HEXOKINASA: ESTRUCTURA, EVOLUCIÓN Y PAPEL EN EL CÁNCER
Redactado por María Arrondo Sánchez y Carolina Amil Zamorano
INTRODUCCIÓN
La hexoquinasa es una enzima transferasa (D-hexose-6-phosphotransferase), del grupo de las quinasas, encargada de fosforilar hexosas. Sin embargo, presenta mayor afinidad por la glucosa, puesto que la Km de esta es menor que la de otras hexosas como la fructosa. Esta proteína presenta cuatro isoformas, que han ido surgiendo de forma gradual. En dicho proceso de evolución ocurren determinados cambios que son claves en la estructura y que han permitido que la hexokinasa en dos de sus isoformas se oligomerice. La estructura de la proteína va a contar con dos dominios de unión a los sustratos (glucosa y ATP) y su actividad va a estar regulada alostéricamente mediante un mecanismo de ajuste inducido provocado por la propia glucosa.
Así mismo, esta enzima va a presentar un papel clave en el cáncer. En este artículo se abordará el normal funcionamiento de la hexokinasa así como su papel tumoral por diferentes vías, profundizando, además, en posibles estudios futuros y nuevos campos que se abren en la investigación contra el cáncer que emiten un rayo de esperanza en el estudio biomédico.
PAPEL BIOLÓGICO
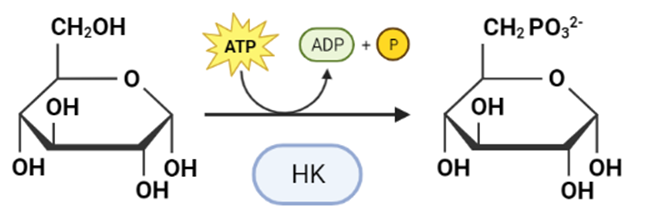
La Hexokinasa participa en la primera reacción irreversible de la glucólisis, que es la primera etapa del metabolismo de la glucosa. Tras hidrolizar el ATP, transfiere el grupo fosfato a la glucosa, para dar glucosa-6-fosfato (G6P), y así manteniendo el gradiente de glucosa que permite que haya un flujo de la misma mediado por los transportadores GLUT. La G6P es un inhibidor competitivo del ATP, por tanto se trata de un fenómeno de feedback negativo, en el que el mismo producto regula alostérica y negativamente su reacción de síntesis. Además, el grupo fosfato (Pi) liberado de la hidrólisis del ATP puede antagonizar la inhibición de G6P o sumarse al efecto inhibidor, según la isoenzima que haya llevado a cabo la reacción.
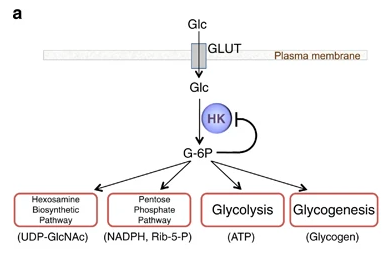
El producto (G6P) puede seguir varias rutas o vías celulares y funcionales:
- Metabolismo catabólico: se introduce la glucosa en la glucólisis, para llevar a cabo un metabolismo oxidativo y obtener energía.
- Metabolismo anabólico: la G6P es destinada a la vía de las pentosas fosfato, para sintetizar NAPDH y Ribulosa-5-Fosfato; o puede ser convertido a sus formas poliméricas (glucógeno), mediante la gluconeogénesis.
ESTRUCTURA
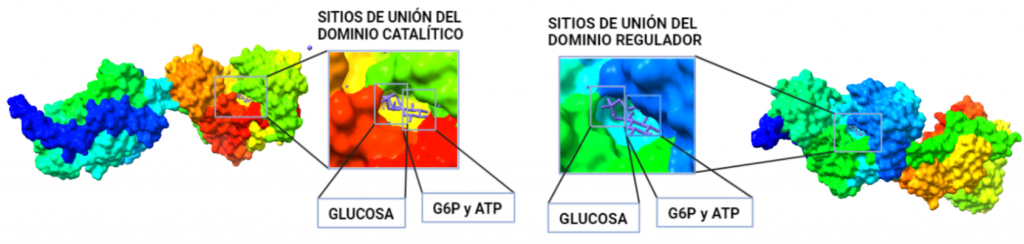
La estructura de las Hexokinasas más comunes (las isoformas I, II y III) cuenta con dos lóbulos muy similares de unos 50KDa cada uno. Algunas de ellas, como la HK I, son monoméricas, pero cuando se une a la membrana externa de la mitocondria se oligomerizan. De esta manera, la Hexokinasa cuenta con dos dominios principales, uno regulador y otro catalítico. La estructura dimérica y por tanto cuaternaria está presente en todas las isoformas salvo en la IV, que es la más ancestral.
