de Alzheimer es un proceso neurodegenerativo de la neocorteza y el hipocampo. En ella se definen dos tipos principales de lesiones: las placas neuríticas y las marañas neurofibrilares. Ambos representan los productos de un trastorno del plegamiento de proteínas que se caracterizan por la proteolisis. Esta acumulación está relacionada con disfunciones de la actividad proteolítica del proteosoma.
La acumulación de presenilina-2 debido al mal funcionamiento del proteosoma en enfermedad de Alzheimer puede ser asociado con la acumulación de β-amioloide, lo que dificultaría la función del proteosoma con el consecuente daño celular.
REFERENCIAS
Pickart, C. M. and Eddins, M. J. (2004) ‘Ubiquitin: Structures, functions, mechanisms’, Biochimica et Biophysica Acta – Molecular Cell Research, 1695(1–3), pp. 55–72. doi: 10.1016/j.bbamcr.2004.09.019.
Strous, G. J. and Govers, R. (1999) ‘The ubiquitin-proteosome system and endocytosis’, Journal of Cell Science, 112(10), pp. 1417–1423.
Tanaka, K. (2009) ‘The proteasome: Overview of structure and functions’, Proceedings of the Japan Academy Series B: Physical and Biological Sciences, 85(1), pp. 12–36. doi: 10.2183/pjab.85.12.
Amm, I., Sommer, T. and Wolf, D. H. (2014) ‘Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system’, Biochimica et Biophysica Acta – Molecular Cell Research. Elsevier B.V., 1843(1), pp. 182–196. doi: 10.1016/j.bbamcr.2013.06.031.
Pick, E., & Berman, T. S. (2013). Formation of alternative proteasomes: Same lady, different cap? FEBS Letters, 587(5), 389–393. https://doi.org/10.1016/j.febslet.2013.01.014
Esser, C., Scheffner, M., & Höhfeld, J. (2005). The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. Journal of Biological Chemistry, 280(29), 27443–27448. https://doi.org/10.1074/jbc.M501574200
Cellular protein degradation occurs in different cellular. (n.d.).
Redondo, C. L. (2015). Papel del inmunoproteosoma en enfermedades neurodegenerativas y cáncer Revisión bibliográfica.
Barrio, R., & Izquierdo, M. (n.d.). Aplicaciones terapéuticas de la ubiquitina.
Ubiquitinación, U., Un Un Sistema Sistema Sistema Sistema Sistema De De De De De Regulación Regulación Regulación, U. U., Manuel Zamudio-Arroyo, J., Teresa Peña-Rangel, M., Rafael Riesgo-Escovar María Teresa Peña-Rangel, J., & Rafael Riesgo-Escovar, J. (n.d.).
Erales, J., & Coffino, P. (2014, January). Ubiquitin-independent proteasomal degradation. Biochimica et Biophysica Acta – Molecular Cell Research. https://doi.org/10.1016/j.bbamcr.2013.05.008
Zelada Valdés, A. (2019). The proteasome: subtypes and involvement in central tolerance. Revista Cubana de Investigaciones Biomédicas (Vol. 38).
Hershko, A., & Ciechanover, A. (1998). THE UBIQUITIN SYSTEM. Annu. Rev. Biochem (Vol. 67).
Un RNA funcional: XIST, lionización del cromosoma X, corpúsculo de Barr
escrito por Grupo 14 | 10 enero, 2022
Ana Belén Alonso Aguado, Irene Chavarría Cubel y Rodrigo Díaz Muñoz.3º Biología Sanitaria, UAH.
La lionización debe su nombre a la genetista Mary Lyon y consiste en el proceso de inactivación de uno de los cromosomas X en las células de las hembras de mamíferos. Para ello, se producen una serie de modificaciones epigenéticas que conducen a la aparición de la cromatina sexual o corpúsculo de Barr, un cuerpo de heterocromatina adherido a la envoltura nuclear.
Estas modificaciones se desencadenan gracias a un RNA no codificante funcional, producido por el gen Xist situado en la región XIC del cromosoma X, el cual permite el reclutamiento de los factores implicados en la reorganización de la cromatina.
El objetivo de este fenómeno esequiparar los niveles de expresión de los genes ligados al sexo, ya que las hembras presentan dos cromosomas X, mientras que los machos poseen sólo uno (XY).
Antecedentes históricos: corpúsculo de Barr e hipótesis de Lyon
En 1949, Bertran y Barr introdujeron el término cromatina sexual o corpúsculo de Barr tras observar en neuronas del asta anterior de la médula espinal de gatos, junto al nucléolo y bien distinguido de éste, un corpúsculo de cromatina densa (heterocromatina), que estaba adherido a la envoltura nuclear. Esta estructura solo aparecía en hembras, por lo que pensaron que estaría relacionado con el sexo.
En 1959, Susumu Ohno demostró que el corpúsculo de Barr se corresponde con un cromosoma X heterocromatizado y propuso que uno de los dos cromosomas X está inactivo en cada célula somática (células epiteliales, fibroblastos, leucocitos, etc.).
En 1966, Mary Lyon propuso la hipótesis de Lyon para la cromatina sexual o corpúsculo de Barr:
La cromatina sexual es genéticamente inactiva.
La heterocromatización ocurre en la embriogénesis temprana (hacia el día 16 del desarrollo embrionario en la mujer).
El proceso de inactivación del cromosoma X en cada célula de una hembra se produce al azar, es decir, puede inactivarse el cromosoma X materno o paterno.
Se trata de un proceso irreversible y heredable, por lo que en un linaje celular se mantendrá el mismo patrón de inactivación.
Como consecuencia, las hembras poseen mezclas de líneas celulares en las que se inactiva el cromosoma X paterno y líneas celulares en las que se inactiva el cromosoma X materno, manifestándose unos u otros factores genéticos ligados al sexo. De esta forma, una hembra heterocigótica para una característica o enfermedad ligada al cromosoma X es un mosaico(ver figura 2).
Además, el corpúsculo de Barr sigue la llamada “regla n-1”(ver figura 1): el número de corpúsculos de Barr de una célula es igual al número de cromosomas X que posee la célula (n) menos 1. Por lo tanto, en todas las células femeninas hay un único cromosoma X activo, mientras que el otro (cariotipo XX) o los otros cromosomas X (cariotipos anómalos) se encuentran heterocromatizados (inactivos). Solo en la meiosis de los ovocitos el cromosoma X heterocromatizado se reactiva, de manera que el patrón de inactivación no se mantiene en la descendencia.
Figura 1. A la izquierda, imagen de Mary Lyon. A la derecha, corpúsculos de Barr al microscopio óptico. (A) Célula femenina con cariotipo 46 XX; presenta un cromosoma X inactivo y, por ende, un único corpúsculo de Barr (señalado con flecha blanca). (B) Célula masculina con cariotipo anómalo 49 XXXXY; presenta tres cromosomas X inactivos y, por tanto, muestra tres corpúsculos de Barr (flechas negras). [13]
Figura 2. Si se ha inactivado el cromosoma X materno (m) o paterno (p) en una célula del embrión en las etapas tempranas del desarrollo embrionario, en todas las células que se originen a partir de ésta se inactivará siempre el cromosoma X materno o paterno, respectivamente. Como resultado, observamos células somáticas formando tejidos mosaicos. [12]
Además, el cromosoma X heterocromatizado no se inactiva por completo (ver figura 3). Se estima que la inactivación afecta al 65% de los genes del cromosoma en todas las células, mientras que un 20% se inactiva solo en algunas células y el 15% consigue escapar de este proceso. Por ello, las mujeres con síndrome de Turner (X0) presentan un fenotipo particular. Si el cromosoma X se inactivara completamente, el fenotipo de estas mujeres sería idéntico al de las mujeres XX.
Figura 3. El esquema muestra los genes que se expresan (azul) y no se expresan (amarillo) en el cromosoma X inactivo (Xi). Estos resultados se obtuvieron mediante RT-PCR. [13]
Ejemplos de fenotipos mosaico ligados al cromosoma X
Color del pelaje en los gatos calicó: el pelaje de las gatas puede ser naranja, negro o parcheado, mientras que el de los gatos es totalmente negro o naranja (ver figura 4).
Enfermedades dermatológicas: algunas de estas enfermedades, como la displasia ectodérmica anhidrótica, presentan un patrón en mosaico.
Isoformas de glucosa-6-fosfato deshidrogenasa: se observó la presencia de una única isoforma en fibroblastos aislados de mujeres heterocigotas para los genes de las isoenzimas A y B. Esto confirmó la hipótesis de Lyon, ya que, si no hubiera inactivación del cromosoma X y ésta no fuese al azar, se deberían observar dos isoformas distintas o la misma en todas las células (suponiendo uno de los alelos dominante).
Figura 4. El color de las gatos calicó depende del gen B que se encuentra localizado en el cromosoma X; el alelo B da lugar a una coloración naranja y el b a una coloración negra. Como los machos solamente tienen un cromosoma X, serán naranjas o negros (B o b). Sin embargo, podremos encontrar hembras homocigotas para el alelo b (todas sus células independientemente del cromosoma que se inactive presentarán el alelo b y serán negras), homocigotas para B (naranjas) o heterocigotas (mosaicos). En este último caso, las zonas naranjas proceden de las células en las que se inactivó el cromosoma X portador del alelo b, mientras que las zonas negras estarán formadas por las células en las que se inactivó el cromosoma X portador del alelo B. Creada con BioRender.
Mecanismo de inactivación del cromosoma X y modificaciones en la cromatina
El proceso de inactivación del cromosoma X consta de cuatro pasos: contaje (cociente entre el número de cromosomas X y autosomas), selección (inactivación del cromosoma X materno o paterno), iniciación de la inactivación y mantenimiento en las siguientes generaciones de un mismo linaje celular.
La heterocromatización se inicia en un punto del cromosoma X llamado XIC (centro de inactivación de X), el cual está situado en el locus multifuncionalXq13 y se extiende hacia ambos extremos del cromosoma (ver figura 5).
Principales elementos de XIC implicados:
Gen Xist (transcrito específico para la inactivación de X): produce un transcrito primario que, tras ser procesado por splicing y poliadenilación, da lugar a un RNA no codificante y funcional de 15-17 kb necesario para iniciar el silenciamiento del cromosoma X. Este RNA se dispone a lo largo del cromosoma X que se debe inactivar y recluta una serie de factores, los cuales producen modificaciones epigenéticas que conducen a la condensación de la cromatina.
Locus Xce (elemento regulador del cromosoma X): los genes incluidos en este locus están implicados en los mecanismos de contaje y selección del cromosoma X que se va a inactivar.
Genes reguladores de la expresión del gen Xist:activadores (Jpx, Ftx, Rnf12) y represores (Tsix en ratones). El gen Jpx y Ftx producen RNAs no codificantes que activan la expresión del gen Xist, mientras que el gen Rnf12 produce una proteína con actividad ubiquitina-ligasa que parece degradar un inhibidor del gen Xist. Por el contrario, el gen Tsix en ratones produce un RNA antisentido no codificante (complementario al Xist RNA) que inhibe la expresión del gen Xist y es fundamental en la selección del cromosoma X que se va a inactivar.
Figura 5. La figura muestra un esquema de la región XIC en ratones. Creada con BioRender.
En el comienzo del desarrollo embrionario, los factores de pluripotencialidad (NanoG, Sox2 y Oct-4) interaccionan con regiones promotoras del gen Xist, manteniendo bajos los niveles de expresión de este gen, tanto en el cromosoma X de origen materno como en el paterno. Además, estos factores regulan positivamente la expresión del gen Tsix (represor).
Llega un momento, aun en la embriogénesis temprana, en el que se produce un apareamiento transitorio entre algunas regiones de las secuencias XIC de dos cromosomas X. Este proceso parece constituir un mecanismo de recuento de cromosomas X y se repetirá hasta que solo quede uno activo (en caso de cariotipos anómalos con más de dos cromosomas X)(ver figura 6).
Como resultado de la interacción, se produce la activación del gen Tsix (represor) al azar en uno de los cromosomas (“competencia”). Esto conducirá al cese de la expresión del gen Xist en uno de los cromosomas X, mientras que aumentará significativamente en el otro (futuro cromosoma X inactivo o Xi).
En humanos también se da este apareamiento, pero Tsix produce un RNA antisentido no codificante truncado en el extremo 5’, incapaz de actuar como represor de Xist (en este caso, Tsix parece ser un vestigio evolutivo).
Figura 6. Apareamiento y papel del gen Tsix en la inactivación del cromosoma X en ratones. La expresión de Xist determinará el futuro cromosoma inactivo (Xi), mientras que la expresión de Tsix determinará el futuro cromosoma activo (Xa). Creado con BioRender.
A continuación, comienza el silenciamiento del cromosoma X seleccionado. Para ello, en el futuro cromosoma Xi, el gen Xist produce un transcrito primario que será procesado mediante splicing y poliadenilación para dar lugar a un RNA no codificante funcional que recubre el cromosoma Xi. Este es el Xist RNA, requerido para iniciar y estabilizar la inactivación del cromosoma X, pero no para mantenerla.
Se ha observado que, al eliminar el gen Xist del cromosoma X, éste no se inactiva. Sin embargo, si se coloca en cualquier cromosoma autosómico, automáticamente se produce la inactivación de dicho cromosoma.
Los elementos LINE (elementos nucleares dispersos largos) son secuencias repetidas a lo largo del cromosoma X, principalmente en regiones cercanas a XIC (forman un 30% del cromosoma). Al parecer, estas secuencias favorecen una rápida expansión del Xist RNA, facilitando el proceso de inactivación.
Una vez situado sobre el futuro cromosoma Xi, el Xist RNA recluta directa o indirectamente una serie de factores, como los complejos proteicos Polycomb PRC1 y PRC2, que reorganizan la cromatina para que ésta adquiera una conformación de heterocromatina facultativa (cromatina condensada transcripcionalmente inactiva).
El mantenimiento de la inactivación del cromosoma X se debe a las siguientes modificaciones epigenéticas que producen la condensación de la cromatina: trimetilación en las lisinas 9 y 27 de la histona H3 (H3K9me3 y H3K27me3, respectivamente), desmetilación de la lisina 4 de la H3 (no H3K4me3), aumento de nucleosomas con la variante macroH2A de la histona H2A, desacetilación de histonas y metilación de las islas CpG (residuos de citosina unidos a guanina) en los promotores de los genes que se van a inactivar (en los genes activos estas islas no están metiladas). Como consecuencia, se producirá una exclusión de la RNA polimerasa II y otros factores, así como una replicación tardía en la fase S(ver figura 7).
