Introducción a la biología molecular del coronavirus SARS-CoV-2
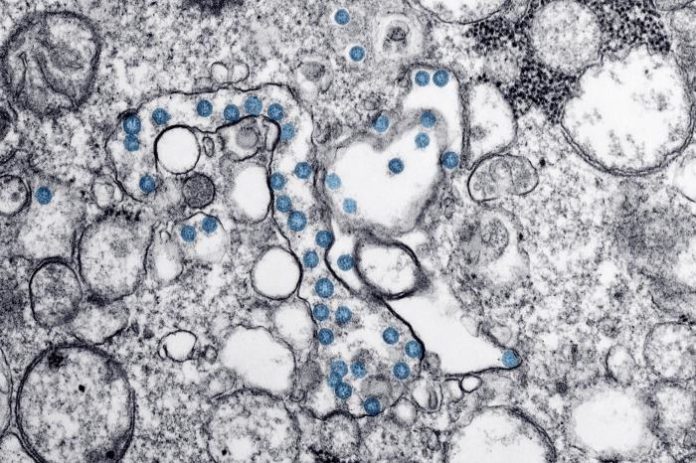
Estructura, replicación y tratamiento del coronavirus SARS-CoV-2
Un aspecto positivo del problema de la pandemia del virus SARS-CoV-2 es que hemos tenido la oportunidad de aprender muchas cosas en diversos aspectos. A mi, como bioquímico especializado en el trabajo con moléculas pequeñas y estructuras moleculares en general, me ha servido para aprender mucho sobre estructuras y biología molecular viral y adquirir una serie de conceptos epidemiológicos. Aún tenemos mucho que aprender. A lo largo de la pandemia, he ido montando una serie de figuras, con intención de su futuro uso en mis propias clases, y que ahora comparto aquí para quienes puedan resultar útiles. En caso de que se vea algún error, por favor, no dejen de comunicármelo.
El SARS-CoV-2
Es un virus de la familia coronaviridae (coronavirus), y el último de los siete coronavirus identificados hasta ahora que infectan a humanos. Dos de ellos, el CoV-229E y el CoV-OC43, provocan aproximadamente el 20% de los resfriados comunes. Se estima que un humano adulto adquiere una infección por coronavirus al menos una vez cada 2 o 3 años. Todos los virus de esta familia son similares: virus con genoma de RNA (¡no confundir con retrovirus! ¡no confundir RNA con DNA!) encapsulados o con envoltura, es decir, virus que tienen su cápside envuelta en una membrana lipídica, «robada» al individuo infectado; esta membrana contiene una serie de proteínas de membrana importantes para el mecanismo del virus. Un poquito más adelante entraremos en detalle con éstos conceptos.
Un virus no es un ser vivo y no se reproduce como las células. Más bien es una especie de «robot» que, una vez activado, ejecuta un programa de replicación determinado en su genoma. Pero, aunque no es un ser vivo, si puede evolucionar, ya que los ácidos nucleicos (en nuestro protagonista, el RNA) son moléculas que van sufriendo cambios en su secuencia y procesos de selección, que van dando lugar a la aparición de cuasiespecies. Esta evolución viral es sumamente importante en la progresión de los brotes epidémicos y en la aparición de nuevas infecciones virales (Recomiendo el artículo de José Campillo, un brillante científico mexicano experto en virus, listado en el apartado de referencias).

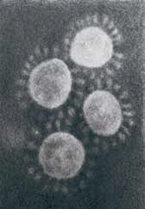
El coronavirus se denomina así por su forma visto con microscopio electrónico; para los científicos que lo veían, esas pequeñas bolitas rodeadas de puntitos se les asemejaba a una pequeña corona mecánica (rueda dentada o piñón) y, de ahí, el nombre. Los virus, en general, están formados fundamentalmente por una cápside, que consiste en una estructura de proteínas que sostienen el genoma viral (que puede ser DNA o RNA).

Algunos virus, como los rhabdovirus, o virus emblemáticos como el del mosaico del tabaco, son «no encapsulados», es decir, solo están formados por la cápside, el complejo de ácido nucleico y proteína que veis en la imagen anterior. El coronavirus, en cambio, es un virus «encapsulado» o «con envoltura»: su cápside está envuelta en una membrana de lípidos (grasa). Esta membrana contiene a su vez una serie de proteínas estructurales esenciales para el funcionamiento y para el montaje y estabilidad de las partículas virales: la glicoproteína de la envoltura, la glicoproteína de membrana y la famosa glicoproteína S o espícula, una gran proteína trimérica que protrude desde la membrana y confiere su aspecto característico a los coronavirus.