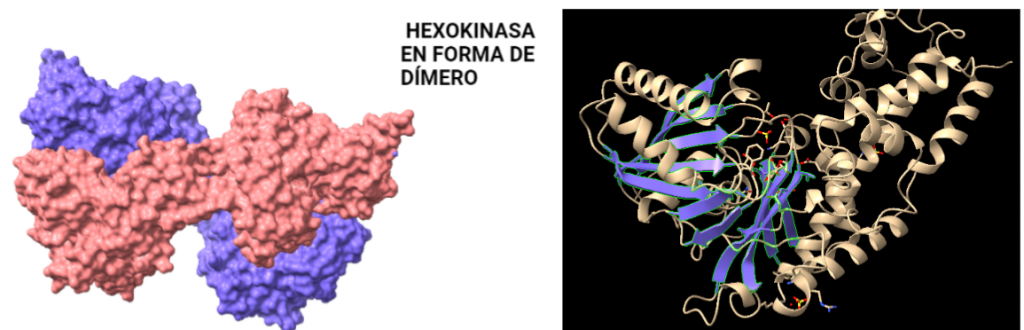
Figura IV: Dominio de unión del ATP, se observan cuatro láminas paralelas y una antiparalela. Hecho con Chimera
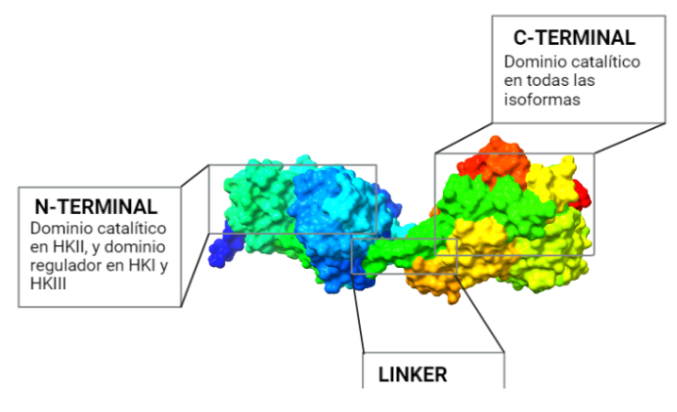
El dominio N-terminal se considera el dominio regulador en las isoenzimas I y II, y contiene el motivo de unión a la mitocondria. Además, está unido al dominio C-terminal (que es el dominio catalítico) a través de una hélice alfa. Ambos dominios presentan sitios de unión con la glucosa, G6P y ATP, y la inhibición de G6P en el dominio regulador se contagia al dominio catalítico por medio del contacto por la hélice alfa entre los dominios. La estructura terciaria de la hexoquinasa se basa en un plegamiento alfa/beta abierto. El dominio de unión al ATP está compuesto por cinco láminas beta y tres hélices alfa en el cual cuatro de las láminas beta son paralelas y una es antiparalela. Por otro lado, la hexoquinasa requiere de iones de magnesio para poder llevar a cabo la actividad catalítica. El magnesio (Mg2+) va a ser el cofactor de la enzima y se encuentra formando un complejo con el ATP (MgATP2-), que estabiliza la catálisis y reduce la energía de activación de la reacción.
EVOLUCIÓN
Todas las isoenzimas de la Hexokinasa provienen de una Hexokinasa de 50 kDa, susceptible a la inhibición por el producto G6P, por tanto, todas las isoenzimas presentan esta característica. A partir de la duplicación y fusión del gen que codificaba esta forma ancestral, surgieron las isoenzimas I, II y III, que ya son moléculas de 100kDa.