Figura 7. Diferencias a escala nucleosomal en las modificaciones epigenéticas del cromosoma X activo (Xa) e inactivo (Xi). RNAPII: RNA Polimerasa II; GTFs: Factores transcripcionales generales; TFs: Factores transcripcionales de unión a intensificadores; CTCF: Proteína aisladora. Enhancer: secuencia intensificadora; Insulator: secuencia aisladora. Nomenclatura modificaciones en el recuadro inferior: ac (acetilación), H (histona), K (lisina), me (metilación), me2 (dimetilación), me3 (trimetilación), ub (ubiquitinación), CpG (islas Cpg), meCpG (metilación islas CpG). [5]
Figura 8. En morado se observan las histonas macroH2A y en azul el DNA, formando un nucleosoma. Las figuras A y B muestran distintas perspectivas. PDB: 2F8N. Creado con UCSF Chimera.
Xist RNA y factores reclutados implicados en la remodelación de la cromatina
El Xist RNA presenta unas regiones formadas por repeticiones en tándem de secuencias consenso, las cuales se clasifican de la A a la F. Las repeticiones A-D y F se encuentran en el exón 1, mientras que las repeticiones E se encuentran en el exón 7(ver figuras 9, 10 y 13).
Figura 9. (A) El esquema superior muestra los genes Xist y Tsix localizados en la región XIC del cromosoma X. El esquema inferior muestra el Xist RNA tras el splicing, donde se ilustran en amarillo las repeticiones en tándem de secuencias consenso (A-E). (B) Tabla que describe las secuencias repetidas. [8]
