RETROTRANSCRIPCIÓN Y RETROVIRUS
Por Miguel Ángel Lendínez y Celia Izurrategui. Biología Sanitaria. Universidad de Alcalá de Henares
Introducción
Retrovirus. Esa palabra es una de las muchas que se nos vienen a la cabeza a los científicos y a los lectores habituales de ciencia cuando toca hablar del síndrome de inmunodeficiencia humana, abreviado como SIDA. Lo cierto es que mucha gente no sabe qué es lo que desencadena exactamente esta enfermedad, y lo que es más, el desconocimiento general de la población de otros organismos de la misma familia que el VIH los hace realmente peligrosos. El objetivo de nuestra entrada será explicar brevemente qué son y cómo funcionan los retrovirus, pero sobre todo y más importante, qué podemos hacer para frenar el avance de estos asesinos invisibles.
¿Qué es un retrovirus?
Lo primero de todo es conocer a nuestro enemigo. Los organismos normalmente codificamos nuestra información genética mediante el ADN porque es una molécula muy estable. Sin embargo, otros seres como los virus especialmente la codifican en forma de ARN, lo que complica la manera de luchar contra ellos ya que su código genético es mucho más inestable. Los retrovirus (pertenecientes a la familia Retroviridae) son un tipo de virus de ARN muy especiales, ya que necesitan traducir ese ARN a ADN para replicarse dentro de la célula a la que infectan. Esta característica tan especial se debe a una enzima esencial del virus, conocida como retrotranscriptasa o transcriptasa inversa, de la que hablaremos más adelante. También debemos tener en cuenta otra familia de virus (Hepadnavirus) que poseen transcriptasa inversa pero que son virus ADN.[1]
Dentro de los virus, debemos distinguir entre envueltos y no envueltos. Los virus envueltos presentan una capa (la envuelta) que procede de las membranas de las células o de un órganulo llamado aparato de Golgi que se encuentra en el interior de las mismas. Los retrovirus pertenecen al segundo grupo, ya que al momento de salir de la célula infectada se rodea de la membrana de la misma, además de llevarse consigo algunas de las proteínas del Aparato de Golgi. [2]
Diferencias aparte, todos los virus poseen una cápside formada por proteínas que rodea al material genético del virus. Por suerte, en este caso los retrovirus no son una excepción. Aún así, por encima de esta capa los retrovirus poseen una matriz proteica.
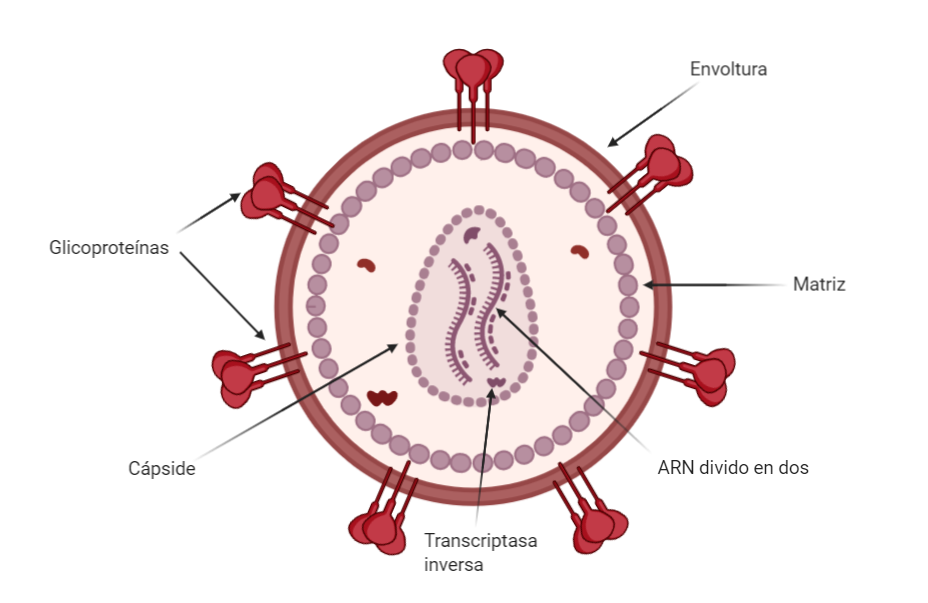
Retrovirus principales
Dentro de la familia de los retrovirus encontramos varios géneros que afectan diversas especies, como por ejemplo virus que producen leucemia y tumores mamarios en ratones u otras enfermedades en diferentes aves y mamíferos. Los que afectan al ser humano pertenecen a dos géneros en concreto: Deltaretrovirus y Lentivirus.
Dentro de los Deltalentivirus encontramos HTLV-I y HTLV-II, virus linfotrópicos (con afinidad por linfocitos) de las células T humanas, que son causantes de leucemias de estas células. El HTLV fue el primer retrovirus humano infeccioso y oncogénico descubierto.
El descubrimiento de este virus comenzó en 1978 cuando un paciente de 28 años fue erróneamente diagnosticado con linfoma cutáneo de células T o micosis fungoide. Tras cultivar células T de una biopsia de nódulos linfáticos y ser sometidas a los estudios estándar de la época se descubrió un retrovirus no descrito anteriormente. Se cultivaron los viriones y cuando estaban parcialmente purificados se realizó la caracterización bioquímica y la de la retrotranscriptasa asociada a estos. Tras esta caracterización se realizaron numerosos estudios que ayudaron a demostrar la naturaleza única del HTLV. [3]
En el género Lentivirus encontramos los virus de la inmunodeficiencia humana, VIH-I y VIH-2, conocidos por producir el síndrome de inmunodeficiencia adquirida o SIDA.