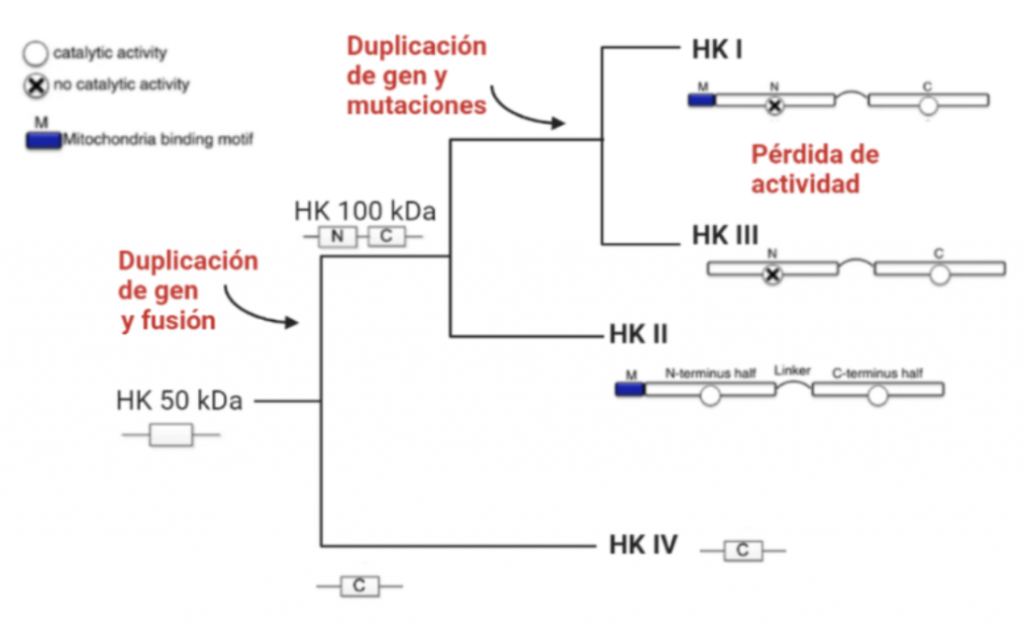
La isoforma más próxima evolutivamente a la Hexokinasa original es la Tipo IV, que no sufrió la duplicación y fusión génica. Una vez que esto ocurrió, la segunda isoenzima que apareció fue la Hexokinasa II, que mantiene la actividad catalítica en ambos extremos terminales de la proteína, al igual que la Hexokinasa ancestral.
Una consecuente duplicación tuvo como resultado la aparición de la isoforma III. Posteriormente, las mutaciones de genes que codificaban la Hexokinasa 100 kDa, produjeron que el extremo N-terminal se diferenciara funcionalmente, perdiendo la actividad catalítica, y adquiriendo una función reguladora (con un sitio de unión para el inhibidor G6P). Esta diferenciación dio lugar a las en las Hexokinasas I y III.
Además, en las HK I y II, el extremo N-terminal presenta un dominio hidrofóbico que permite a estas integrarse en la membrana de la mitocondria. Concretamente, se unen a las porinas (VDAC) de la membrana mitocondrial externa, las cuales interaccionan con los ANT (Translocadores de Nucleótidos de Adenina). Esto es esencial para el mecanismo enzimático de la HK, puesto que es el sitio de salida del ATP producto de la fosforilación oxidativa (que usará la HK), y el sitio de entrada del ADP resultante de la reacción enzimática de la hexokinasa. Por tanto, existe una coordinación entre la introducción de la glucosa al metabolismo glucolítico y las últimas etapas de este en la mitocondria (la fosforilación oxidativa), para que se den a un ritmo adecuado a las necesidades celulares.
Hay cuatro isoenzimas de la Hexokinasa (HK) en los tejidos de mamíferos, con una estructura similar, pero expresión en diferentes tejidos:
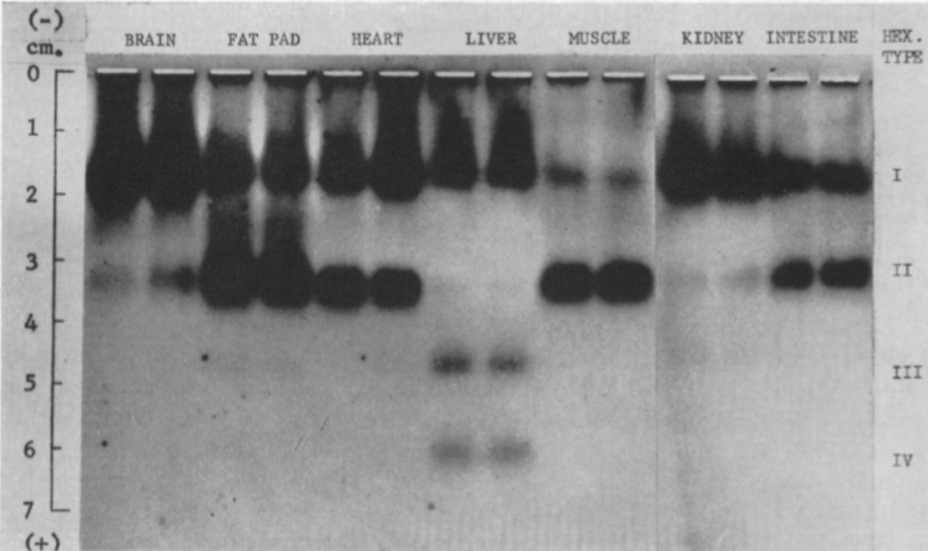
- HEXOKINASA I (HKI)
Fundamentalmente en el cerebro, donde la tasa metabólica es muy exigente, pero expresada de manera general.