Figura 10. Tetraloops AUCG en las repeticiones A de Xist RNA. (A)Estructura secundaria en solución de las repeticiones A del Xist RNA en humanos. PDB: 2Y95. El círculo negro marca el tetraloop AUCG. Las letras de colores indican los nucleótidos del tetraloop, que se corresponden con los indicados en los esquemas B y C.(B)Esquema de la estructura ilustrada en A. (C) Esquema del tetraloop AUCG, rodeado en la ilustración A con un círculo negro. Creada con BioRender y [15].
Como ya sabemos, Xist RNA inicia el silenciamiento del cromosoma X mediante el reclutamiento directo e indirecto de distintos factores implicados en la remodelación de la cromatina. Estos factores interaccionan con las repeticiones mencionadas anteriormente y se conocen como proteínas de unión al RNA (RBPs). Actualmente, se sabe que las principales RBPs que participan en el silenciamiento son: SPEN (SHARP en humanos), RBM15, hnRNPK y LBR.
SPEN (SHARP en humanos)
SPEN es una proteína con capacidad de interacción con el RNA y otras proteínas gracias a cuatro dominios RRM presentes en el extremo N-terminal y un dominio SPOC en el extremo C-terminal, respectivamente (ver figura 11).
Por un lado, los RRM 2-4 (sobre todo RRM 3) interactúan directamente con las repeticiones A del Xist RNA, mientras que el dominio SPOC recluta el complejo de desacetilación de histonas (NCoR-HDAC3).
De esta forma, se llevará a la desacetilación de histonas en el cromosoma X que se está inactivando. Como ya sabemos, esta desacetilación es una de las modificaciones que favorecen la condensación de la cromatina y, por tanto, la disminución de la accesibilidad de la RNA polimerasa II a los promotores de los genes.
Parece que otra función de SPEN es secuestrar un complejo coactivador de la metiltransferasa H3K4 (KMT2D) mediante el dominio SPOC, de manera que disminuirá la actividad de la metiltransferasa H3K4 (recordemos que la metilación de H3K4 es característica del cromosoma X activo)(ver figura 7).