Los primeros casos del SIDA fueron detectados en Nueva York y Los Angeles en 1981. Se observó en pacientes jóvenes, sobre todo homosexuales, previamente sanos el desarrollo de infecciones oportunistas como neumonía, infecciones de mucosas por Candida albicans y la aparición de sarcoma de Kaposi. Algunos pacientes presentaban linfadenopatía generalizada precediendo el desarrollo de estas manifestaciones infecciosas. Se asociaron estas manifestaciones con una inmunodeficiencia celular adquirida no descrita hasta ese momento. Estamos hablando del inicio de la epidemia. Poco tiempo después se describieron otros grupos de riesgo que incluían pacientes con hemofilia, usuarios de drogas intravenosas, haitianos y receptores de hemoderivados. La evidencia epidemiológica apuntaba hacia un agente infeccioso transmisible por vía sexual o sanguínea.
A pesar de estos importantes antecedentes y de que Gallo sospechaba desde un inicio que el agente causal de esta nueva enfermedad era un retrovirus, tuvo dificultades para aislar, en un principio, el virus causante del SIDA y otro grupo de investigadores se adelantaría. En 1985 se llevó a cabo la clonación y secuenciación del genoma del virus, y una caracterización precisa de las proteínas de su envoltura.[4]

Enfermedades que provocan y sintomatología
Vamos a comenzar con el VIH. Dentro de los primeros días de la adquisición del VIH ocurre una enfermedad transitoria, en ocasiones sintomática asociada a altos niveles de replicación del VIH y a una rápida caída de los linfocitos T CD4 (también llamados linfocitos T helpers, que se encargan de ayudar en la respuesta inmune). Inmediatamente después de la exposición y transmisión, el virus se replica en la mucosa y en la submucosa, que son la parte más externa del tejido infectado (ya sea el tejido vaginal, rectal, uretral… etc) y drena hacia el tejido linforeticular, donde se encuentran muchos linfocitos, y ya no puede ser detectado en sangre. Esta fase es denominada “fase de eclipse” y dura entre 7 a 21 días. Se comienza a considerar infección aguda cuando encontramos una gran cantidad del RNA del virus en el plasma sanguíneo.
La respuesta inicial es inespecífica, y llevada a cabo por anticuerpos no neutralizantes y no selectivos. Los anticuerpos que neutralizan el virus son encontrados después de tres meses de la infección aguda. Tanto los anticuerpos específicos como inespecíficos atacan al virus destruyendo diferentes glicoproteínas de la envoltura. [5]
Debemos diferenciar entre 2 tipos de pacientes: sintomáticos y asintomáticos. Los primeros presentan un cuadro clínico similar a la mononucleosis que consisten en fiebre elevada, malestar general y fatiga. Esto suele estar acompañado por el rash cutáneo (erupción de la piel característica de la infección con VIH). Para controlar la propagación del virus en el caso de pacientes asintomáticos, se deben realizar pruebas de detección a las personas que pertenezcan a algún grupo de riesgo de contraer el virus, por ejemplo, consumidores de droga mediante vía parenteral, prostitutas, etc. [6]
En cuanto al HTLV-I, el virus de las células-T linfotrópico humano es el primer virus identificado en causar leucemia en células adultas linfoides T (CD4+ y CD8+) y más raramente la paraparesis tropical espástica (TSP). Esta última provoca una debilidad corporal, específicamente en las piernas, incontinencia de la orina, causadas por lesiones ocasionadas en la médula espinal por la acción de citoquininas producidas por linfocitos T citotóxicos sanos que destruyen a los infectados con HTLV-I, los cuales dañan los nervios encontrados en ésta.
La Organización Mundial de la Salud según la clasificación de tumores de tejidos hematopoyéticos y linfáticos logró, en 2008, dividir la severidad de las patologías causadas por los retrovirus “transformadores” y su incidencia mundialmente, en: agudo 60%(versión temprana de la leucemia, donde los linfocitos T se infectan) , linfomatosa 20%(o linfoblástica, donde la médula ósea produce linfocitos inmaduros), crónica 15%(crecimiento anormal de linfocitos T) y latente 5%(la médula ósea no funciona normalmente, también se le llama preleucemia).[7]
Retrotranscripción
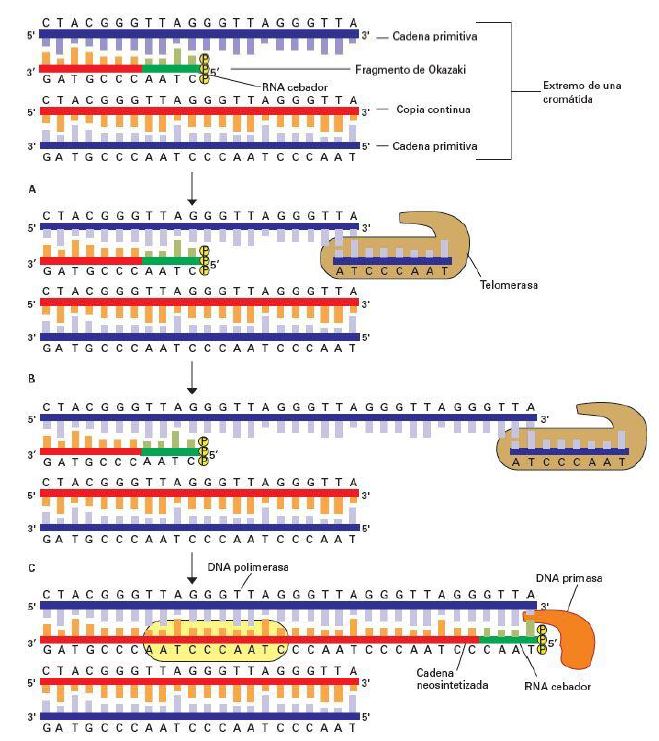
La retrotranscripción es un proceso por el que una enzima determinada hace una copia de ADN a partir ARN. La enzima más reconocida y que generalmente hace la copia de ADN se llama retrotranscriptasa y se encuentra sobre todo en retrovirus como el VIH que llevamos mencionando. Este proceso tiene la ventaja de que lo podemos realizar además en un laboratorio para llevar a cabo distintas pruebas. Vamos a pasar a describir la enzima que lleva a cabo el proceso y el proceso en sí.