- HEXOKINASA II (HKII)
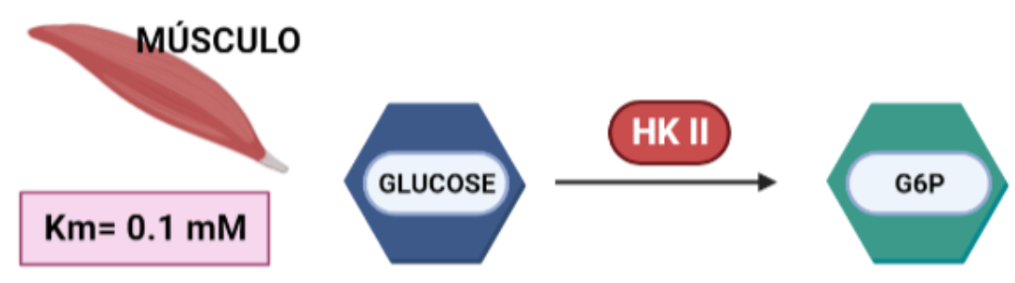
Más limitada en su expresión, aparece en tejidos sensibles a insulina, como es el caso del tejido adiposo y el músculo esquelético. En el músculo, es necesaria una alta tasa de glucólisis, y por tanto, lo que ocurre es que esta isoforma presenta una gran afinidad por la glucosa (menor Km), para que con concentraciones muy bajas de glucosa, se alcancen altas velocidades de la enzima.
La insulina aumenta la actividad de esta isoforma, induciendo la transcripción del gen que la codifica, y de esta forma, favoreciendo la metabolización y eliminación de glucosa. Por esta razón, en los individuos con diabetes de tipo II, la expresión de la HKII se ve reducida, acentuando la hiperglucemia.
Al igual que la isoenzima Tipo I, incluye un dominio hidrofóbico en el extremo N-terminal que permite que se inserte en la membrana externa mitocondrial, y también usa ATP intramitocondrial.
Cabe destacar su predominancia en células tumorales, puesto que estas se caracterizan por presentar un aumento anormal del metabolismo, en el que la reacción que lleva a cabo la Hexokinasa es esencial para la obtención de energía.
- HEXOKINASA III (HKIII)
A diferencia de las dos isoenzimas anteriores, la Hexokinasa III no está unida a la mitocondria, puesto que carece del dominio hidrofóbico en el extremo N-terminal. Se piensa que se expresa en el citoplasma, o que incluso tiene una localización perinuclear, en células del hígado.
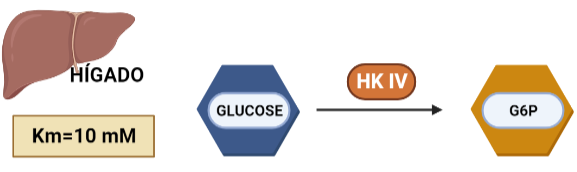
- HEXOKINASA IV (HK IV)
En hepatocitos y células beta pancreáticas, y es conocida como glucoquinasa. La G6P que produce está destinada a la síntesis de glucosa. Esto es un proceso que se lleva a cabo cuando la cantidad de sustrato (glucosa) es alta, por tanto, tiene sentido que esta isoenzima presente una mayor Km, porque necesitará altas concentraciones de glucosa para realizar la reacción a una velocidad alta.