Además, algunos estudios sugieren que SPEN podría ser responsable de los bajos niveles de RNA polimerasa II (RNAPII) sobre el cromosoma Xi, ya que parece interactuar con esta enzima y algunos cofactores asociados, secuestrándolos.
Figura 11. (A) Dominios SPOC (PDB: 1OW1) y (B)RRM 2-4 de la proteína SPEN (PDB: 4P6Q). Creado con UCSF Chimera y BioRender.
RBM15
RBM15 es una proteína perteneciente a la misma familia que SPEN, por lo que presenta tres dominios conservados RRM (unión a RNA) en el extremo N-terminal y un dominio SPOC (unión a otras proteínas) en el extremo C-terminal.
Los dominios RRM, al igual que en la proteína SPEN, interactúan con las repeticiones A del Xist RNA. Sin embargo, el dominio SPOC va a interactuar con el complejo proteico METTL3/14 que lleva a cabo la metilación del N6 de adenosinas (m6A). Esta modificación parece estabilizar el RNA, siendo muy abundante en RNAs no codificantes, en este caso Xist RNA, y también en mRNAs.
Asimismo, al igual que en SPEN, el dominio SPOC de RBM15 puede secuestrar otro complejo coactivador de la metiltransferasa H3K4 (SET1B o KMTD2G). De esta forma, disminuye la metilación de la lisina 4 en la histona 3 del cromosoma Xi.
hnRNPK
hnRNPK es otra de las RBPs que interacciona con Xist RNA, en este caso con las repeticiones B/C a través de tres dominios KH (KH1, KH2, KH3) (ver figura 12). Asimismo, a través de su dominio KI cercano al extremo C-terminal, recluta el complejo Polycomb PRC1.
Los complejos multienzimáticos Polycombcatalizan las modificaciones de histonas que conducen a la remodelación de la cromatina.