Transcriptasa inversa
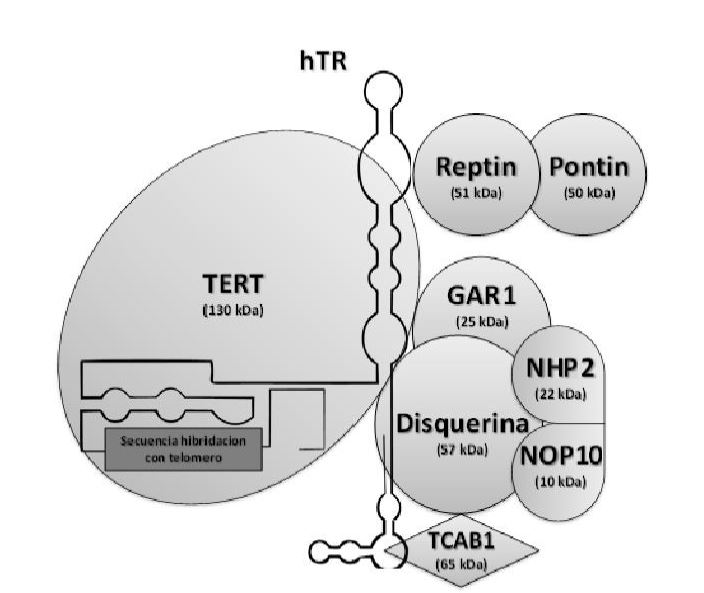
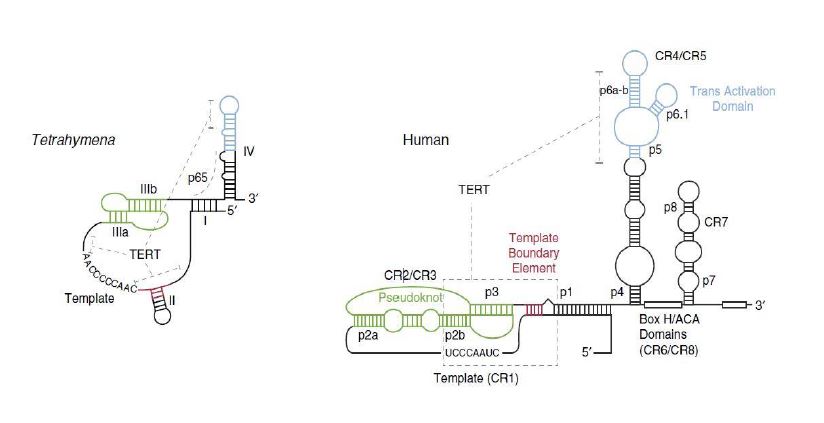
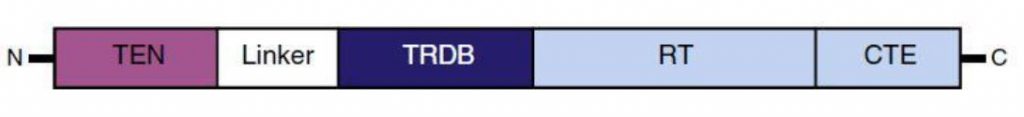
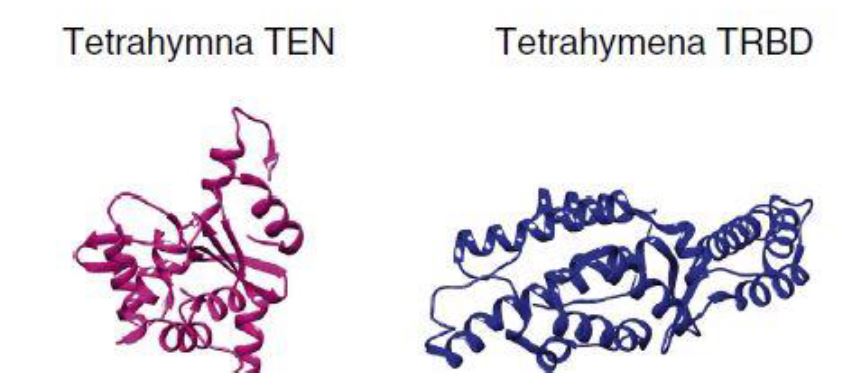
La transcriptasa inversa es una enzima característica de retrovirus que permite la replicación del material genético viral, el ARN, y lo transforma en ADN, simplificando mucho el proceso. También ayuda en la formación de una doble hélice de ADN una vez que el ARN ha experimentado una transcripción inversa en una sola cadena de cDNA. Esta enzima fue descubierta por David Baltimore y Howard Temin en 1970, aunque la descubrieron por separado. [8]
Bien, ¿y cómo actúa esta enzima? Cuando el ARN de una sola hebra del retrovirus entra en la célula, lleva consigo la transcriptasa inversa. La transcriptasa primero sintetiza una hebra de ADN complementaria al ARN y se forma un híbrido ARN-ADN. A continuación, se degrada la hebra de ARN y se reemplaza por ADN, así el resultado es una doble hebra de ADN que normalmente se integra en el genoma de la célula infectada. Este genoma integrado utiliza la maquinaria celular para transcribir sus genes y proteínas. [9]
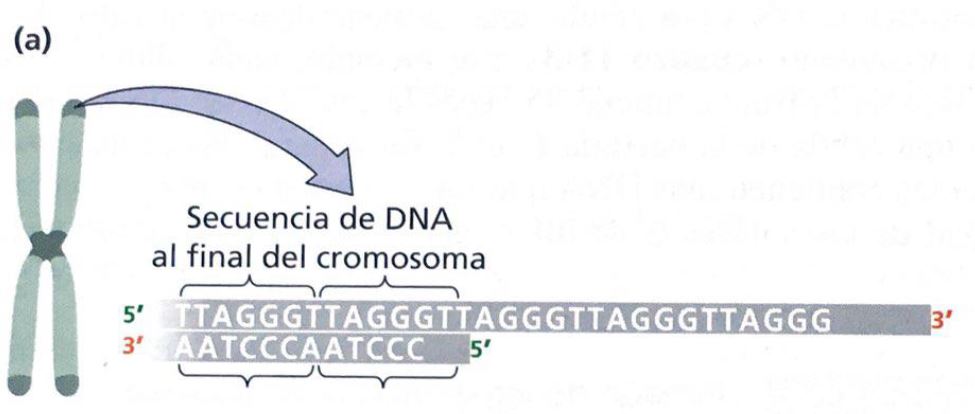
Sin embargo, la transcriptasa inversa no es una enzima exclusiva de virus. Nosotros, los humanos, al igual que otros seres vivos poseen en sus cromosomas una enzima llamada telomerasa que actúa como una transcriptasa inversa. A partir de un molde de ARN que lleva consigo, alarga el ADN de los telómeros con el fin de evitar la senescencia celular antes de tiempo. Aun así, esta enzima no está activa durante toda la vida de la célula (sólo en los estadíos de desarrollo tempranos, ya que, si lo estuviera toda la vida celular, la célula no moriría y se transformaría en una célula cancerosa).