MECANISMO
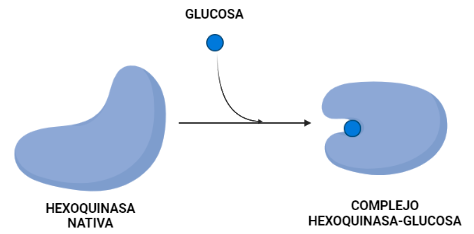
La hexoquinasa sufre un cambio conformacional que es regulado por la propia glucosa que va a ser esencial para la catálisis. En este proceso se observa que la superficie en contacto con el solvente del complejo hexoquinasa-glucosa es más pequeña que la hexoquinasa nativa.
Utilizando dicho cambio en el área de superficie que se encuentra expuesta se ha podido estimar la contribución hidrofóbica a los cambios de energía libre tras la unión de la glucosa. De esta manera se descubre que el efecto hidrofóbico por sí solo favorece la conformación activa de la hexoquinasa en presencia y ausencia de azúcar. La estabilidad observada de la conformación inactiva de la enzima en ausencia de sustratos puede resultar de una deficiencia de interacciones complementarias dentro de la cavidad que se forma cuando los dos lóbulos se unen.
El cambio conformacional que sufre la hexoquinasa mantiene la estructura terciaria prácticamente igual excepto por un gran cambio en la orientación de los dos lóbulos. Para demostrar este cambio lo que se hizo fue superponer los carbonos alfa de cada lóbulo usando un procedimiento de mínimos cuadrados y tratando a los carbonos como cuerpos rígidos. Esta superposición mostró que cada lóbulo se comporta como un cuerpo rígido durante el cambio conformacional entre la forma nativa de la proteína y el complejo con la glucosa.
Tal y como venimos viendo, la superficie accesible de la hexoquinasa se reduce cuando se une la glucosa para formar el complejo enzima-sustrato (ES) y se reduce aún más por el cambio a E’(inactiva)-S. Por lo tanto, se puede esperar que las fuerzas hidrofóbicas favorezcan la conformación activa en presencia de azúcar, asumiendo que todos los donantes y aceptores de enlaces de hidrógeno están satisfechos en E’-S. Sin embargo, el área superficial también se reduce cuando la conformación activa se forma en ausencia de azúcar (E’ – E). Sin embargo, debido a que hay menos de un factor de dos diferencias entre los cambios en la superficie accesible para las transiciones E-S → E’-S y E’-E, el efecto hidrofóbico no puede explicar la gran diferencia en las constantes de equilibrio conformacional K2 y K3 en presencia y ausencia de azúcar.
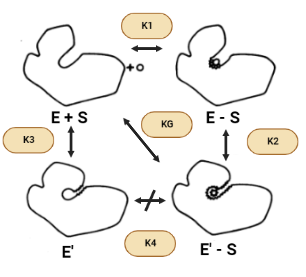
Otra cuestión que surge es por qué la enzima no permanece en el estado activo, E, en ausencia de ligandos sabiendo que el efecto hidrófobo, de manera individual, predice que la estructura E debería ser más estable. La respuesta a esta pregunta reside en que cuando la enzima carece de la presencia de la glucosa contiene una cavidad en la que entran las moléculas de agua y donde quedan encerradas. Además, tanto los puentes de hidrógeno como las fuerzas de Wan der Waals contribuyen muy poco a la estabilidad de la proteína y del complejo proteína-ligando. El hecho de no obtener estas interacciones complementarias dentro de la cavidad daría como resultado entalpías desfavorables causadas por la pérdida de los puentes de hidrógeno o fuerzas de Van der Waals en relación con los que se producen en la estructura abierta. También puede haber alguna pérdida de entropía traslacional al atrapar una pequeña cantidad de moléculas de agua en la cavidad.
Presuntamente, el agua misma desestabiliza la forma activa mediante la creación de interacciones favorables con la estructura abierta inactiva. Solamente el ligando correcto puede proporcionar las fuerzas de Van der Waals y los puentes de hidrógeno necesarios para que se active la estructura.
Con ello, concluimos que hay al menos dos posibles funciones para el cambio conformacional inducido por la glucosa: permitir un «mecanismo de acogida» o proporcionar especificidad.
PAPEL BIOMÉDICO: HEXOKINASA II EN CÁNCER
El metabolismo de las células tumorales se caracteriza por una alta actividad glucolítica: metabolizan anaeróbicamente grandes cantidades de glucosa en ácido láctico, incluso en presencia de oxígeno, aumentando la velocidad de la glucólisis y de la síntesis de ATP. Esto es lo que se conoce como el efecto Warburg. Por tanto, la actividad de cualquier enzima glucolítica como la HK será esencial en un tumor.