El complejo PRC1 cataliza la ubiquitinación de la lisina 119 de la histona H2A (H2AK119ub1). Esta modificación promueve la concentración del complejo PRC2 sobre el cromosoma Xi, así como de otros complejos Polycomb. En concreto, el complejo PRC2 produce la trimetilación de la lisina 27 de la histona 3 (H3K27me3).
El mecanismo de acción de los complejos Polycomb aún se desconoce, pero parece que el silenciamiento génico podría ser resultado de:
La ubiquitinación y metilación de histonas podría reclutar otras proteínas implicadas en el silenciamiento de los genes.
Como resultado de la modificación de las histonas, la cromatina se reorganiza de manera que limita la accesibilidad a los factores de transcripción y la RNA polimerasa II.
Figura 12. Dominio KH de hnRNPK. PDB: 1KHM. Creado con UCSF Chimera.
LBR
LBR es un receptor de la lámina nuclear que interactúa directamente con Xist RNA, por lo que se incluye dentro de las RBPs. Esta unión se produce principalmente entre el dominio SR (tramo de arginina-serina) de LBR y los sitios LBS en las repeticiones F de Xist RNA.
Por lo tanto, la interacción de LBR con Xist RNA permite la unión del cromosoma Xi a la lámina nuclear (por ello, el corpúsculo de Barr se observa en la periferia nuclear). En estudios recientes, se ha observado que esto podría mejorar la tasa de silenciamiento.
Figura 13. Esquema resumen de las interacciones de las RBPs con las repeticiones de Xist RNA (A, F, B, C, D, E), así como los factores reclutados por cada una de ellas que ejercen diversos efectos sobre la estructura de la cromatina y la localización del cromosoma Xi en el núcleo celular. [5]