El encapsulamiento viral hizo pensar que el virus podría tener un comportamiento estacional: a las altas temperaturas veraniegas, la membrana lipídica se desestabiliza (al fin y al cabo es grasa y se funde, como la mantequilla, a alta temperatura). Es una de las razones por las que los virus de gripes y resfriados (también virus encapsulados) tienen comportamiento estacional. Sin embargo, nada es tan sencillo: las membranas se estabilizan gracias a las proteínas que contiene y a su composición, por lo que el SARS-CoV-2 se muestra más resistente al calor veraniego que el virus de la gripe. No obstante, aún no he visto claro este tema en la literatura científica y la temperatura bien podría ser una de las causas de que el rebrote de COVID-19 tiene una capacidad para contagiarse menor que en invierno, resultando en un número reproductivo inferior. O tal vez, el efecto de la temperatura se compensa con la mutación prevalente observada en la proteína S que aumentó su infectividad (ver figura de la arquitectura de la proteína S, mas abajo) Intentaré actualizar este punto conforme vea resultados científicos al respecto, porque de momento he sido muy especulativo. Pero siempre hay que considerar que hay una suma de factores en estos fenómenos tan complejos.

Cómo se replica el virus
El virus inicia su programa cuando se une a su receptor en un sujeto humano: la proteína ACE-2 (angiotensin-converting enzyme 2), que se expresa sobre todo en el tejido pulmonar, aunque también se encuentra en otros tejidos. Por ello, la infección afecta principalmente al aparato respiratorio, pudiendo ser desde prácticamente asintomática hasta producir una neumonía potencialmente letal; sin embargo, si se vuelve sistémica, puede afectar a otros tejidos provocando diversos síntomas y daños que, incluso, pueden dejar secuelas. En estos diversos efectos juega un papel fundamental la respuesta inmune del paciente. La inmunología de la infección es uno de los aspectos más complejos en todo el proceso. No soy experto en inmunología, por ello no voy a abundar en esta parte.
En el inicio de su programa de replicación es esencial la proteína S o espícula, un complejo proteico en la membrana del virus que se une con gran afinidad y especificidad a la proteína ACE-2 humana. Este proceso es un reconocimiento puramente químico: las estructuras proteicas son compatibles y se unen químicamente la una a la otra mediante enlaces por puentes de hidrógeno. Una vez que el virus se ha unido a su receptor, comienza el proceso de replicación.

El genoma viral del SARS-CoV-2 se conoce públicamente desde principios de enero de 2020 (gracias a ello existen pruebas diagnósticas como la PCR) y codifica para las diversas proteínas virales, que incluyen las cuatro proteínas estructurales que se ensamblan para construir nuevas partículas virales, así como proteínas no estructurales o nsp, que se expresan en el interior de la célula infectada y constituyen la maquinaria replicativa. El coronavirus tiene un genoma de RNA (nosotros tenemos un genoma de DNA) de algo más de 30 kilobases, es decir, algo más de 30000 «letras». El de los coronavirus es el genoma de RNA más grande conocido. Esto es debido a la inestabilidad del RNA respecto al DNA. Cuando, durante la evolución, se produjo la retrotranscripción del genoma RNA original de la primera protocélula al genoma DNA de la primera célula ancestral (al menos este es paradigma que manejamos actualmente), la mayor estabilidad del DNA condujo a la construcción de genomas más grandes, lo que aumentó la complejidad y diversidad biológica.
Durante la evolución viral, tras años y años de replicaciones en hospedadores vivos, las especies virales van desarrollando adaptaciones que les permiten ‘sobrevivir’. Una de ellas es la endoribonucleasa nsp15. Esta proteína protege al coronavirus de la respuesta inmune innata, una defensa antiviral del hospedador. Esta se produce gracias a que, durante la replicación, el virus genera un PAMP o patrón molecular asociado a patógeno. Este patrón es reconocido por receptores celulares y desencadena la síntesis de interferón tipo 1 y la respuesta antiviral innata. Sin embargo, el coronavirus produce la ribonucleasa nsp15, que rompe el patrón molecular asociado a su patogenicidad, inhibiéndose la síntesis del interferón, la síntesis de citoquinas proinflamatorias y la eliminación del virus. Es decir, el propio virus está modulando la respuesta inmune de su hospedador.