La Hexokinasa II aparece sobreexpresada en células tumorales, satisfaciendo estas altas velocidades de la glucólisis. La introducción de la glucosa en el metabolismo glucolítico es crucial para la producción de energía, y la síntesis de precursores de nucleótidos (derivados de glucosa) por la vía de las pentosas fosfato, destinados a la síntesis de ADN para la proliferación del tumor.
Imagen: Annette Rempel
La regulación por AKT de la HKII es el factor determinante para que esta isoforma sea la esencial en el metabolismo tumoral, puesto que controla la unión de esta a la mitocondria, y con ello, fija la función de la HK, que varía en función de si aparece unida a la mitocondria o no.
Con el objetivo de crecimiento del tumor, y cuando hay disponibilidad de nutrientes, AKT une la HKII a la mitocondria (fosforilando su residuo Thr-473), conectándola con VDAC. Esto le permite a la enzima tener un acceso privilegiado al ATP que sintetiza la mitocondria, y la célula sigue un metabolismo de proliferación y de producción de energía, mediante la glucólisis.
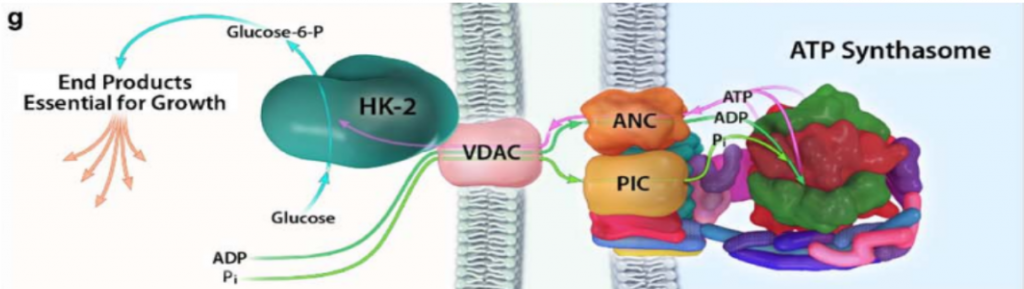
Imagen: Pedersen, P.L.
La HK II mitocondrial, además, lleva a cabo una función protectora en tumores con acceso a nutrientes, promoviendo la supervivencia celular: inhibe a los miembros pro-apoptóticos de la familia Bcl2, e introduce la G6P en la ruta de las pentosas fosfato, cuyos productos son antioxidantes que reducen las ROS (especies reactivas de oxígeno).
Sin embargo, en una situación de isquemia, en la que no llegan suficientes nutrientes y oxígeno a la célula, disminuye la actividad de AKT y HKII mitocondrial, aumentando la HKII citoplásmica. Entonces, esta isoforma se une a mTORC (mediante el motivo TOS), inhibiéndola, e induciendo la vía autofágica, y la conservación de energía y homeostasis en ausencia de glucosa, pensando en el “bien mayor” del tumor.