Xist RNA antagonista del complejo SWI/SNF
El complejo SWI/SNF se encarga de la remodelación de la cromatina y, normalmente, actúa cuando se requiere una activación de la transcripción. En estudios recientes, se ha propuesto el posible papel de Xist RNA como antagonista de este complejo.
Curiosamente, la deleción del gen Xist produce un aumento de la accesibilidad de la cromatina regulado por BRG1, una subunidad ATPasa del complejo remodelador de la cromatina SWI/SNF.
Por tanto, parece que la la unión de Xist RNA al cromosoma Xi inhibe la actividad de remodelación del nucleosoma de BRG1 y da como resultado la expulsión del complejo SWI/SNF del Xi.
Bibliografía
1. Jégu T, Blum R, Cochrane JC, Yang L, Wang C-Y, Gilles M-E, et al. Xist RNA antagonizes the SWI/SNF chromatin remodeler BRG1 on the inactive X chromosome. Nat Struct Mol Biol. 2019 Feb 21;26(2):96–109.
2. Balderman S, Lichtman MA. A History of the Discovery of Random X Chromosome Inactivation in the Human Female and its Significance. Rambam Maimonides Med J. 2011 Jul 29;2(3).
3. Acosta Lobo ME, Vásquez Araque NA, Londoño Franco LF. Inactivación del cromosoma X en el desarrollo embrionario mamífero. CES Med Vet y Zootec. 2013;8(2):108–19.
4. Migeon BR. Is Tsix repression of Xist specific to mouse? Nat Genet. 2003 Mar 1;33(3):337–337.
5. Brockdorff N, Bowness JS, Wei G. Progress toward understanding chromosome silencing by Xist RNA. Genes Dev. 2020 Jun 1;34(11–12):733–44.
6. Maclary E, Hinten M, Harris C, Kalantry S. Long nonoding RNAs in the X-inactivation center. Chromosom Res. 2013 Dec 3;21(6–7):601–14.