La ruptura de los dsRNA por la ribonucleasa viral es sólo uno de los mecanismos de adaptación generados por el coronavirus tras generaciones. Otro importante desarrollo evolutivo del SARS-CoV-2 se encuentra en su proteína S o espícula: el escudo de glicósidos. La proteína S es una glicoproteína, es decir, una proteína a la que se unen moléculas de diversos azúcares en sitios específicos y siguiendo un patrón determinado. Estos azúcares forman un escudo que protege a aproximadamente el 40% de la proteína de la unión de anticuerpos neutralizantes. Además tienen otras funciones importantes, tales como ayudar al plegamiento correcto de la proteína, proteger la proteína viral de las proteasas del hospedador y modular el proceso de entrada del virus en la célula.

El escudo de glicósidos es una adaptación evolutiva viral, es no sólo específico de cada virus, sino también cambia con el hospedador. Es una consecuencia de las relaciones entre virus y el sistema inmune de organismos infectados. El conocimiento de la composición de azúcares de la glicoproteína es importante para conocer la evolución del virus y es un posible punto de ataque en tratamientos antivirales. La glicosilación de la proteína y otros aspectos adaptativos y estructurales, así como la filogenia del virus, son elementos que, de haber sido el virus un producto creado en laboratorio, como algunas voces conspiranoicas afirman (y que implicaría, además, un enorme reto tecnológico cercano a la ciencia ficción), habrían permitido descubrirlo rápidamente por científicos de todo el mundo.
¿por qué el escudo de glicósidos evita la unión de anticuerpos neutralizantes? Porque los anticuerpos son proteínas, que interaccionan con puntos específicos (secuencias específicas de aminoácidos) de las proteínas del virus al que están atacando. Esos puntos específicos se llaman epítopos. Los anticuerpos se unen a los epítopos por interacciones químicas específicas, resultado de las cadenas de aminoácidos que forman las proteínas. Por ello, un anticuerpo es específico para un epítopo concreto. El sistema inmune puede generar anticuerpos contra diferentes epítopos, pero dependiendo de dónde esté el epítopo, el anticuerpo puede ser no neutralizante o neutralizante. Imaginad que la superficie de una proteína está cubierta de velcro. Los anticuerpos son dardos con el velcro complementario. Cuando lanzáis el dardo, el velcro se pega, pero no necesariamente se pega en un punto que bloquea la proteína, sino que puede pegarse en puntos no vitales. El escudo de glicósidos sería como cubrir parte del velcro con un film de plástico: el dardo no puede pegarse.

Los virus están en constante evolución, que podemos ver en muy poco tiempo. En la figura previa en la que vemos la estructura de la espícula, comento la importante mutación del aminoácido 460 de la cadena (aminoacido 614 de la secuencia de la proteína): un ácido aspártico ha cambiado por una glicina. Esta mutación da lugar a una variante del virus, la G460, que presenta una ventaja: mayor infectividad. Este sencillo cambio mejora la afinidad del virus por su receptor y la fusión de la membrana viral con la célula receptora, lo que le aporta una ventaja. El resultado es que se ha seleccionado la variante G14, desapareciendo prácticamente la original. Curiosamente, no se asocia la nueva variante con un aumento de la agresividad e, incluso, podría ser lo contrario. En una infección viral, infectividad no es sinónimo de agresividad. Un virus puede ser muy contagioso y poco agresivo. Aún está pendiente de demostración el efecto de éste cambio sobre la sintomatología y clínica de los pacientes.

En un resultado reciente, aún publicado en «preprint» y pendiente de validación y mas trabajo (Weissman et al., julio de 2020), los científicos sugieren que ésta mutación confiere al virus más vulnerabilidad ante los anticuerpos neutralizantes del hospedador, lo cual podría explicar la aparente paradoja por la que la mayor infectividad no se manifiesta como mayor agresividad.
¿por qué el SARS-CoV-2 es más grave y afecta a más tejidos que el SARS-CoV de la pandemia de 2003? La hipótesis de la furina.
Esta es una pregunta habitual: Si los virus son tan parecidos, ¿por qué el SARS de la pandemia de 2003 fue mucho menos severo que COVID-19? Una de las posibles razones se halla en el mecanismo por el cual el virus entra en la célula que infecta. Los coronavirus son virus encapsulados o con envoltura, es decir, su cápside viral está envuelta en una membrana lipídica que «robó» a la célula hospedadora anterior. Para introducir el genoma viral en la célula y poder activar su programa, primero debe activarse un mecanismo llamado fusión de membrana. Las dos membranas, la del virus y la de la célula, se integran, y la cápside viral pasa al citoplasma celular; allí se activa la replicación como vimos antes.
Para este proceso, el coronavirus dispone de la proteína S o espícula. Esta proteína de membrana está formada por tres subunidades iguales (es un trímero) y cada subunidad está diferenciada en dos dominios: el dominio S1 (formado fundamentalmente por láminas beta) y el dominio S2 (formado por hélices alfa, y con la estructura clásica de una proteína de membrana). Cuando la espícula reconoce al receptor, sufre un cambio de conformación: el motivo estructural de unión al receptor se despliega, para «engancharse» bien. Al conectarse al receptor, ocurre algo fundamental: una enzima proteasa (una especie de tijera molecular que corta proteínas) corta la espícula en la zona donde S1 y S2 se unen. Al cortarse, el dominio S2 comienza su trabajo: se inserta en la membrana de la célula y sufre un cambio conformacional que facilita que las dos membranas se peguen y se disuelvan una en la otra, creándose el poro de fusión. Este poro se agranda y la cápside queda dentro del citoplasma celular. Es decir, en el proceso de fusión es fundamental la intervención de una proteasa celular, que activa el proceso.