Aplicaciones, RT-PCR
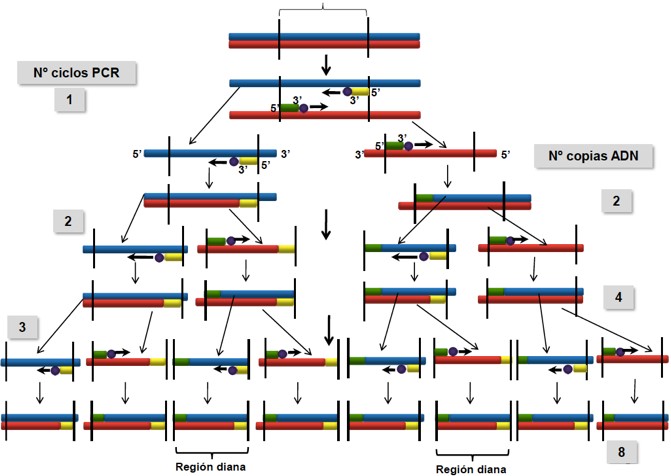
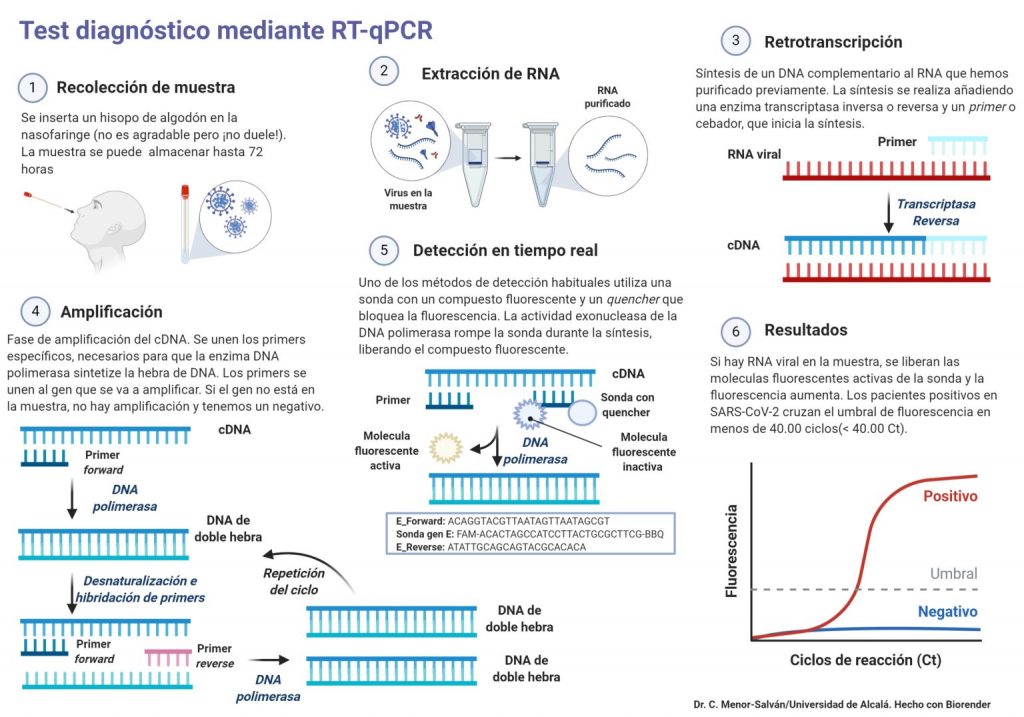
Sin embargo como hemos mencionado antes, podemos aprovechar las características de la transcriptasa inversa a nuestro favor. La podemos usar para identificar cantidades muy pequeñas de ARN en la prueba conocida como RT-PCR (Reverse transcription polymerase chain reaction o reacción en cadena de la polimerasa con transcripción inversa). Si queremos cuantificar y detectar la cantidad de ARN de una muestra, tenemos que realizar además una PCR cuantitativa. Esta prueba por ejemplo la usamos en el momento de detectar el Sars-CoV-2, ya que es un virus de ARN.
¿Cómo se lleva a cabo esta reacción? Vamos a poner como ejemplo el caso del coronavirus. Primero debemos obtener el ARN viral. Para ello, tomamos una muestra (de la garganta de la persona infectada a la que le vamos a realizar la prueba en nuestro caso) y la llevamos al laboratorio. Esa muestra contiene fragmentos de ADN también, por lo que lo primero es aislar el ARN del virus y degradar ese ADN residual.
Una vez hayamos hecho esto, es el momento de amplificar (aumentar la cantidad) el ARN viral. En esta ocasión, añadimos a la muestra de ARN nuestra transcriptasa inversa, cebadores para el inicio de la transcripción (primers) y dNTPs (desoxinucleótidos trifosfato para incorporar a la cadena de nueva síntesis). Finalmente obtenemos nuestro ADNc (copia) a partir de ARN.