7. Froberg JE, Yang L, Lee JT. Guided by RNAs: X-Inactivation as a Model for lncRNA Function. J Mol Biol. 2013 Oct 9;425(19):3698–706.
8. Pontier DB, Gribnau J. Xist regulation and function eXplored. Hum Genet. 2011 Aug;130(2):223–36.
9. Duszczyk MM, Wutz A, Rybin V, Sattler M. The Xist RNA A-repeat comprises a novel AUCG tetraloop fold and a platform for multimerization. RNA. 2011 Nov;17(11):1973–82.
10. Arieti F, Gabus C, Tambalo M, Huet T, Round A, Thore S. The crystal structure of the Split End protein SHARP adds a new layer of complexity to proteins containing RNA recognition motifs. Nucleic Acids Res. 2014 Jun 2;42(10):6742–52.
11. Paniagua R. Biología celular y molecular. 4ª ed. Madrid: McGraw-Hill; 2017.
12. Jorde L, Carey J, Bamshad M. Medical genetics. 5th. ed. Sant Lake City: Elsevier; 2015.
13. Strachan T, Read A. Human molecular genetics. 5th. ed. Florida: Garland Science; 2019.


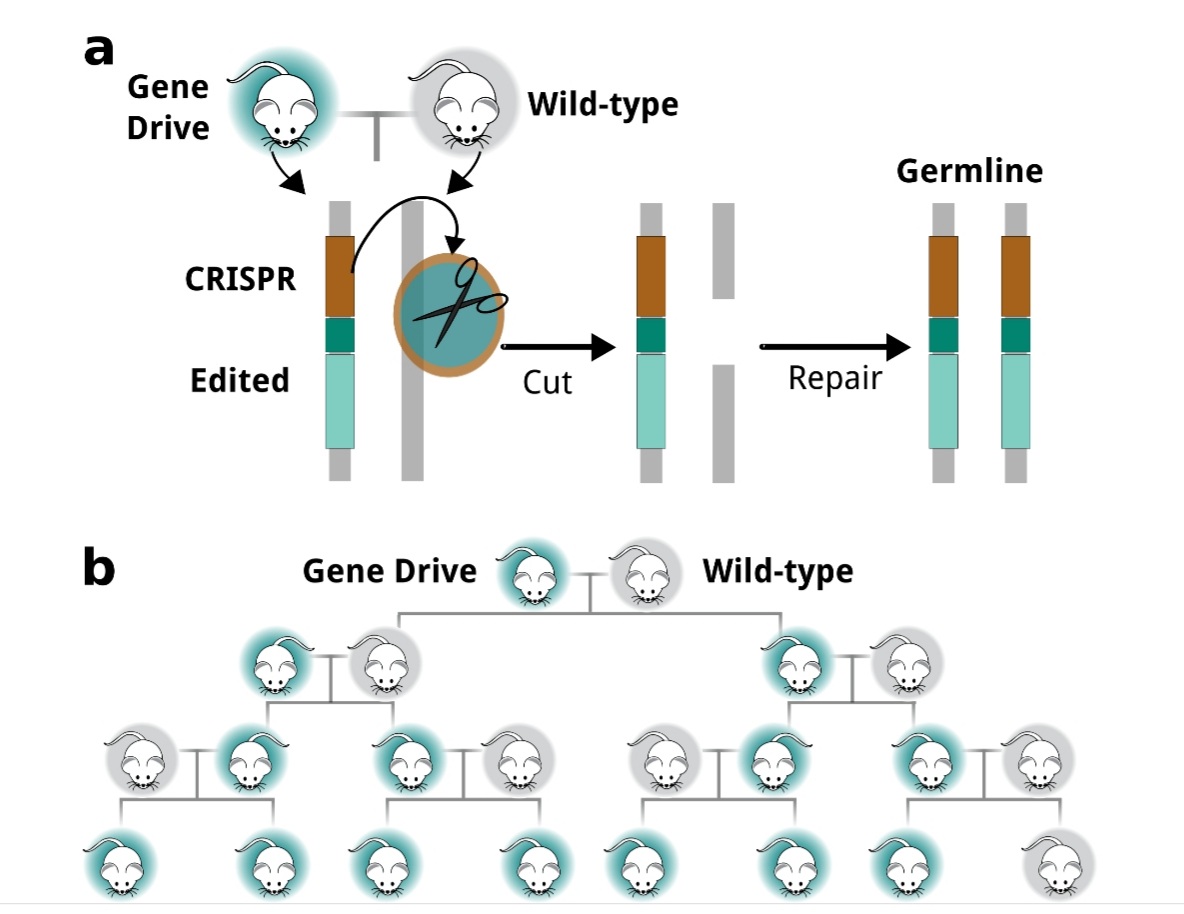
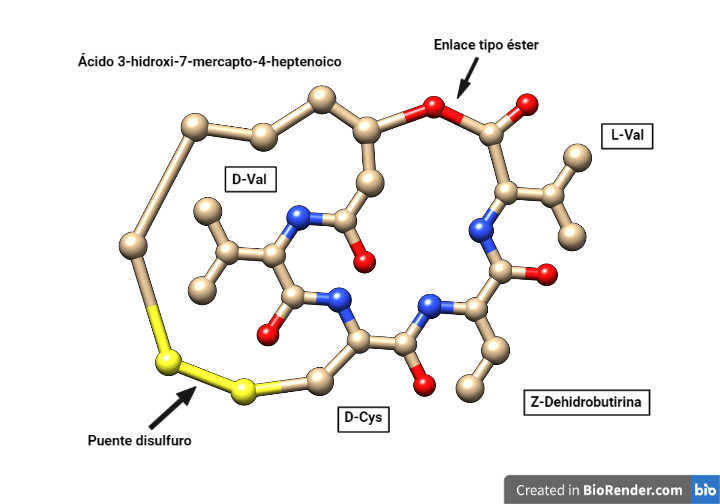
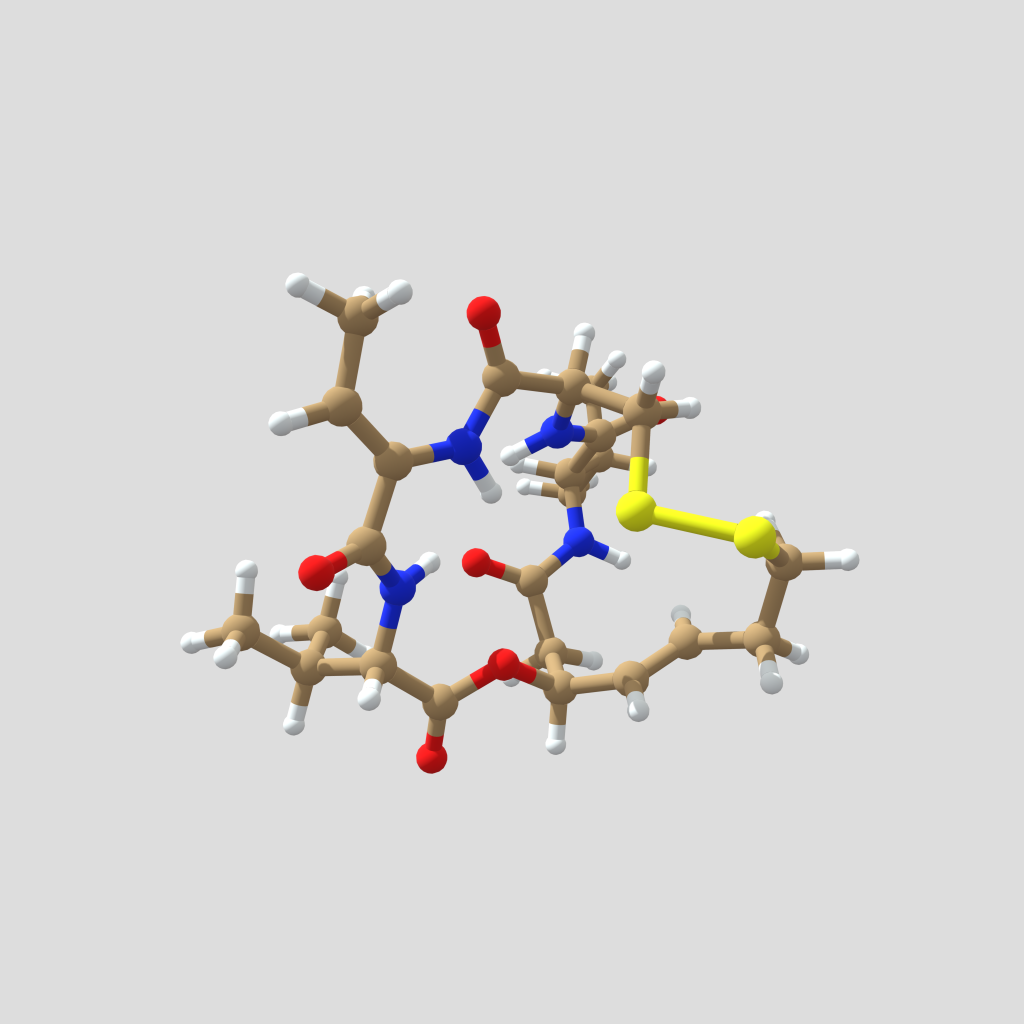
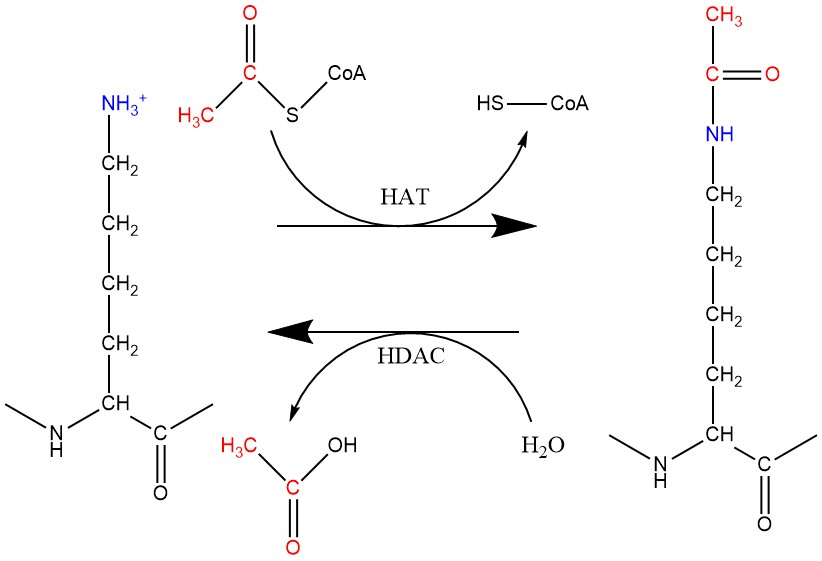
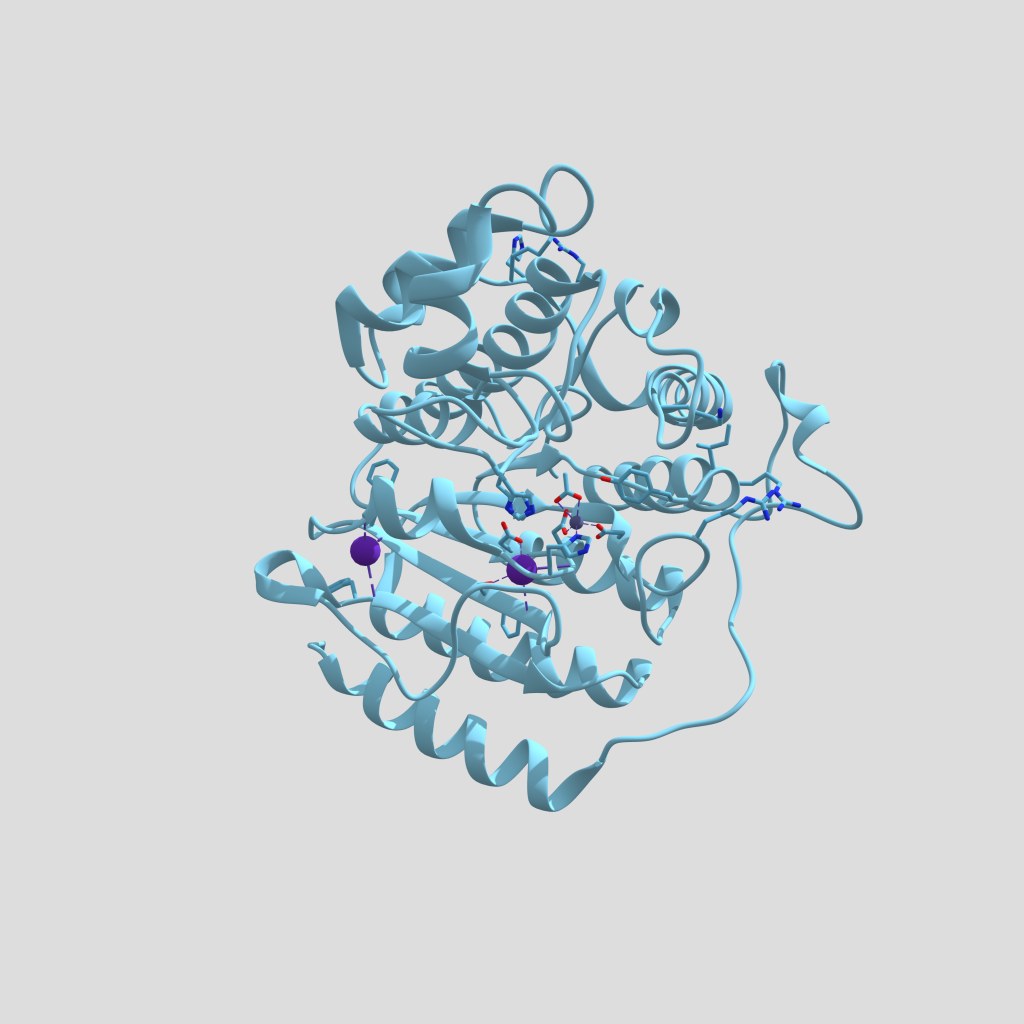

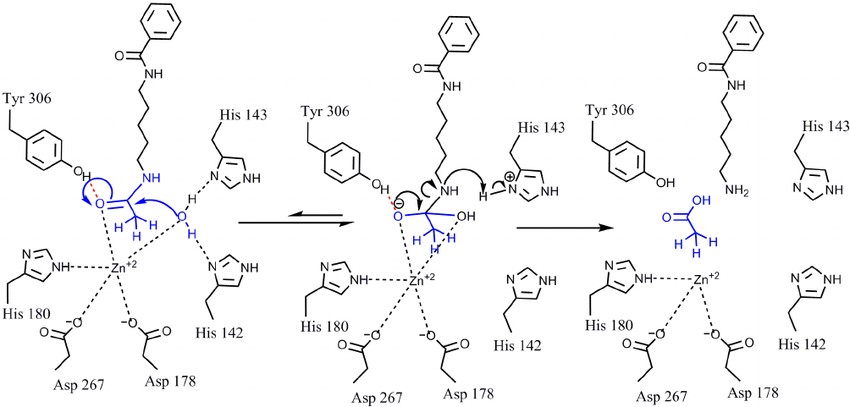
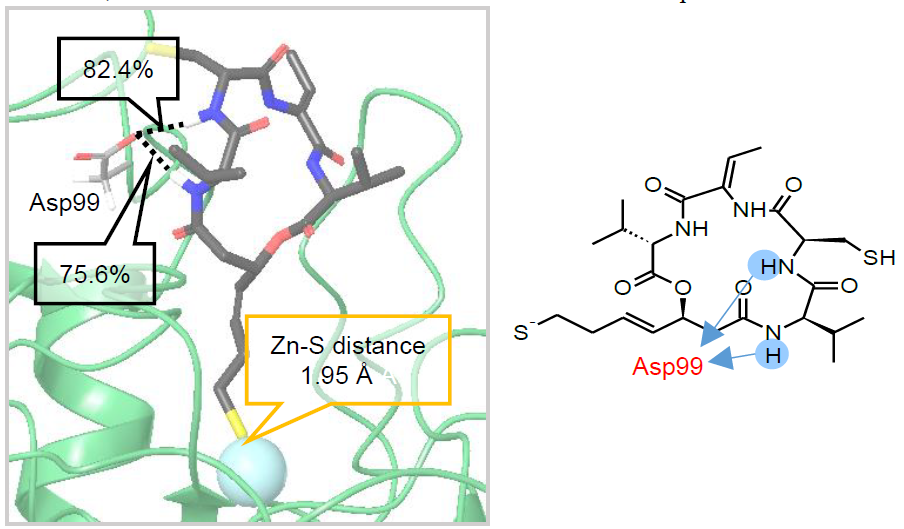
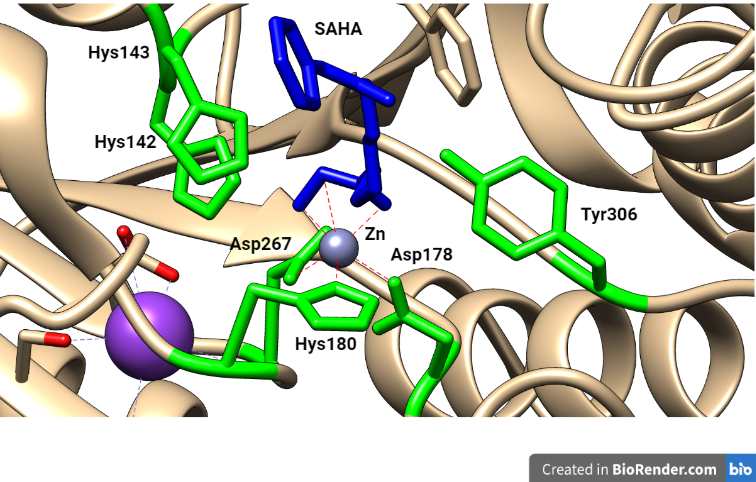
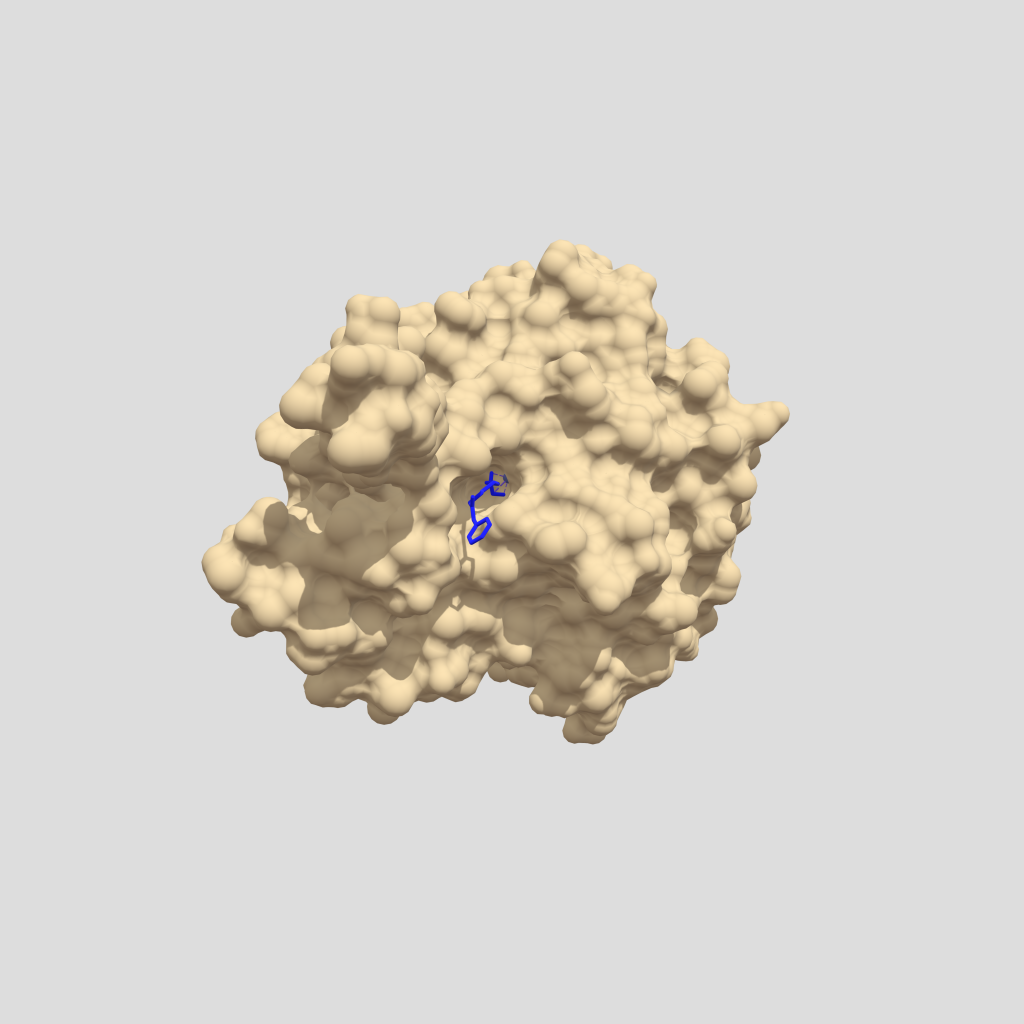
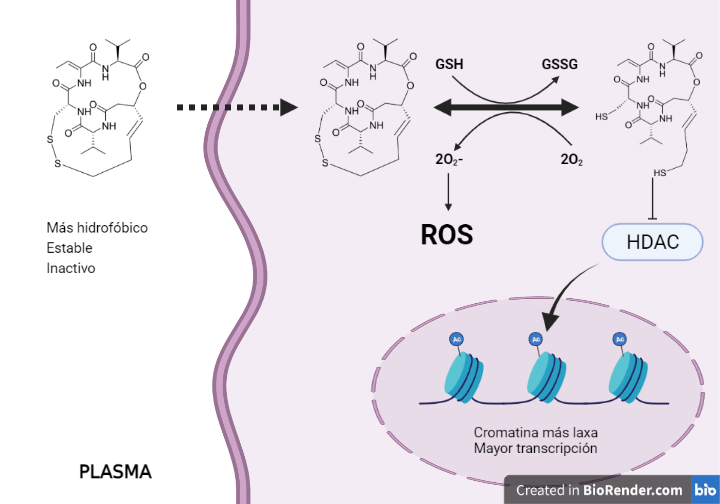
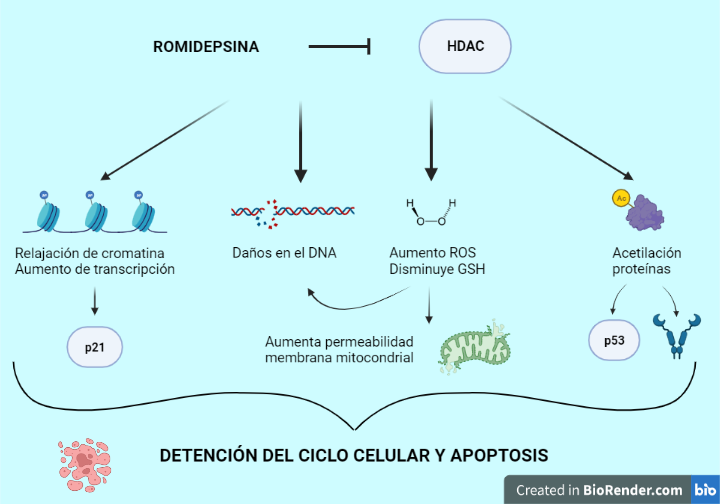


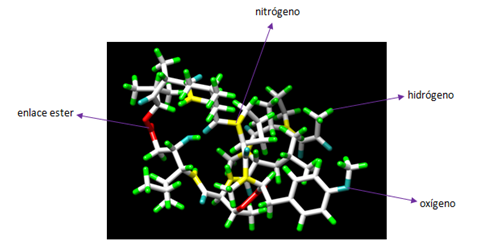
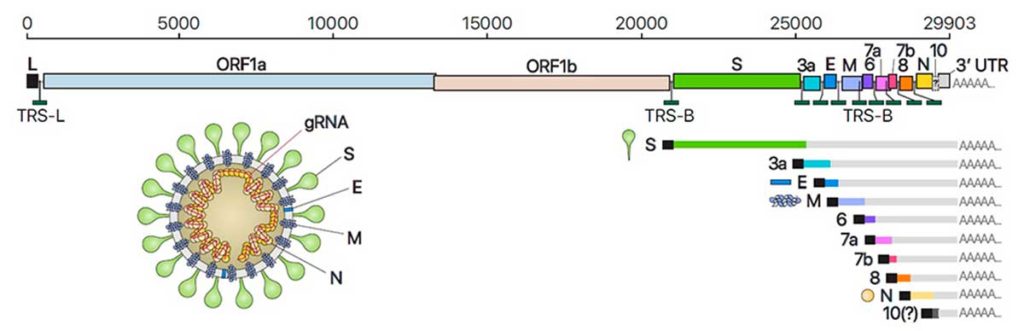
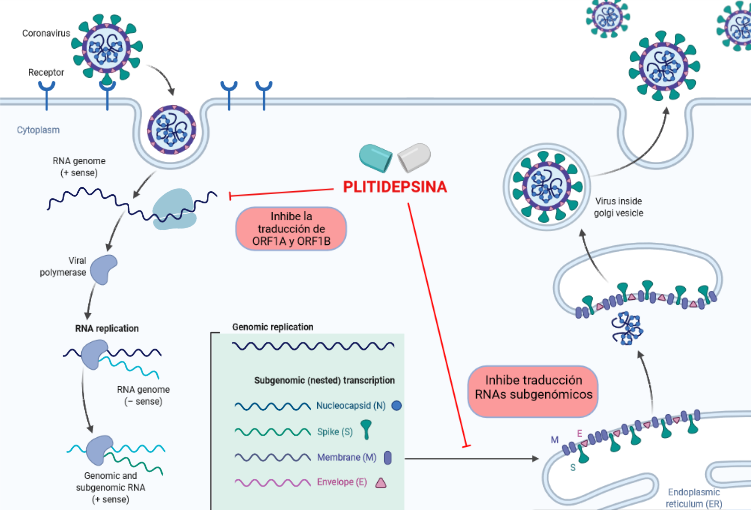
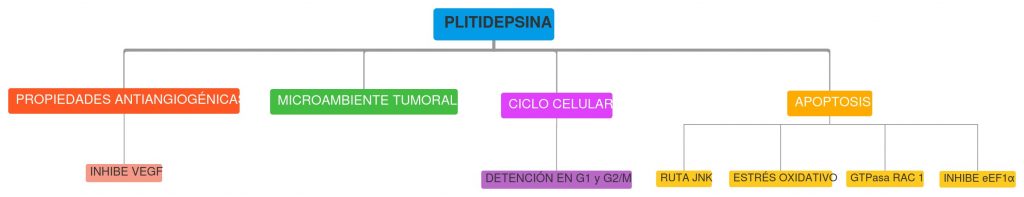

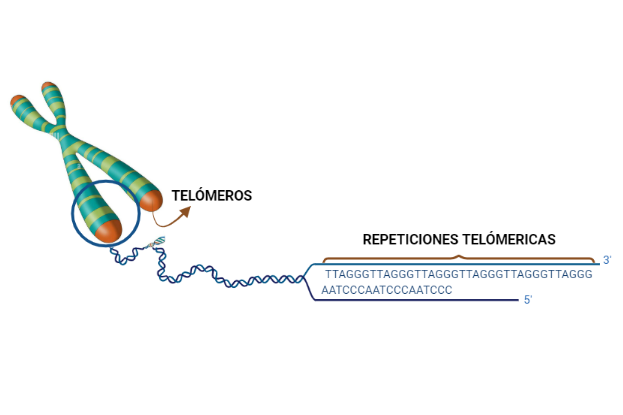
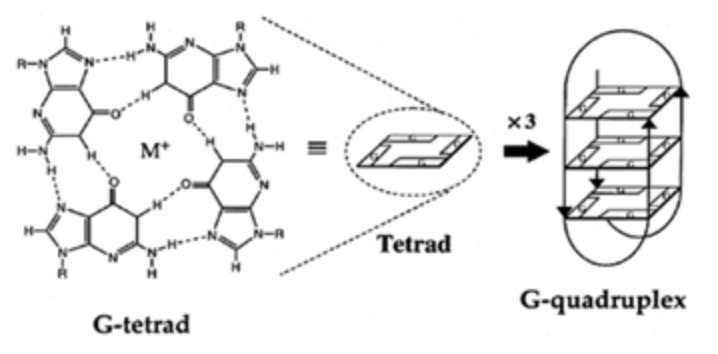
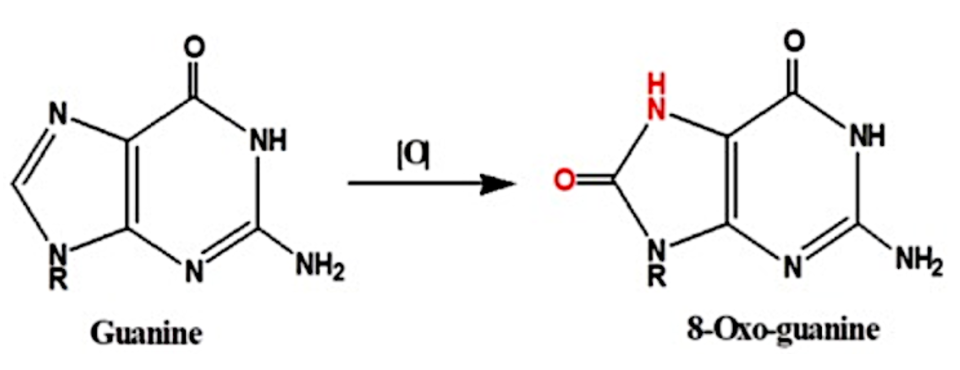