El corte por proteasa es clave. La invasión de la célula por coronavirus como SARS-CoV o los coronavirus de resfriados está mediada por una proteasa de membrana llamada TMPRSS2. Esta proteasa, cuya función fisiológica no se conoce bien, activa la fusión de la membrana y la entrada del genoma viral en la célula. Aquí tenemos una diferencia fundamental entre el SARS-CoV de 2003 o los coronavirus de resfriados y el coronavirus SARS-CoV-2: en éste aparece un sitio de corte por la proteasa furina. Este sitio de corte prepara a la proteína para que la TMPRSS2 active la fusión, realizando un segundo corte. La furina es una proteasa muy importante en diversos procesos celulares. Algunos investigadores lanzaron la hipótesis de que el corte por furina podría ser responsable de la mayor infectividad del SARS-CoV-2 respecto del virus de la pandemia de 2003, y también de su extensión a tejidos con menos expresión del receptor ACE-2, a diferencia de otros coronavirus respiratorios. Sin embargo, esto no está claro aún. De hecho, parece que el sitio de corte por furina no es esencial para la activación del virus, aunque al perderse, disminuye la infectividad. Un efecto curioso es que el sitio de corte por furina se pierde cuando el virus se cultiva in vitro. No se sabe qué ventaja aporta al virus perder el sitio de furina al replicarse en cultivos in vitro, pero, desde luego, este es otro argumento contra la paranoia del «virus creado en laboratorio».
Lo que sí parece más claro es que la mutación G614, mayoritaria ahora en las poblaciones virales, supone una ventaja evolutiva para el virus; la glicina 614 disminuye la interacción entre esa zona de la proteína y el dominio S2, aumentando la eficiencia de la activación por corte con proteasas. Ello mejora la infectividad viral, pero no necesariamente su agresividad. De hecho, es posible que, al debilitarse una interacción molecular (el ácido aspártico original, al tener carga, interaccionaba más fuertemente con el dominio S2, estabilizando la estructura de la espícula) el virus sea más vulnerable a ataques con anticuerpos o a degradación. Veremos si esto finalmente se demuestra.
En cualquier caso, a modo de resumen, las causas estructurales que hacen al SARS-CoV-2 más infectivo que otros coronavirus son: mayor flexibilidad y mejores interacciones entre aminoácidos de la proteína S con el receptor, lo que mejora la afinidad con la célula que va a ser infectada, además de mejoras en la activación de la fusión entre la membrana viral y la membrana celular.
Tratamientos farmacológicos: el nafamostat y el remdesivir
Lo que sí resultaba claro al estudiar es que las proteasas podrían ser un target farmacológico: si las inhibimos, bloqueamos la fusión del virus con la célula. Esta es la estrategia que se ha propuesto con algunos fármacos, como el nafamostat, un fármaco para el tratamiento de la pancreatitis y que actualmente está en fase de ensayo clínico para el tratamiento de COVID-19. A fecha de hoy no se que tal está resultando.

Otro importante target farmacológico está en la que, para mi, es una de las proteínas más interesantes del coronavirus y, en general, de todos los virus RNA: la enzima que replica el genoma del virus o RNA polimerasa dependiente de RNA. Esta enzima está altamente conservada en la evolución de todos los virus RNA y, quizá, nos conecta con el ¡mismísimo origen de la vida!