En este momento, cuando tengamos el ADN copia, procedemos a hacer una PCR normal, por lo que añadimos de nuevo primers, dNTP y por último en lugar de transcriptasa inversa usamos la Taq polimerasa, la enzima usada en esta prueba para sintetizar nuevas cadenas de ADN y así poder cuantificar. Además, si queremos cuantificar el amplificado de ADN debemos añadir un componente fluorescente para así poder detectar el número de cadenas amplificadas en los ciclos que repitamos.
La reacción de la PCR se puede resumir en 4 pasos fundamentales:
- Desnaturalización del DNA molde mediante altas temperaturas (a unos 95ºC).
- Adición de los cebadores previa síntesis en el laboratorio.
- Hibridación (annealing o anillamiento) de cebadores al ADN molde que depende de cada tipo de cebador, aunque se baja la temperatura a 50-70ºC, para que los cebadores se puedan unir a la cadena molde. El tiempo de hibridación debe ser muy corto para evitar que las cadenas se puedan volver a unir.
- Aumento de la temperatura hasta 72ºC (temperatura óptima de la Taq polimerasa) que provoca una extensión del cebador mediante la adición de nucleótidos, es decir, se producirá la síntesis de ADN mediante la Taq polimerasa. Esta etapa de extensión durará dependiendo de la dimensión de la muestra, teniendo en cuenta que se sintetizan unos 1000 nucleótidos por minuto.
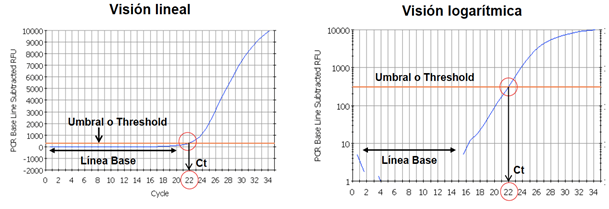
Si queremos cuantificar para obtener un positivo en la prueba, se dice que realizamos una PCR cuantitativa. Esto quiere decir que establecemos 3 parámetros para medir los ciclos que tarda una PCR en dar positivo: la línea base (que determina la fluorescencia basal detectada por el aparato) el umbral de fluorescencia (línea que deben cruzar las cadenas amplificadas para que se detecte positivo) y el valor Ct (cantidad de ciclos necesarios para cruzar el umbral) Cuantos menos ciclos necesite la prueba para dar positivo, mayor es la cantidad de ARN que tenemos en la muestra. [10]
Vamos a poner un ejemplo: si una muestra necesita 40 ciclos para dar positivo, se dice que hay poca cantidad viral, en cambio si otra muestra necesita tan solo 10, hay mucha más cantidad de ARN del virus que en la primera muestra.
ORIGEN, TRANSMISIÓN Y TRATAMIENTOS DEL VIH
Origen y replicación vírica
Bueno, ahora que ya sabemos qué es un retrovirus y qué es la transcriptasa inversa, podemos explicar un poco más a fondo la naturaleza del VIH.
La hipótesis más aceptada establece que el VIH es producto de una zoonosis, una enfermedad transmitida de animales al hombre. En concreto, gracias a técnicas de sampling y analizando ADN mitocondrial de los chimpancés, se cree que este virus surgió al mutar el SIV (virus de inmunodeficiencia en simios) a VIH-1. Lo que resultó curioso es que se pensaba que el SIDA (que provoca una bajada de linfocitos T helpers) comenzó a detectarse con la aparición del VIH, pero en realidad la enfermedad se detectó ya en estos chimpancés, cuando en unos análisis de su sangre pudieron confirmar una presencia de linfocitos Th 3 veces menor comparado con el número normal de estas células.[11]
El virus del VIH-2 también se cree que procede del SIV, pero en este caso el estudio que plantea la hipótesis utiliza un análisis filogenético (un análisis «de parentesco» entre distintas especies) para llegar a la conclusión de que a partir de uno de los genes del SIV se derivaba otro del VIH-2. [12]
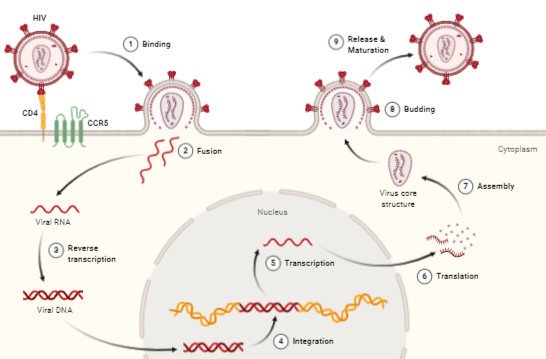
Ya puestos en contexto, vamos a hablar sobre cómo se replica el virus del VIH. Recordando un poco la estructura del virus, mencionamos que había dos glucoproteínas, que se llaman gp120 y gp41. Gp120 facilita el reconocimiento del receptor de la célula diana, que es el receptor CD4 presente en linfocitos Th sobre todo (aunque también se encuentra en otras células como macrófagos, monocitos y células dendríticas), aunque también se ha visto que el virus necesita reconocer a otros correceptores adicionales para permitir la fusión. Una vez reconocido el receptor, la proteína gp41 provoca que la membrana de la célula se fusione con la membrana vírica y el virus entre por fusión al interior de la célula.[13]
Gracias a la transcriptasa inversa, el ARN del virus forma un híbrido ARN-ADN que a su vez terminará formando un ADN duplexo debido a que la transcriptasa inversa elimina el ARN viral. Una vez hecho esto, el ADN penetrará en el núcleo para integrarse dentro del cromosoma y pasar a un estado latente dentro de la célula o por el contrario iniciar un ciclo productivo.
La ARN polimerasa dependiente de ADN se encargará de transcribir los genes virales para formar los mensajeros y dar lugar a las proteínas necesarias para los nuevos viriones mediante su traducción en los ribosomas. Los genes que lleva el virus se conocen como gag, pol y env. El gen env codifica las proteínas de la envoltura (gp120 y gp41), gag codifica las proteínas estructurales (las que forman parte de la matriz, cápside y nucleocáside) y pol las enzimas principales del virus (transcriptasa inversa, integrasa para la integración en el cromosoma celular y proteasa para cortar poliproteínas).