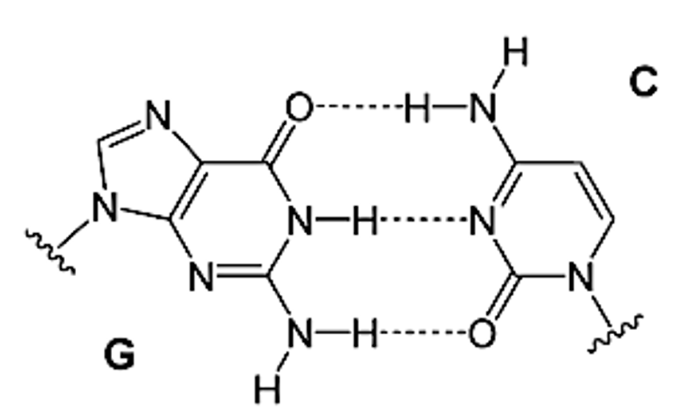
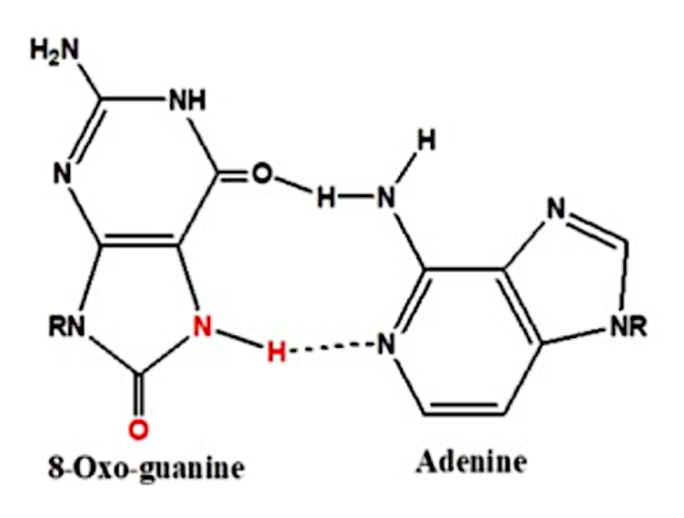
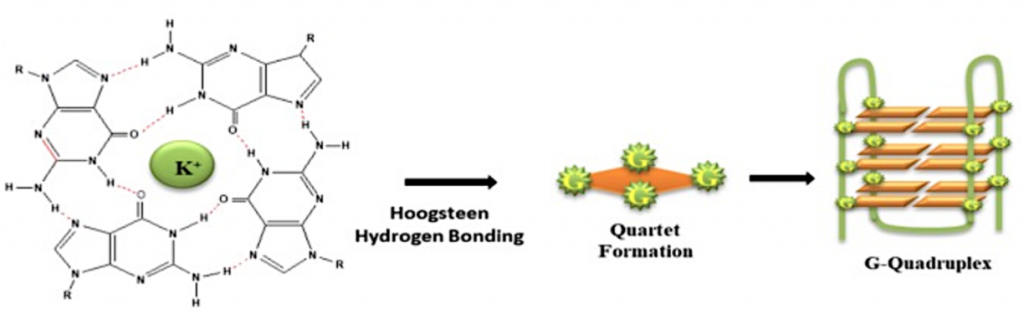
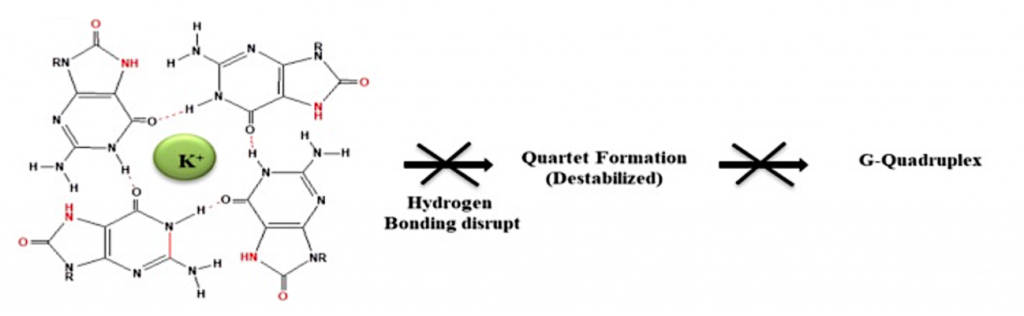
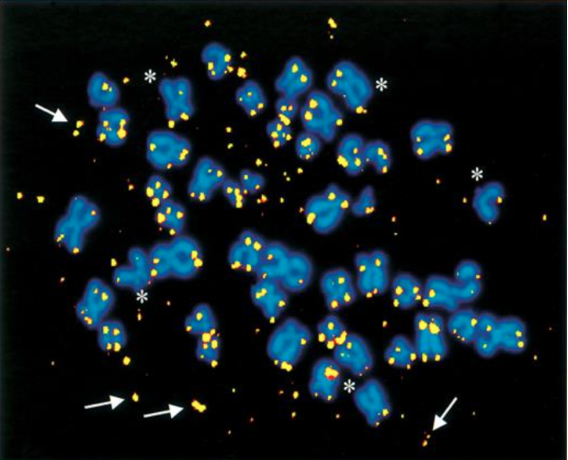



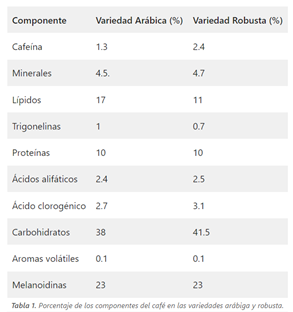
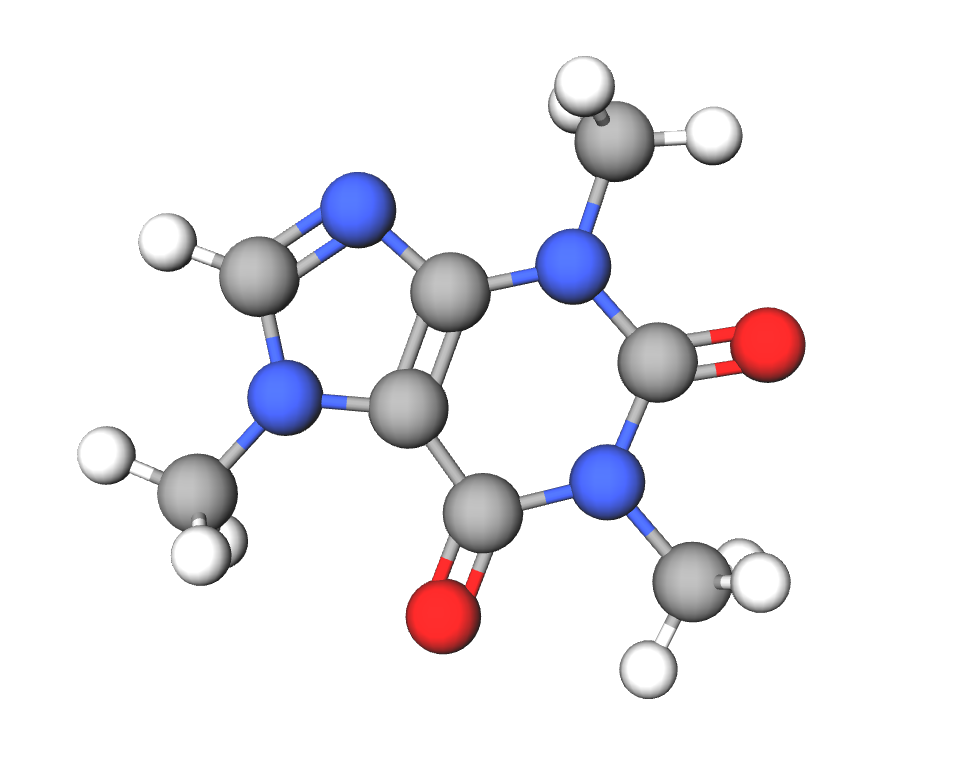
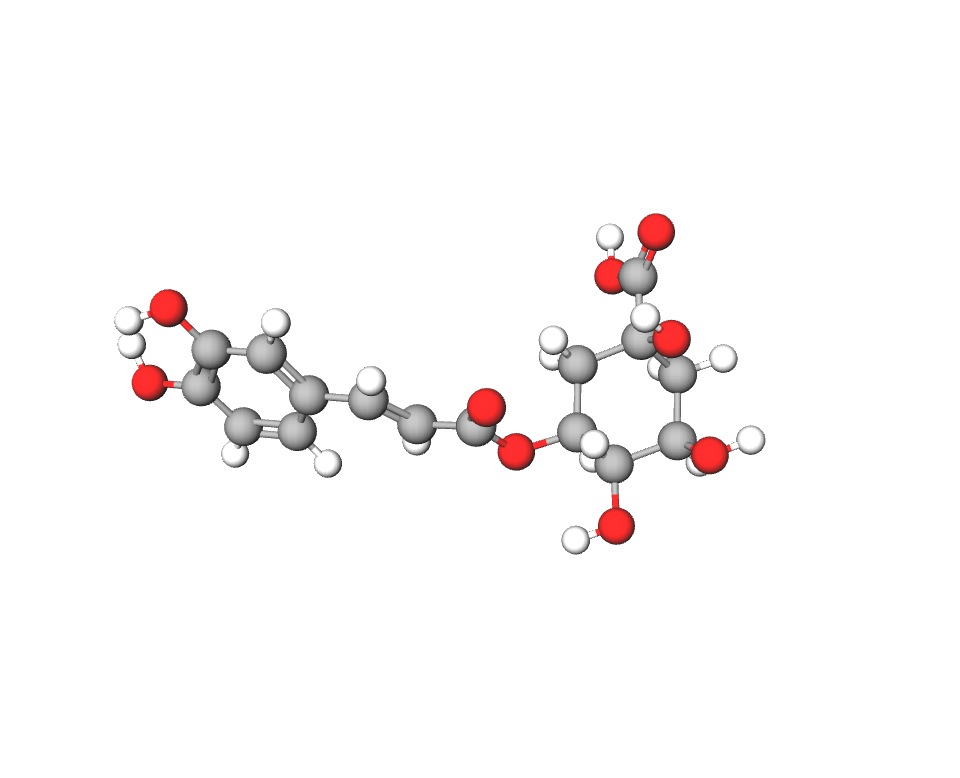
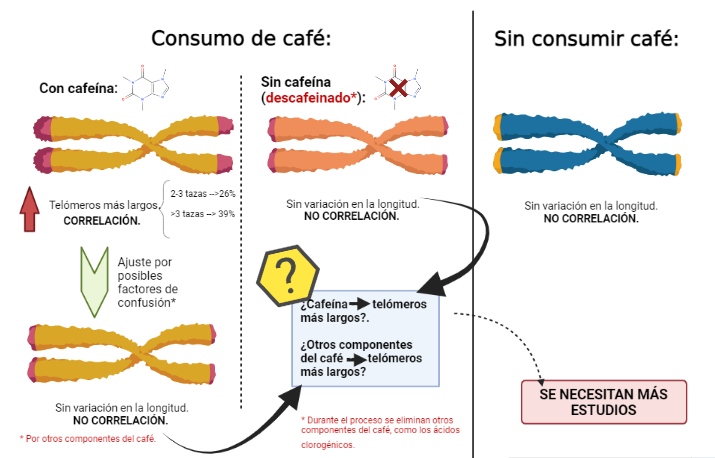
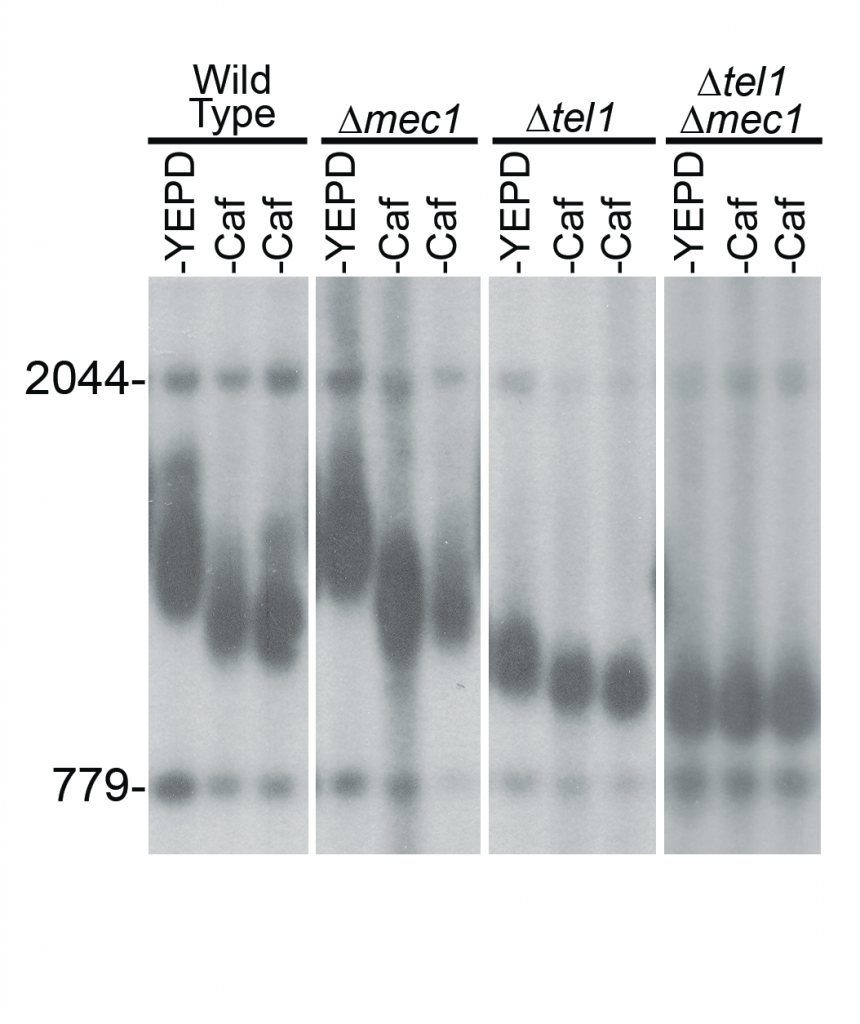
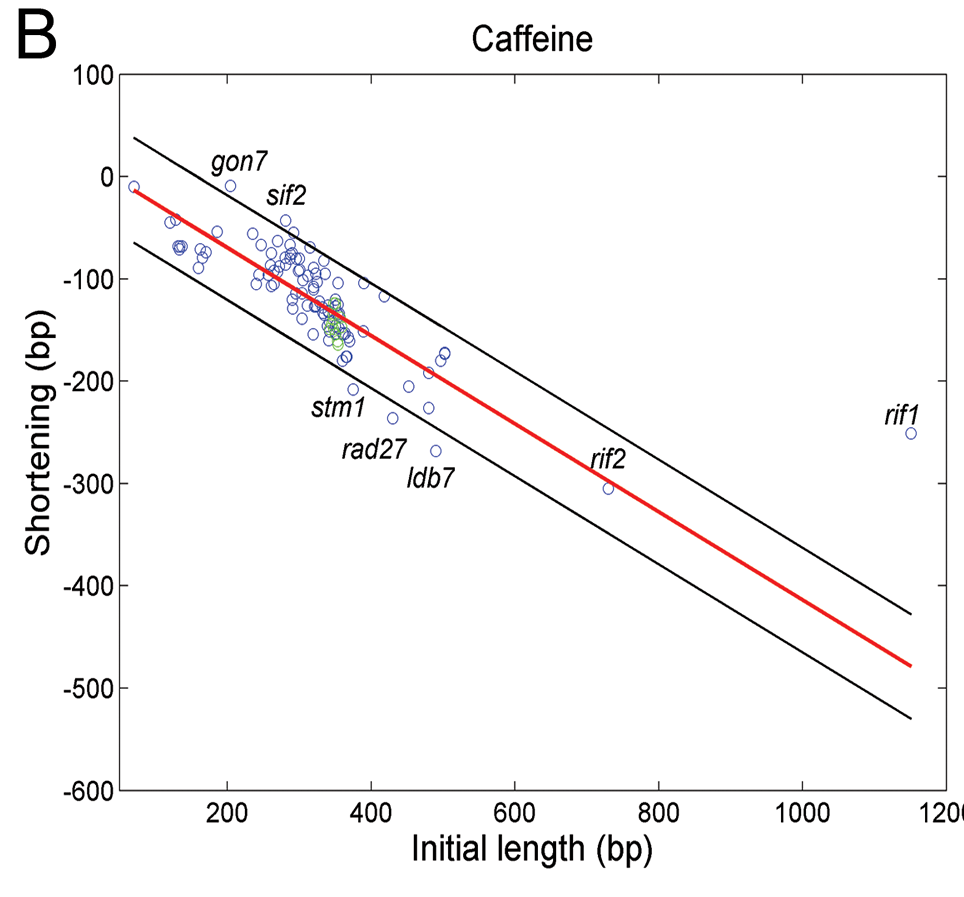
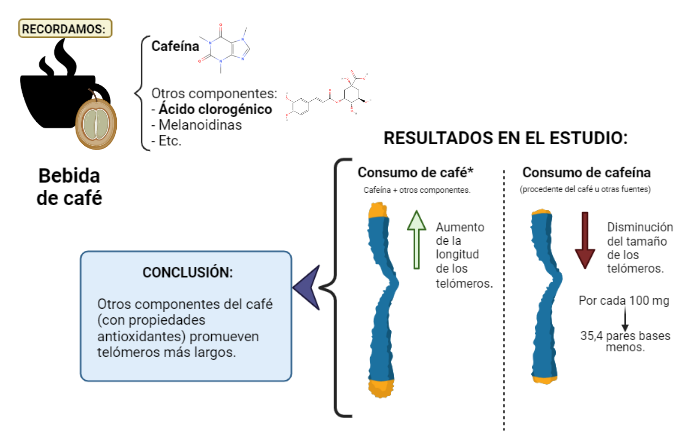
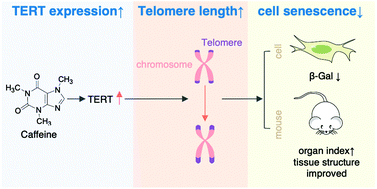














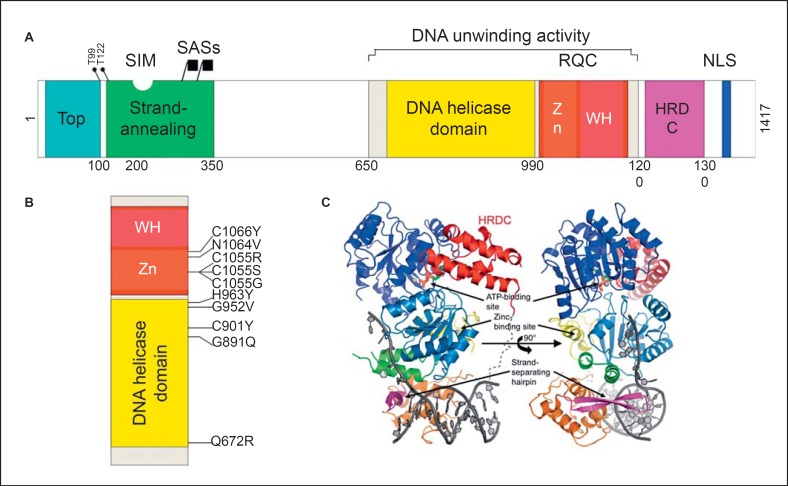
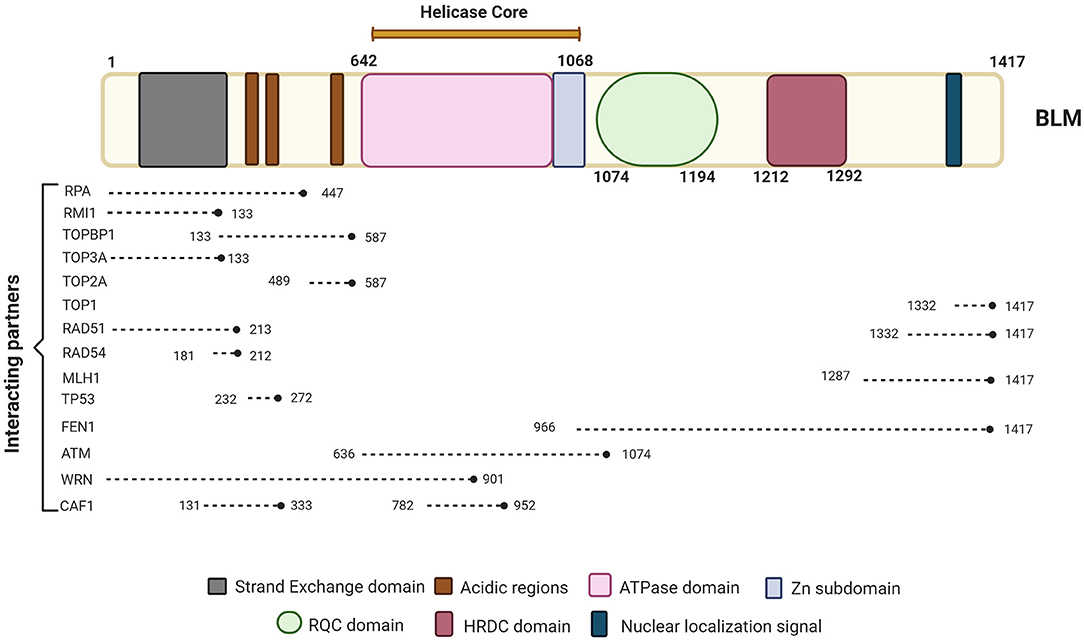
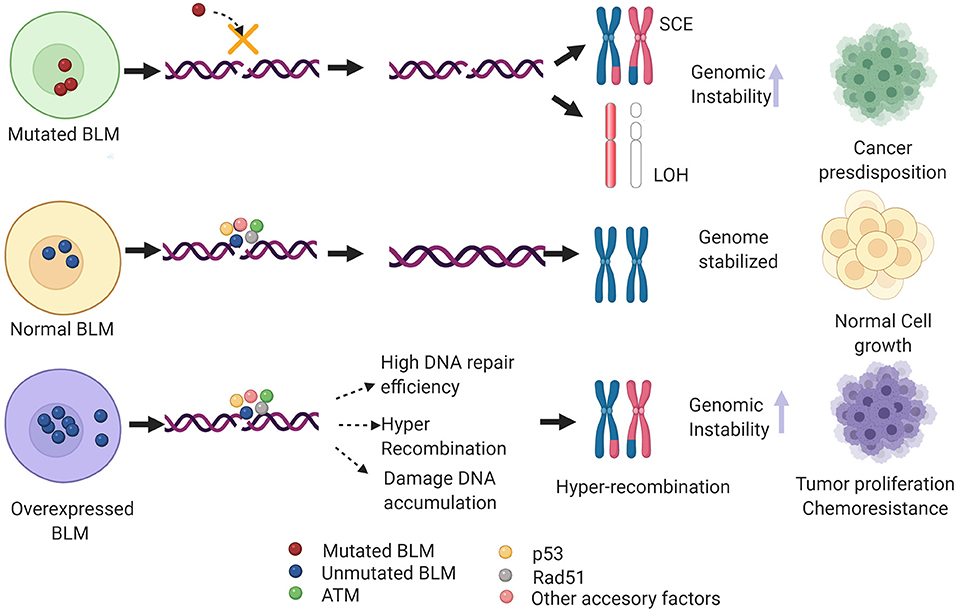



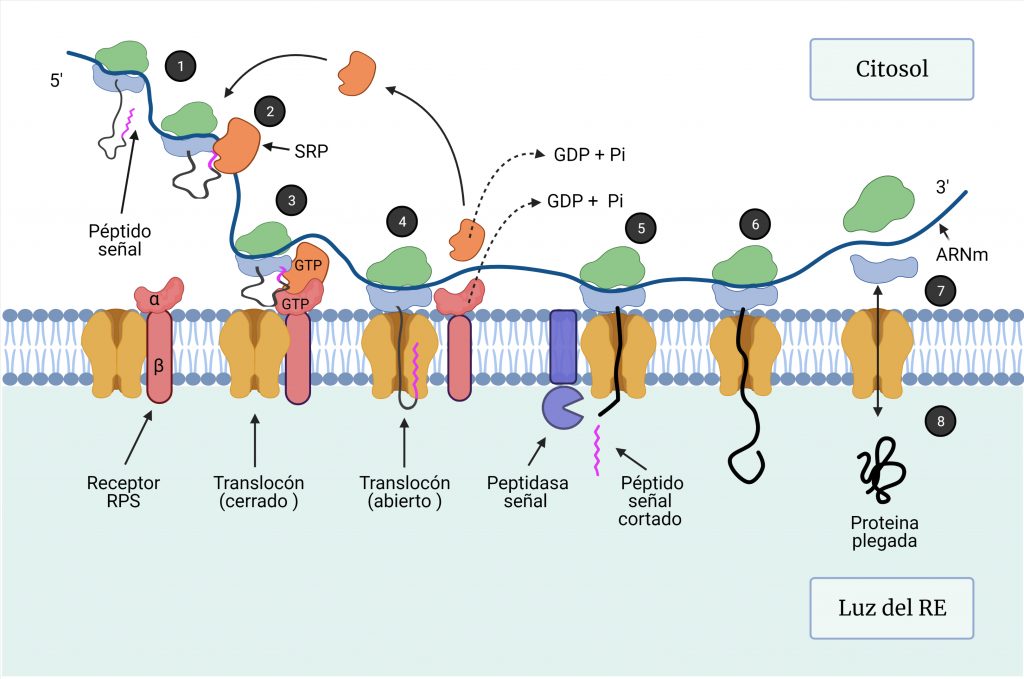
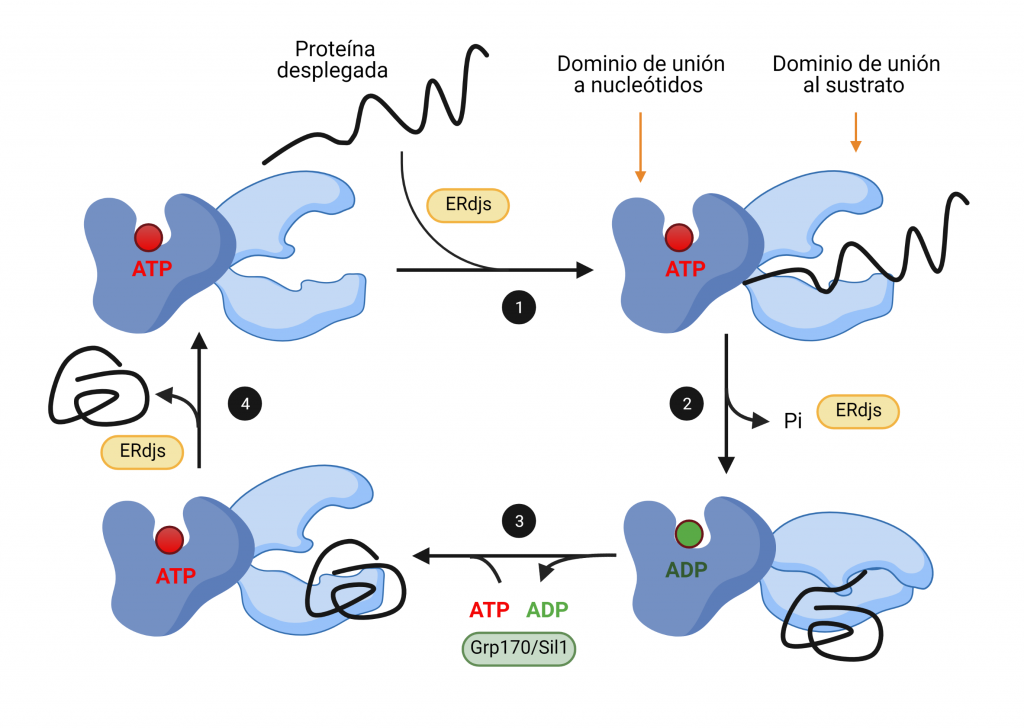
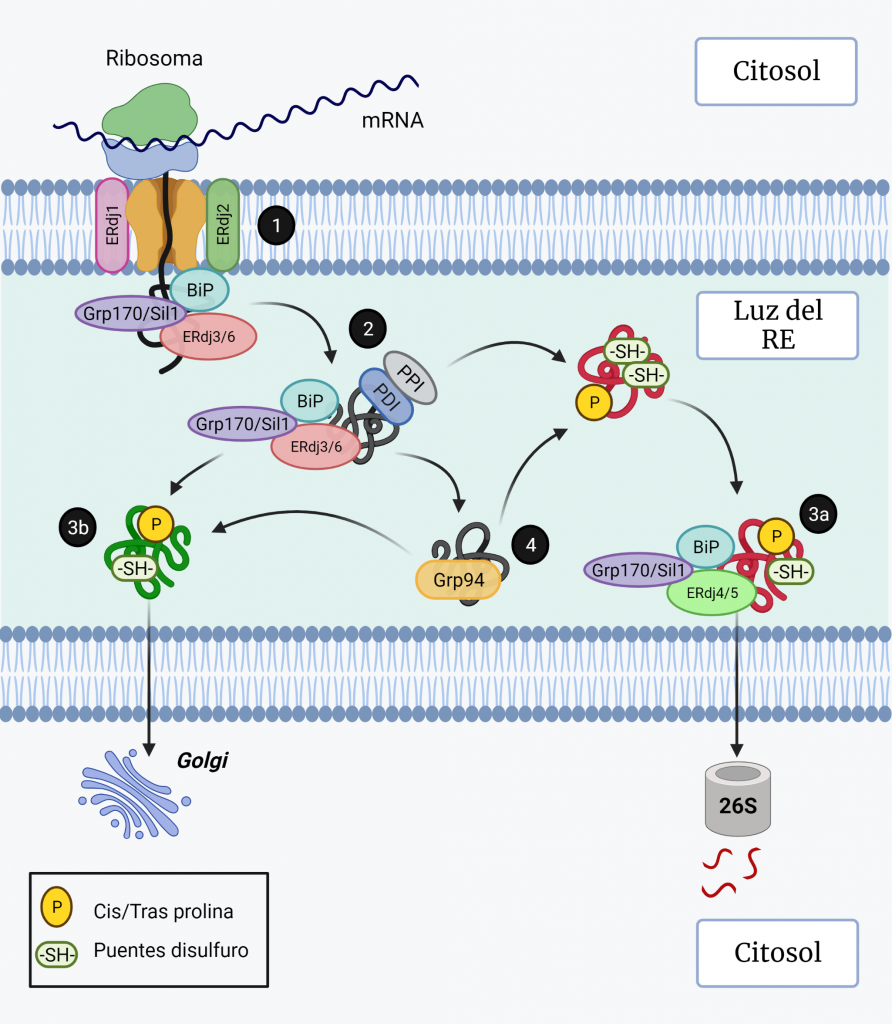
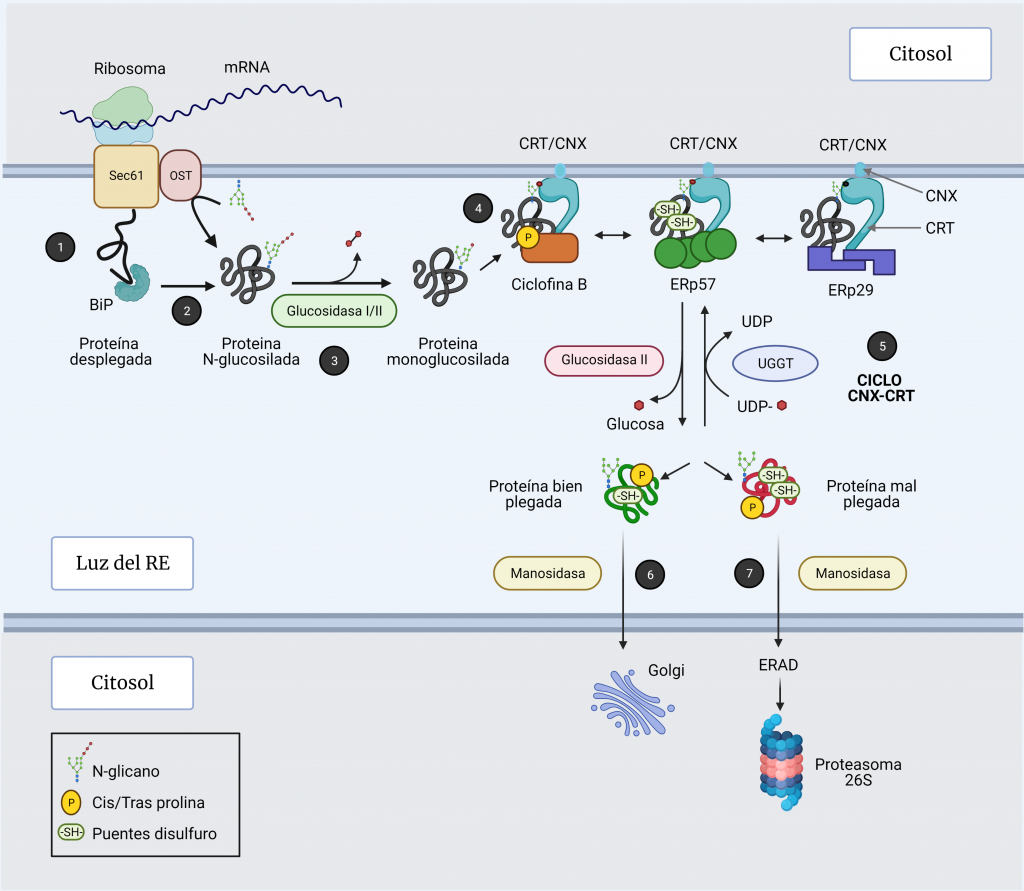
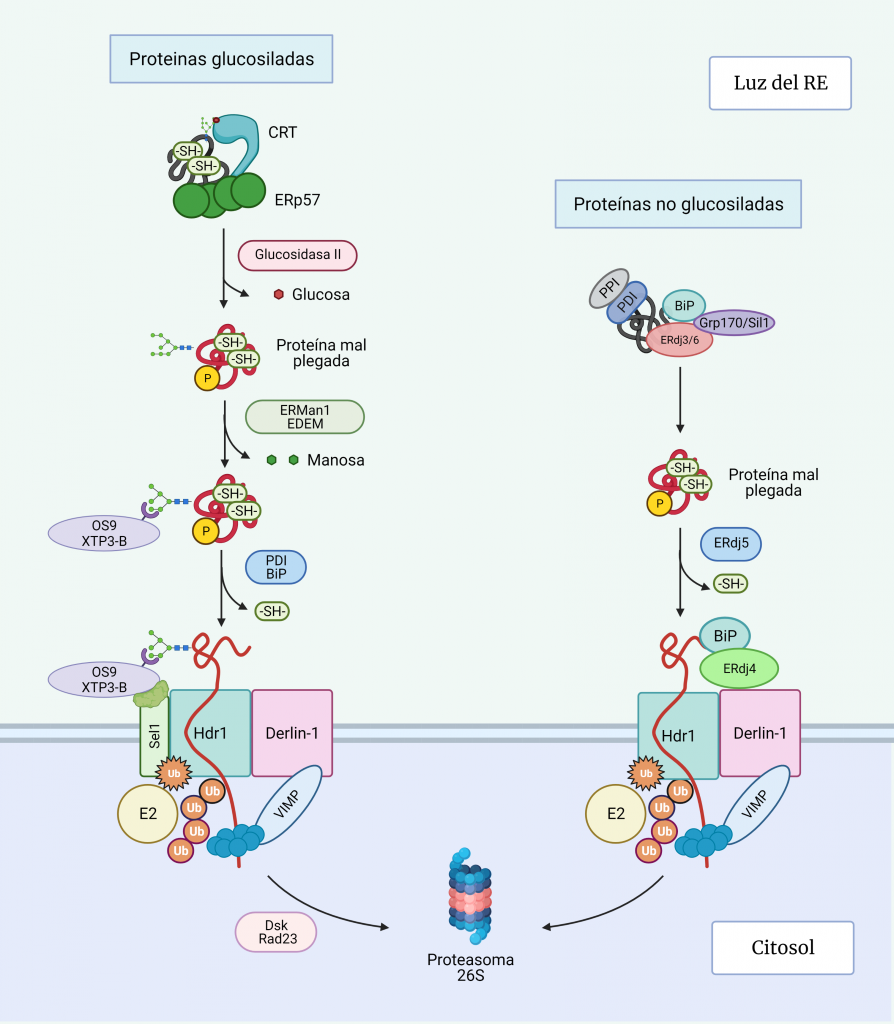
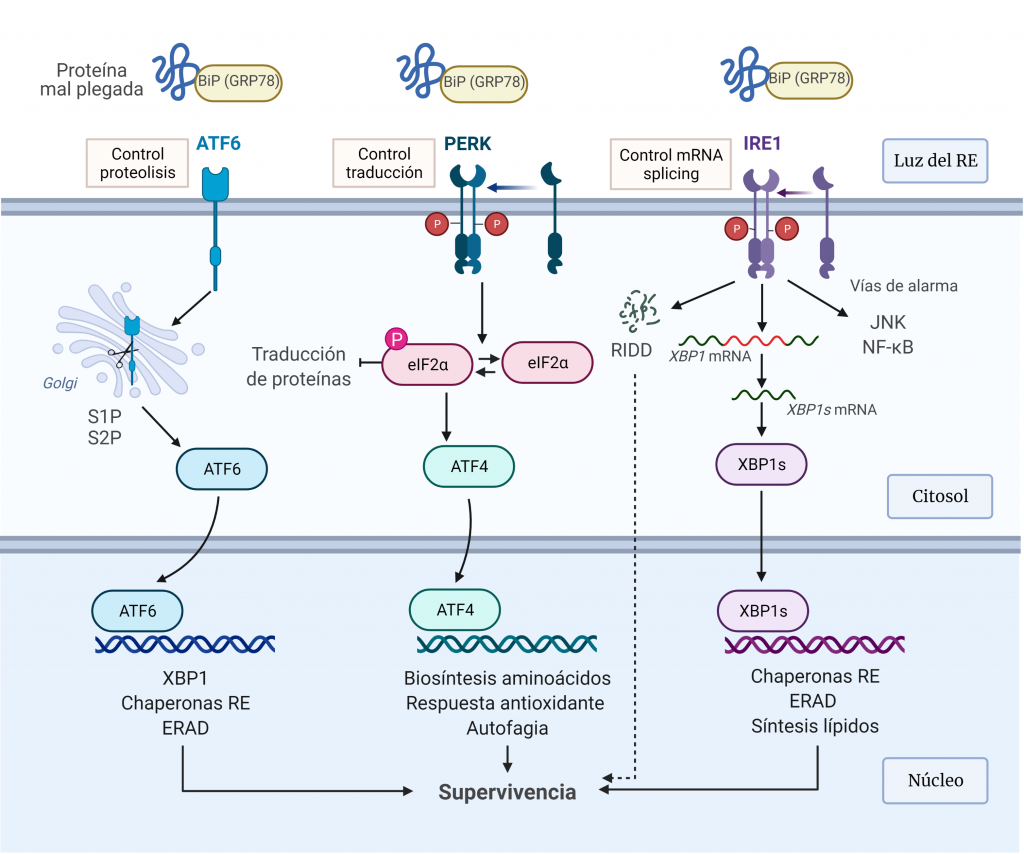
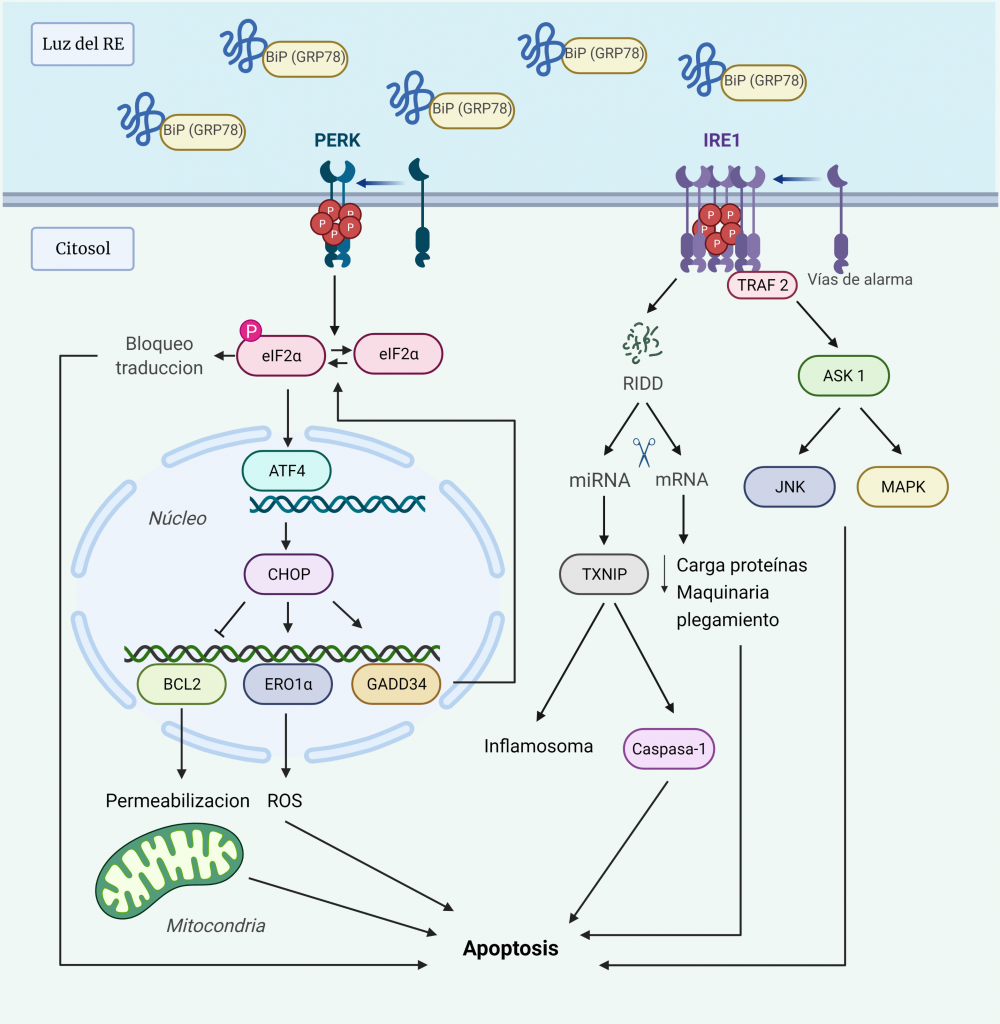
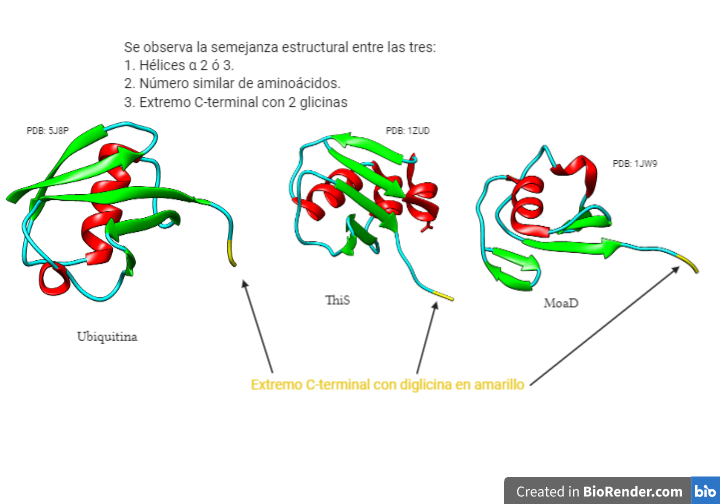
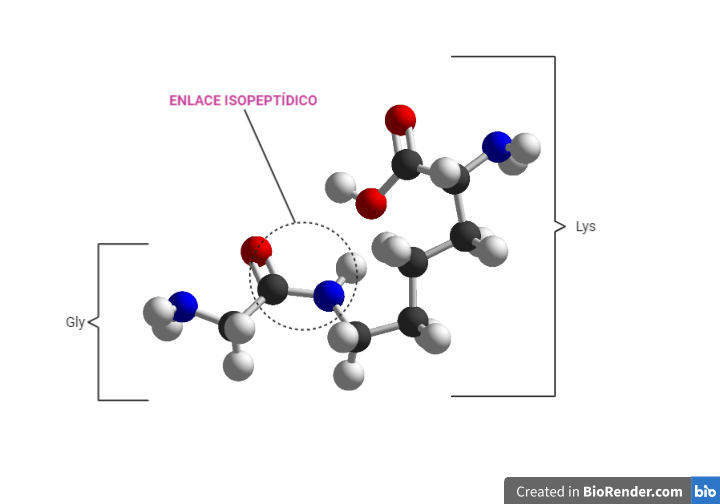
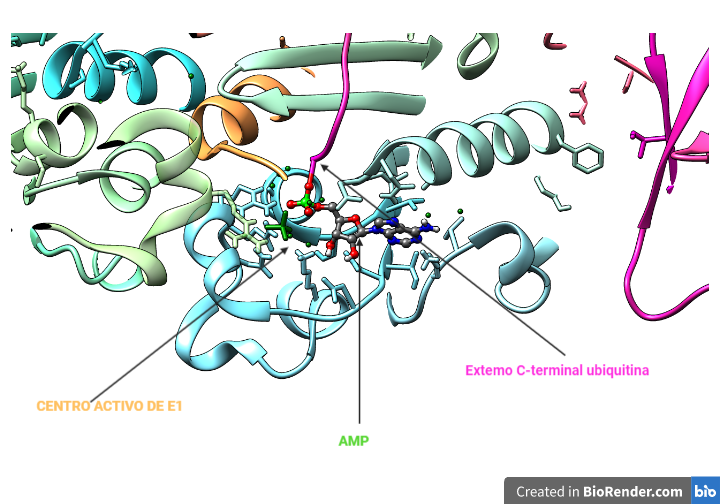
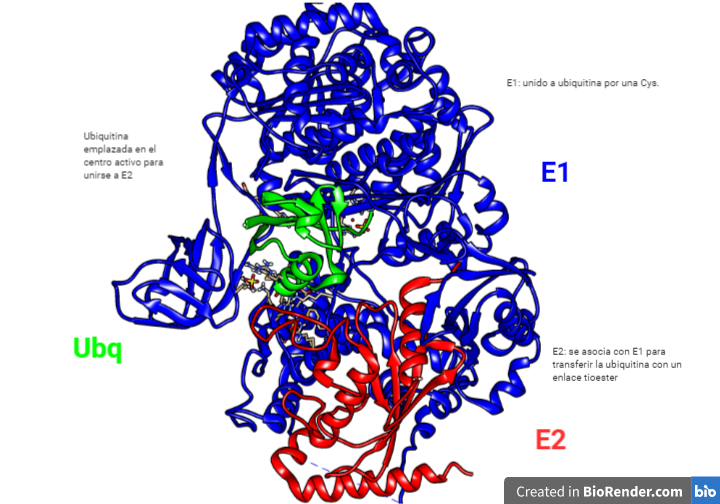
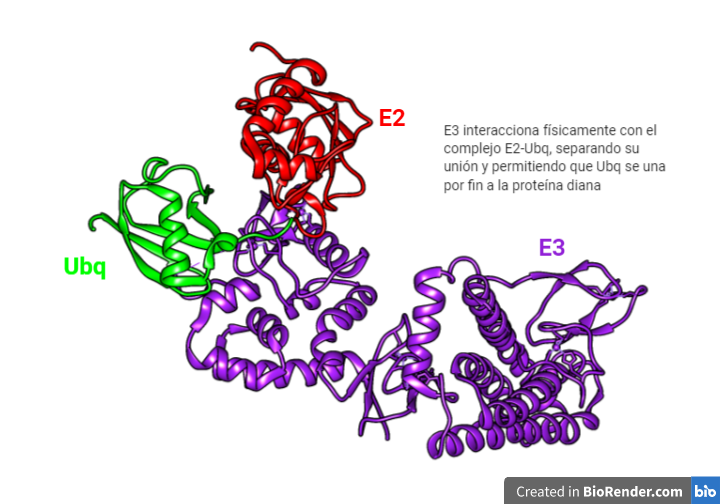
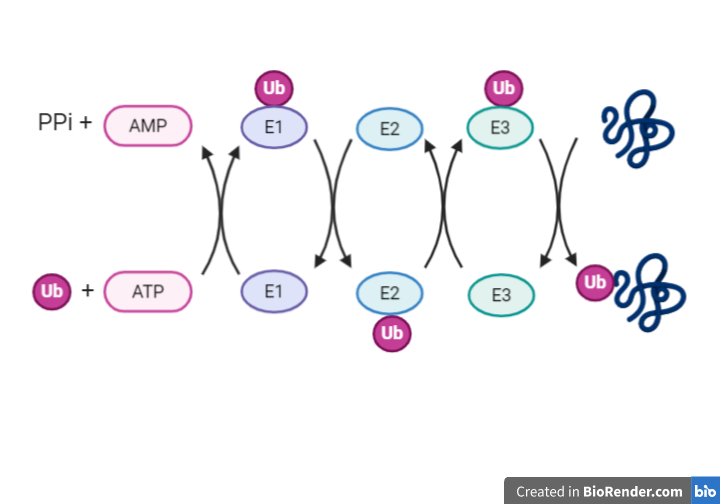
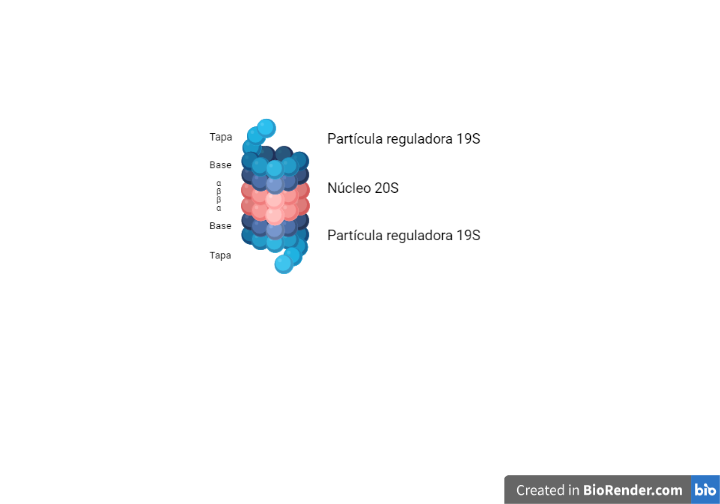
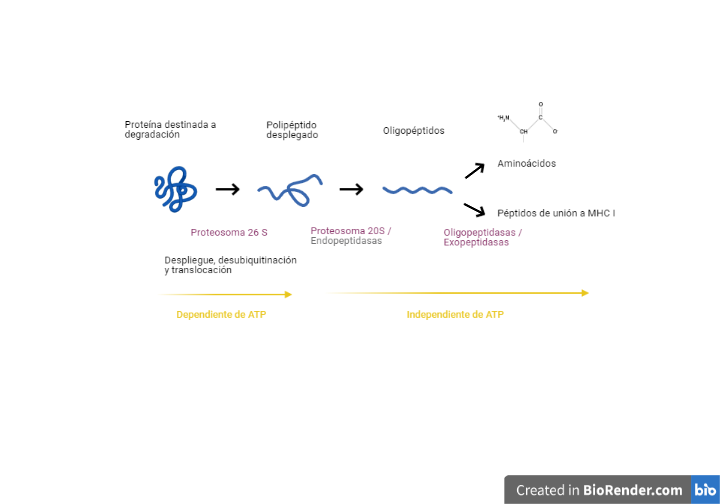
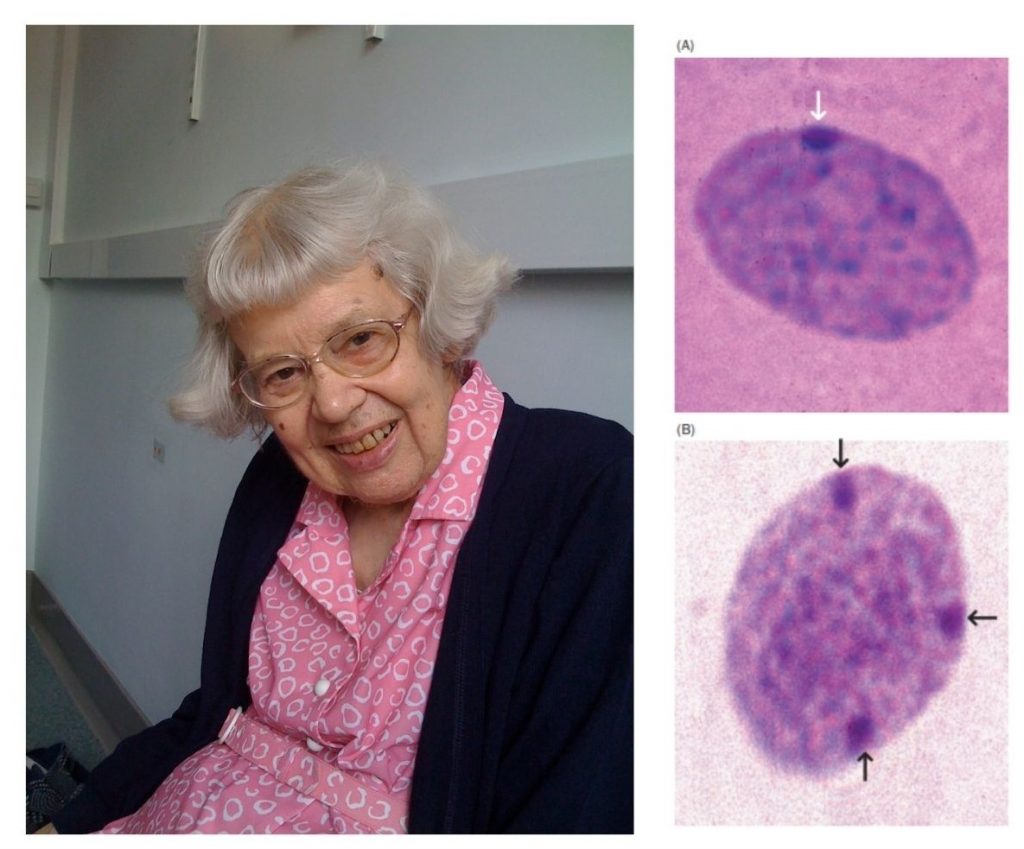
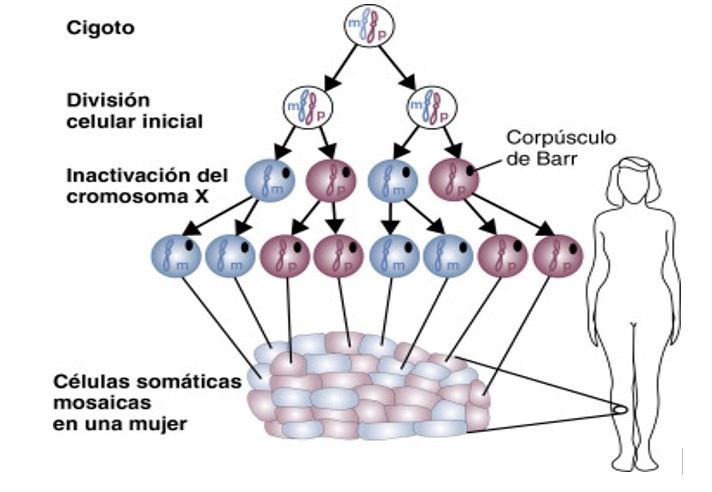
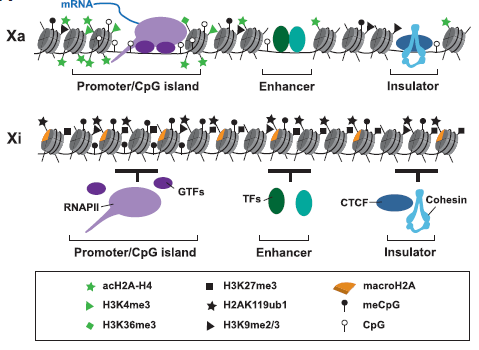
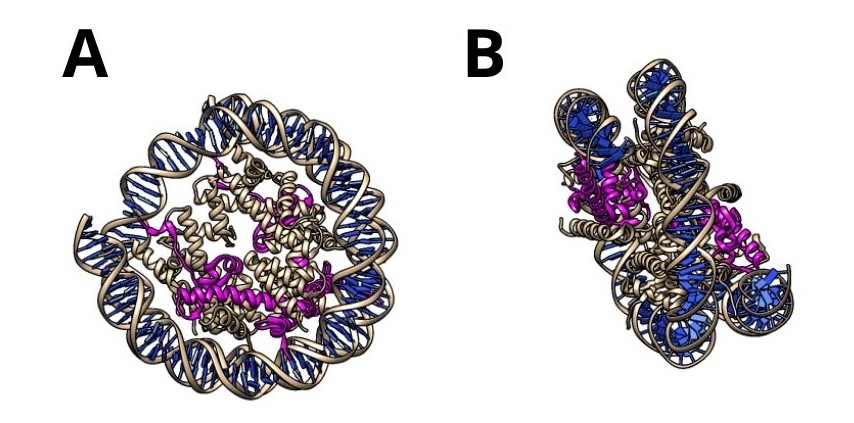
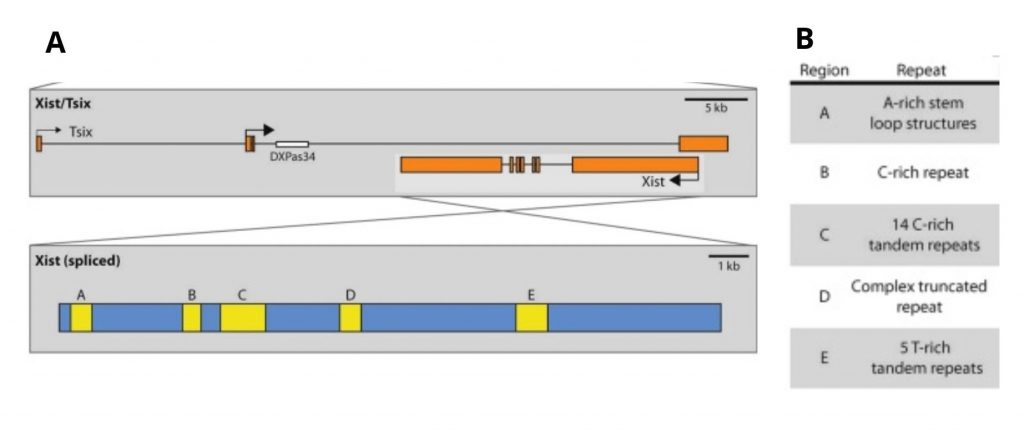
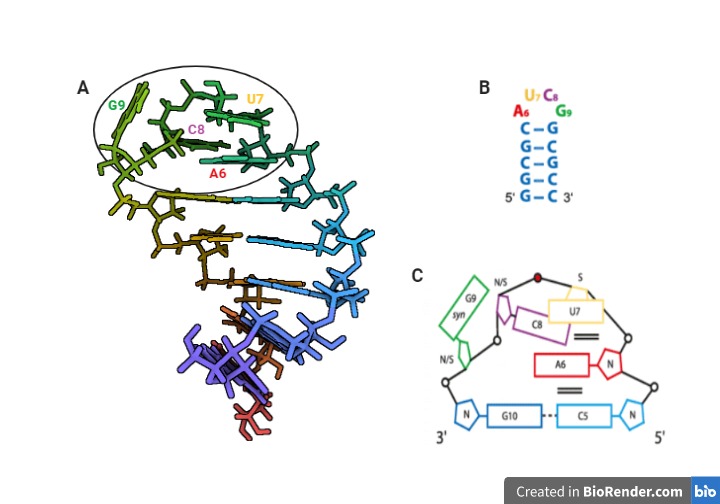
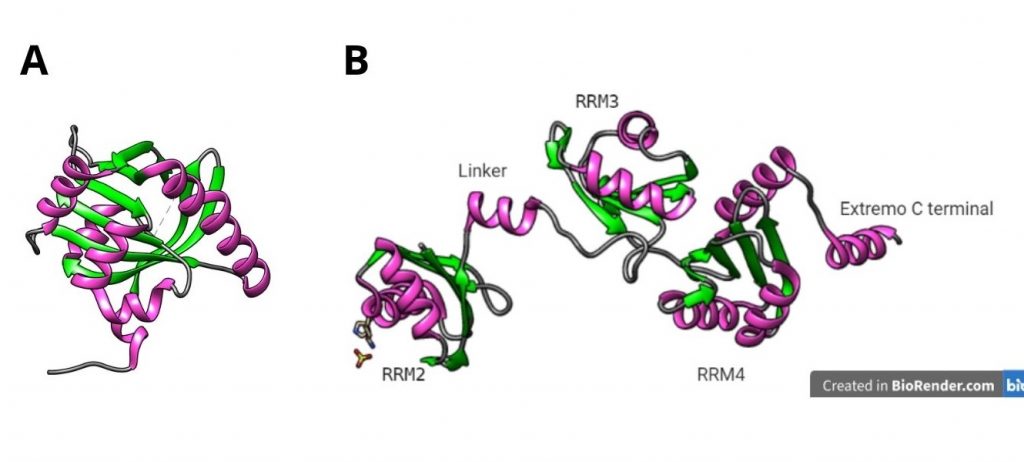
{kind=link}
{kind=link}
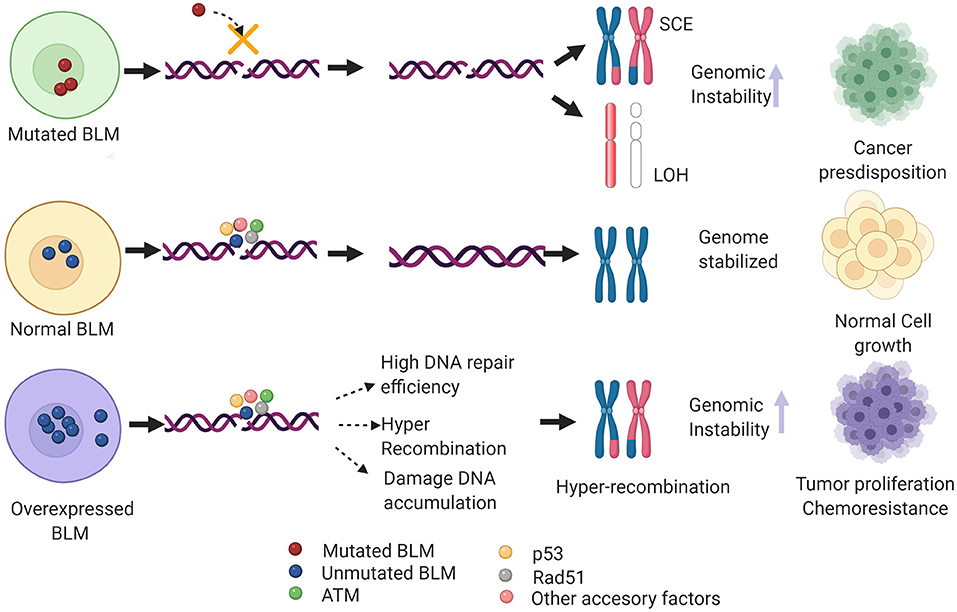{kind=link}