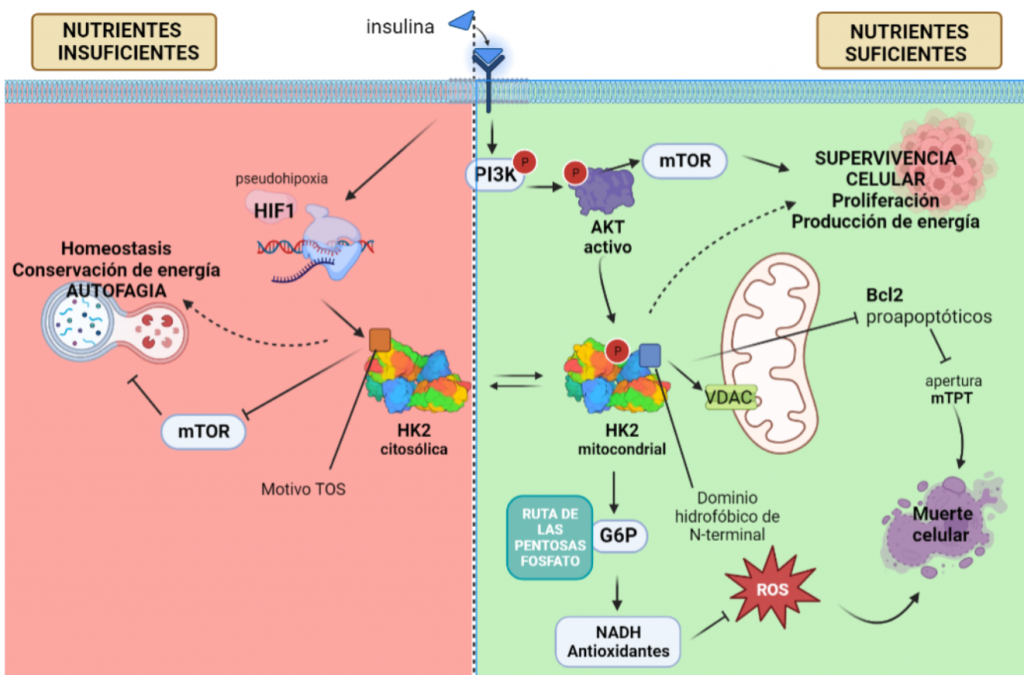
Esta regulación por AKT posibilita esta compleja acción de la HKII, que puede ser proapoptótica (HKII citoplásmica) o antiapoptótica (HKII mitocondrial), según la disponibilidad de recursos. Sin embargo, AKT no regula la HKI, ya que esta no presenta una secuencia de consenso para esta enzima, y por ello, esta isoenzima no se encuentra prevalentemente en tumores. Además, la HKI no puede satisfacer la alta demanda energética, al perder la actividad catalítica en el extremo N-terminal.
Según todas las vías beneficiosas para el tumor mencionadas anteriormente en las que participa la HKII, la eliminación de esta isoforma perjudicaría a la progresión del tumor, por lo tanto, es un frente esperanzador en terapias oncogénicas. Lo ideal sería encontrar un modo de inhibir únicamente esta isoforma, pero es difícil puesto que todas ellas son bastante similares.
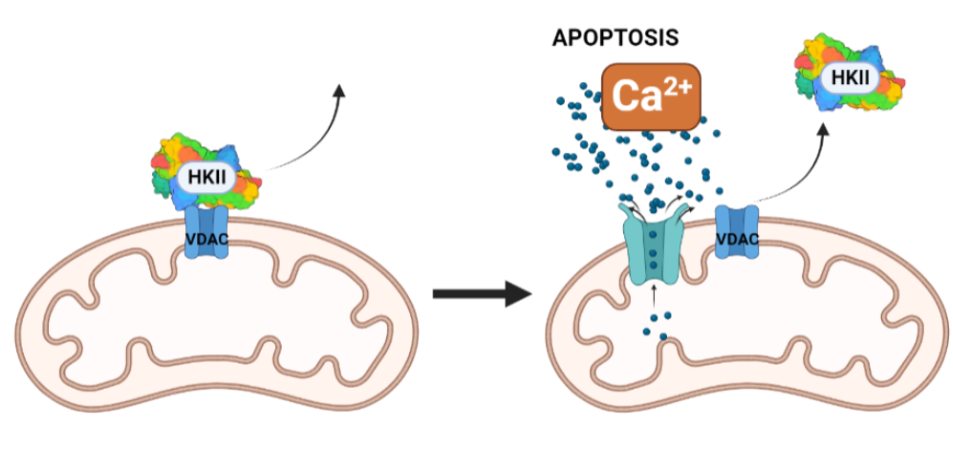
La inhibición de la HKII en células cancerosas puede darse por p53, o por un sustrato análogo a la glucosa, la 2-desoxiglucosa, que favorece la apoptosis. Una combinación de estos dos factores podría ser favorecedora. Otro posible campo a investigar sería la inhibición de la HKII por fosfato inorgánico, que sensibiliza la inhibición por G6P en esta, pero la antagoniza en HKI. Incluso, una alternativa más podría ser el uso de determinados péptidos que desplazaran la HKII de la mitocondria. Este desplazamiento parece producir un aumento de la concentración de Ca2+ citosólica, lo que abriría poros en la membrana mitocondrial e induciría a la célula a apoptosis.