Finalmente, el virus termina de madurar en el citoplasma y sale de la célula por exocitosis, llevándose parte de la membrana celular que tendrá proteínas glicosiladas procedentes del aparato de Golgi. [13]
Dependiendo del tipo de célula el virus decide hacer un ciclo u otro:
- En linfocitos T queda latente hasta que decide iniciar un ciclo lítico. Este efecto se corresponde con la inmunosupresión.
- En otras células como macrófagos el virus realiza un ciclo persistente, es decir, libera los virus poco a poco sin matar a la célula, lo que sirve al virus como reservorio. Esto puede llevar a alteraciones neurológicas si los macrófagos infectados viajan al sistema nervioso.
Formas de transmisión
El virus se transmite de muchas maneras, y las vamos a mencionar de mayor a menor riesgo de contagio[14]:
- Sexo anal sin protección: Es la forma más fácil de contagiarse del VIH. Es más peligroso ser la persona pasiva que la activa. Esto es debido a que la piel que tapiza el recto es muy fina, entonces durante la penetración puede desgarrarse fácilmente y permitir que el virus entre en el cuerpo. También el virus se puede introducir en el cuerpo de la persona activa a través de desgarros en el pene, pequeñas heridas…
- Sexo vaginal sin protección: Ambos en la pareja pueden contagiarse de VIH durante el sexo. Las mujeres se contagian y pueden contagiar a través de la mucosa vaginal y sangre que pueda quedar en la vagina y el cérvix. El hombre, como en el caso anterior se contagia por heridas en el pene.
- Vía parenteral: El virus se transmite si se comparten jeringuillas o agujas usadas por seropositivos, porque siempre quedan restos de su sangre. También es alto el riesgo de contraer otras ETS, además de la hepatitis B y C.
- Transmisión vertical: Una madre puede transmitir el VIH a su hijo durante el embarazo, nacimiento o lactancia. Es la forma más común de transmisión de VIH en niños. De todos modos, si la madre toma antirretrovirales durante el embarazo y la lactancia, y da medicina al niño durante 4-6 semanas después de nacer, el riesgo de contraer el VIH es menor al 1%.
Estas son las formas principales de contagio, pero hay otras más raras como el sexo oral, lugar de trabajo (al rozarse con objetos afilados o agujas contaminadas), mordeduras de otras personas infectadas, besos con lengua… Pero todas estas formas de contagio requieren sangre de la persona infectada.
Se transmite a través de fluidos corporales como la sangre, semen, líquido preseminal, fluido rectal, fluido vaginal y leche materna.
Ahora bien, también hay otros factores que incrementan el riesgo de padecer la enfermedad, como puede ser el abuso de drogas intravenosas, tener otras ETS o la carga viral.
Tratamientos actuales contra el VIH
Tras comunicar a una persona que ha dado positivo en VIH, se le transmite información sobre los posibles tratamientos a seguir para paliar los efectos del virus del que, desafortunadamente, no conocemos la cura. Se informa al paciente de los riesgos y efectos secundarios que provocan estos medicamentos.
Los medicamentos más utilizados actualmente son[15]:
- Los inhibidores de la transcriptasa inversa no análogos de nucleósidos (ITINN), que bloquean una proteína que el VIH necesita para replicarse.
- Los inhibidores de la transcriptasa inversa análogos de nucleósidos o nucleótidos (ITIN) son versiones defectuosas de los componentes básicos que el VIH necesita para replicarse.
- Los inhibidores de la proteasa (IP) inactivan la proteasa del VIH, otra proteína que el VIH necesita para replicarse.
- Los inhibidores de la integrasa funcionan inhibiendo a una proteína llamada integrasa que el VIH utiliza para insertar su material genético en los linfocitos T CD4.
- Los inhibidores de entrada o fusión bloquean la entrada del VIH en los linfocitos T CD4.
El tratamiento inicial consiste en la combinación de dos NRTI (inhibidores de la transcriptasa inversa análogos de nucleótidos/nucleósidos) combinados con un tercer agente, que puede ser un inhibidor de integrasa, un ITINN o bien un inhibidor de la proteasa. Se ha visto que los inhibidores de integrasa son muy efectivos contra el virus, además de que apenas presentan efectos secundarios.[16]
Como curiosidad, se ha visto que en las zonas o comunidades con gran prevalencia de SIDA, el uso de terapias antirretrovirales disminuye enormemente la detección de nuevos casos de la enfermedad, así como la disminución de la carga viral dentro de dicha zona.
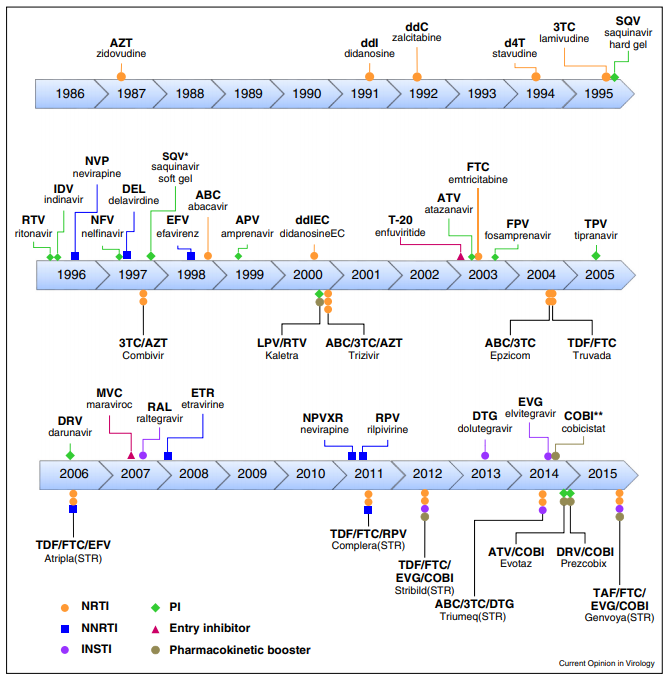
Una mirada al futuro
Proyectos de vacunas contra el VIH
Una vacuna terapéutica contra el VIH se administra a las personas seropositivas con el objetivo de reforzar su respuesta inmunitaria. En la actualidad existen muchas limitaciones en los conocimientos que tenemos de los mecanismos inmunológicos de control de replicación viral del VIH y la eficacia de las vacunas terapéuticas ha sido modesta en el mejor de los casos.