BIBLIOGRAFÍA
Anja Schindler, Edan Foley (2013). Hexokinase 1 blocks apoptotic signals at the mitochondria. https://www.sciencedirect.com/science/article/abs/pii/S0898656813002726
Annette Rempel, Saroj P. Mathupala, Constance A. Griffin, Anita L. Hawkins, Peter L. Pedersen (1996); Glucose Catabolism in Cancer Cells: Amplification of the Gene Encoding Type II Hexokinase1. Cancer Res 1. https://aacrjournals.org/cancerres/article/56/11/2468/502209/Glucose-Catabolism-in-Cancer-Cells-Amplification
Bennett, W. S., & Steitz, T. A. (1978). Glucose-induced conformational change in yeast hexokinase. Proceedings of the National Academy of Sciences, 75(10), 4848-4852. https://www.pnas.org/doi/abs/10.1073/pnas.75.10.4848
Ciscato, F. (2021.). Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. MDPI. https://www.mdpi.com/1422-0067/22/9/4716
Gahr, M. (1980). Isoelectric Focusing of Hexokinase and Glucose-6-Phosphate Dehydrogenase Isoenzymes in Erythrocytes of Newborn Infants and Adults. British Journal of Haematology, 46(4), 529-535. https://doi.org/10.1111/j.1365-2141.1980.tb06009.x
Haruhiko Osawa, Calum Sutherland, R. Brooks Robey, Richard L. Printz, Daryl K. Granner (1996). Analysis of the Signaling Pathway Involved in the Regulation of Hexokinase II Gene Transcription by Insulin. https://www.sciencedirect.com/science/article/pii/S002192581831932X
John E. Wilson (2003); Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol 15 ; 206 (12): 2049–2057. https://journals.biologists.com/jeb/article/206/12/2049/9262/Isozymes-of-mammalian-hexokinase-structure
Mulichak, A., Wilson, J., Padmanabhan, K. et a (1998)l. The structure of mammalian hexokinase-1. Nat Struct Mol Biol 5, 555–560. https://pubmed.ncbi.nlm.nih.gov/9665168/
Pastorino, J. G. (2008). Regulation of hexokinase binding to VDAC. SpringerLink. https://link.springer.com/article/10.1007/s10863-008-9148-8?error=cookies_not_supported&code=334cd9ac-6023-4e88-a330-afab2ff91f76
Patrona, K. C., & Hay, N. (2013). Hexokinase 2 as oncotarget. Oncotarget, 4(11), 1862–1863. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3875752/
R.L. Printz, S. Koch, L.R. Potter, R.M. O’Doherty, J.J. Tiesinga, S. Moritz, D.K. Granner (1993). Hexokinase II mRNA and gene structure, regulation by insulin, and evolution. https://www.sciencedirect.com/science/article/pii/S0021925818535213
R.L. Printz, S. Koch, L.R. Potter, R.M. O’Doherty, J.J. Tiesinga, S. Moritz, D.K. Granner (1993). Hexokinase II mRNA and gene structure, regulation by insulin, and evolution. Journal of Biological Chemistry. Volume 268, Issue 7 https://www.sciencedirect.com/science/article/pii/S0021925818535213
Reference for PDB-101: PDB-101: Educational resources supporting molecular explorations through biology and medicine. Christine Zardecki, Shuchismita Dutta, David S. Goodsell, Robert Lowe, Maria Voigt, Stephen K. Burley. (2022) Protein Science 31: 129-140 doi:10.1002/pro.4200
Reference for RCSB PDB: The Protein Data Bank H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne (2000) Nucleic Acids Research, 28: 235-242. doi:10.1093/nar/28.1.235
Rempel, A., Mathupala, S. P., Griffin, C. A., Hawkins, A. L., & Pedersen, P. L. (1996). Glucose Catabolism in Cancer Cells: Amplification of the Gene Encoding Type II Hexokinase1. American Association for Cancer Research. https://pubmed.ncbi.nlm.nih.gov/11557773/
Roberts, D., Miyamoto, S. (2015) Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ 22, 248–257. https://www.nature.com/articles/cdd2014173
UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE. Protein Sci. 2018 Jan;27(1):14-25.
UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. Protein Sci. 2021 Jan;30(1):70-82.
Zawacka-Pankau, Joanna & Grinkevich, Vera & Hünten, Sabine & Nikulenkov, Fedor & Gluch, Angela & Li, Hai & Enge, Martin & Kel, Alexander & Selivanova, Galina. (2011). Inhibition of glycolytic enzymes mediated by pharmacologically activated p53: Targeting Warburg effect to fight cancer. https://www.researchgate.net/publication/51591216_Inhibition_of_glycolytic_enzymes_mediated_by_pharmacologically_activated_p53_Targeting_Warburg_effect_to_fight_cancer