Una de las vacunas más estudiadas ha sido Remune, la cual contiene el virus completo inactivado mediante la extracción de una proteína de la envoltura. Se administró a más de 3000 personas que tenían el virus controlado por el tratamiento TAR. Los resultados mostraron que era capaz de inducir respuestas específicas de los linfocitos Th (los CD4), pero no se observó la capacidad de control inmunológico de la replicación viral.
La capacidad de las vacunas terapéuticas se demostró con un estudio en el que se utilizó una vacuna de células dendríticas en el modelo animal con infección por SIV en el que se obtuvieron increíbles resultados. El problema llegó a la hora de probarlo en 12 pacientes humanos los resultados fueron bastante peores. La vacuna no provocó efectos adversos importantes.
Otros candidatos a ser usados como vacunas terapéuticas son las basadas en ADN que incluyen algunas proteínas. Presentan todo el genoma del VIH con pérdida del gen de la integrasa y se han administrado intradérmicamente en modelos de monos con resultados prometedores. [17]
También podemos encontrar proyectos de vacunas basados en bacterias lácticas para producir inmunidad en las mucosas. esto podría resultar efectivo teniendo en cuenta que las mucosas la puerta de entrada del VIH en el caso de que se transmita por contacto sexual. El tratamiento consiste en la administración de Lactobacillus lactis modificada para expresar determinadas proteínas que inducen la formación de anticuerpos específicos a nivel de las mucosas. Solo ha sido probada en ratones, pero puede representar una estrategia tecnológica para desarrollar una vacuna contra el SIDA efectiva y segura. [18]
Casos de curación completa del VIH
Aunque mediante los tratamientos con antirretrovirales se consigue que el sistema inmunológico de las personas que padecen VIH se restablezca parcialmente y se retrase la progresión de la enfermedad, la cura de la infección por este virus sigue siendo inalcanzable con los métodos de los que disponemos actualmente. Esto se debe principalmente al establecimiento de depósitos de VIH en células de larga vida que permiten que siga existiendo replicación del virus tras retirar los tratamientos antirretrovirales.
Los estudios enfocados a la cura del VIH actualmente se basan en la mutación Δ32 del gen CCR5 que provoca que las células sean resistentes a diferentes cepas del virus. De hecho, se ha demostrado la viabilidad del trasplante de células madre hematopoyéticas de donantes que tienen esta mutación en pacientes infectados con VIH y que padecen leucemia mieloide aguda en recaída, documentándose la ausencia de viremia durante los primeros 20 meses tras retirar el tratamiento antirretroviral. Con estos resultados favorables, se cree que el camino hacia la cura de la infección por el virus de la inmunodeficiencia humana está en la investigación del gen y la mutación anteriormente mencionados, aunque exista la incertidumbre de si realmente ese paciente está realmente curado. De hecho, en otros pacientes, tras el trasplante con células madre que tenían esta mutación, se retiraron los tratamientos para el VIH y existieron rebrotes virales debido a la existencia de los reservorios del virus dentro del organismo, demostrando que no se podía obtener la eliminación completa del virus.
El estudio del que obtenemos la información sobre este tema trató de evaluar la capacidad de reconstrucción de los linfocitos T colaboradores, que contaban con la mutación Δ32, en un seguimiento de 3 años y medio tras en trasplante tanto a nivel sistémico como en el sistema inmunológico de la mucosa. En sus resultados se demostró la exitosa reconstrucción de estas células y además se encontró una reducción del tamaño de los reservorios de VIH. Si bien las células recuperadas seguían siendo susceptibles de infección por el virus, el paciente permanecía sin evidencia de infección tras 3 años y medio de la retirada de los tratamientos antirretrovirales. A partir de esto resultó razonable concluir que el paciente había sido curado de la infección por VIH. [19]
Campañas de prevención
La primera campaña en España no tuvo lugar hasta el año 1987, lo que resultan un tanto tardío, teniendo en cuenta que existían casos desde 1982, y fue diseñada por Mariscal. En esta se trataba de informar de las vías de contagio del VIH a una población en ese momento muy desinformada, pero la forma de hacerlo fue bastante infantil. A favor de esta campaña se puede decir que trataba de esquematizar todas las formas de contagio, información que era necesaria e imprescindible. el problema era que la importancia del peligro del SIDA y la necesidad urgente del cambio en el comportamiento de las personas resultó mermada por la forma en la que era transmitido el mensaje: unos dibujos animados que emitían repetidamente el juego de palabras «Si da, no da».

Si se observa la campaña de 1988 (que se siguió utilizando hasta 1992), nos damos cuenta de que tanto la imagen como el lema «El sida te engancha por el pico» pueden dar lugar a equivocación porque en realidad el virus del sida no está en la heroína: si uno consume una dosis con su jeringuilla y destruye ésta después, no coge el SIDA. Por lo tanto las primeras campañas dirigidas a las personas drogodependientes cayeron en confundir el mensaje de prevención del sida con el de las campañas antidrogas, y sería necesario aclarar que el SIDA no se encontraba ni en la heroína ni en una jeringuilla nueva.

Una de las campañas que mejor funcionó fue «Protégete» (2002) creada en el 20 aniversario de los primeros casos de sida en España. Hace hincapié en la necesidad de utilizar el preservativo como estrategia fundamental de prevención y es lo suficientemente clara y directa para no dar lugar a segundas interpretaciones. El motivo utilizado, el preservativo-salvavidas, se convirtió en símbolo que acompañaba a otras campañas, como la del año 2003, con el lema «rompe la cadena, protégete». [20]

En cuanto a la campaña que se está llevando actualmente en España, el Misterio de Sanidad afirma que el VIH está produciendo una oleada de nuevas infecciones en hombres homosexuales, ya que uno de cada tres diagnósticos de VIH en nuestro país debe a prácticas sexuales entre hombres. En los estudios realizados, más del 10% de los hombres que tienen relaciones homosexuales están infectados por el VIH.
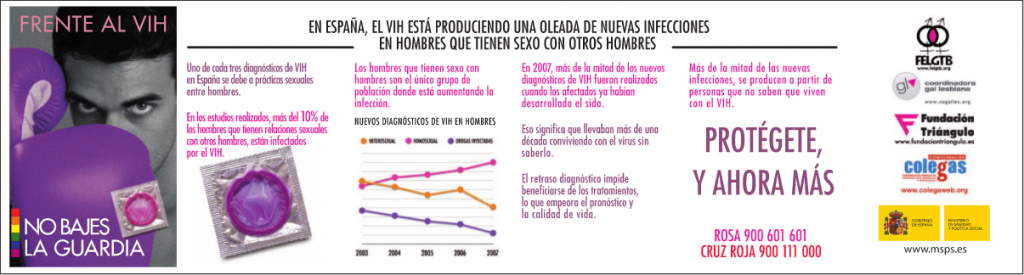

La conclusión que se puede sacar de este apartado es que los responsables de las políticas de prevención de SIDA deberían de ser conscientes de la importancia que tiene diseñar campañas cuyo mensaje resulte coherente, contundente, suficientemente informativo y científicamente correcto.
Bibliografía
- Gallo, R. C. and Montagnier, L. (2003) ‘The Discovery of HIV as the Cause of AIDS’, New England Journal of Medicine, 349(24), pp. 2283–2285. doi: 10.1056/nejmp038194.
- Domingo, E., Parrish, C. and Holland, J., 2011. Origin And Evolution Of Viruses. Amsterdam: Academic Press.
- Coffin, J. M. (2015) ‘The discovery of HTLV-1, the first pathogenic human retrovirus’, Proceedings of the National Academy of Sciences of the United States of America, 112(51), pp. 15525–15529. doi: 10.1073/pnas.1521629112.
- Carrillo Maravilla, Eduardo, & Villegas Jiménez, Armando. (2004). El descubrimiento del VIH en los albores de la epidemia del SIDA. Revista de investigación clínica, 56(2), 130-133.
- Pintos Pascual, I., Muñez Rubio, E., & Ramos Martínez, A. (2018). Diagnosis of acute and chronic HIV infection and their evolutionary stages. Medicine (Spain), 12(56), 3329–3331.
- Esteban, C. S. (2014). VIH: Infeccion aguda, pesquisa y manejo. Revista Médica Clínica Las Condes, 25(3), 419–424.
- Fragoso García, Erick, Rosenthal, Jareth Lassard (2015) ‘Retrovirus que ocasionan cáncer en humanos’ Colegio Indoamericano, S.C. (6779), México.
- Baltimore, D. (1970) ‘Viral RNA-dependent DNA polymerase: RNA-dependent DNA polymerase in virions of RNA tumour viruses’, Nature, pp. 1209–1211. doi: 10.1038/2261209a0.
- Lehninger, A., Nelson, D. and Cox, M., 2013. Principios De Bioquímica.
- Real García, M., Rausell Segarra, C. and Latorre, A., 2017. Técnicas De Ingeniería Genética. Madrid: Síntesis.
- Sharp, P. M. and Hahn, B. H. (2010) ‘The evolution of HIV-1 and the origin of AIDS’, Philosophical Transactions of the Royal Society B: Biological Sciences, 365(1552), pp. 2487–2494. doi: 10.1098/rstb.2010.0031.
- Santiago, M. L., Range, F., Keele, B. F., Li, Y., Bailes, E., Bibollet-Ruche, F., Fruteau, C., Noe, R., Peeters, M., Brookfield, J. F. Y., Shaw, G. M., Sharp, P. M. and Hahn, B. H. (2006) ‘Simian Immunodeficiency Virus Infection in Free-Ranging Sooty Mangabeys (Cercocebus atys atys) from the Tai Forest, Cote d’Ivoire: Implications for the Origin of Epidemic Human Immunodeficiency Virus Type 2’, Journal of Virology, 80(9), pp. 4645–4645. doi: 10.1128/jvi.80.9.4645.2006.
- Freed, E. O. (2001) ‘HIV-1 replication’, Somatic Cell and Molecular Genetics, 26(1–6), pp. 13–33. doi: 10.1023/A:1021070512287.
- https://www.cdc.gov/hiv/basics/hiv-transmission/ways-people-get-hiv.html
- https://www.mayoclinic.org/es-es/diseases-conditions/hiv-aids/diagnosis-treatment/drc-20373531
- Cihlar, T. and Fordyce, M. (2016) ‘Current status and prospects of HIV treatment’, Current Opinion in Virology. Elsevier B.V., 18, pp. 50–56. doi: 10.1016/j.coviro.2016.03.004.
- García, F., Ruiz, L., López-Bernaldo de Quirós, J. C., Moreno, S., & Domingo, P. (2005). Inmunoterapia y vacunas terapéuticas en la infección por VIH. Enfermedades Infecciosas y Microbiología Clínica, 23, 84–94. https://doi.org/10.1016/s0213-005x(05)75164-8
- Luna Cruz, I., Rodríguez Padilla, C., Tamez Guerra, R., & Alcocer González, J. (2003). Desarrollo de vacunas basadas en bacterias lácticas para inducir inmunidad en mucosas contra VIH. Ciencia UANL, 6(1).
- Church, J. A. (2011). Evidence for the cure of HIV infection by CCR5δ32/Δ32 stem cell transplantation. Pediatrics, 128(SUPPL. 3), 2791–2799. https://doi.org/10.1542/peds.2011-2107IIII
- Hernánez, R. M. (2009). El Sida Ante La Opinión Pública : El Papel De La Prensa Y Las Campañas De. Revista de Humanidades, 15(2009), 237–268.