La homología y evolución de las polimerasas es un tema muy complejo como para tratarlo aquí. En base a las estructuras y a las ideas de algunos autores, podríamos proponer esta línea de la evolución de las RNA y DNA polimerasas en la vida terrestre.

Según esta línea evolutiva, las polimerasas de virus RNA como el coronavirus pudieron haberse originado hace 4000 millones de años, con el nacimiento del genoma. Es una hipótesis, y aún queda mucho por estudiar para llegar a comprender todo este proceso de evolución.
Volviendo a nuestro coronavirus, las RNA polimerasas son metaloproteínas o metaloenzimas, es decir, son enzimas que ejercen su acción catalítica gracias a la presencia de iones metálicos. En éste caso, iones de manganeso (las proteínas humanas similares utilizan magnesio). El hecho de que esta enzima tenga un funcionamiento optimo con manganeso o con iones de hierro, podría ser una consecuencia de su origen ancestral, en tiempos pre-oxigenación de la atmósfera, cuando el manganeso y hierro eran aún móviles y no habían sido precipitados en forma de óxidos y otros minerales (pero esto es un poco especulativo). Otro aspecto que podría apuntar a un origen ancestral es que es una polimerasa que no requiere cebador.
Un aspecto importante es que los iones de zinc pueden sustituir al manganeso en la proteína, pero hacen que no funcione. Es decir, altas cantidades de zinc actuarían como inhibidores de la polimerasa, y, por tanto, bloquearían la replicación del genoma del virus. Esta es la base de que se intentara tratar la infección con hidroxicloroquina . Este fármaco contra la malaria es un ionóforo de zinc, es decir, aumenta la concentración intracelular de zinc. El exceso de zinc favorecería su unión a la polimerasa durante la síntesis de la proteína, frente al manganeso. La polimerasa con zinc resultante sería inactiva, disminuyendo la síntesis de nuevas copias del genoma viral. El tratamiento con éste fármaco ha sido muy polémico y, finalmente, parece que hay un consenso en torno a su falta de eficacia real en el COVID-19. Sin embargo, me sigo preguntando si el balance de metales y las variaciones en los niveles intracelulares de zinc podrían explicar en parte la gran variabilidad que muestra la infección por el coronavirus.
Otro tratamiento antiviral que parece que ha funcionado relativamente bien en pacientes hospitalizados graves es el remdesivir. Éste fármaco es un inhibidor de la RNA polimerasa viral, actuando como un «imitador» de los componentes del RNA. Cuando la RNA polimerasa está sintetizando la nueva copia de RNA y se encuentra con el fármaco, éste bloquea la enzima, deteniéndose la copia.

¿que vemos entonces en la figura de arriba? El Remdesivir es un antiviral (no confundir con antibiótico ni con anticuerpo) que se basa en una estrategia clásica de ataque: para que el virus pueda replicarse y generar nuevos virus, necesita replicar su RNA genómico y fabricar las copias de RNA que necesita para generar las proteínas del virus. El RNA es un polímero formado por nucleótidos de las bases adenina, guanina, uracilo y citosina. Para fabricar el RNA necesita utilizar una «fotocopiadora»: la polimerasa. Ésta toma un RNA molde y hace copias. La «fotocopiadora» usa lo que voy a fotocopiar como modelo, va cogiendo folios en blanco y me saca la copia. Esos folios en blanco son los nucleótidos. Seguramente veréis totalmente lógico ésto: si yo le proporciono un nucleótido fake, diseñado de modo que sea parecido al nucleótido natural, pero que «atasque la fotocopiadora», igual consigo detener la replicación del virus. Es como si estoy haciendo fotocopias y, entre el taco de folios en blanco (nucleótidos), inserto uno que entre en la fotocopiadora, pero que sea muy rígido, de modo que ésta se atasque.
Pues esto mismo es lo que hace el fármaco remdesivir. El fármaco es como un misil (lo que llamamos profármaco). El misil, una vez disparado, suelta la primera fase y libera su cabeza de guerra (warhead, el fármaco propiamente dicho) que impacta en la polimerasa. La polimerasa captura la warhead activa, la confunde con el nucleótido natural adenosina trifosfato (ATP) y la inserta en el nuevo RNA que está construyendo. Esto hace que la polimerasa se atasque y no pueda seguir fabricando RNA.
Si os fijáis en la figura, veis que la «warhead» del fármaco tiene dos características químicas fundamentales que la diferencian de la adenina. La primera es que tiene un grupo ciano (CN) en la posición 1′ de la molécula. Este grupo voluminoso es lo que provoca el «atasco de la fotocopiadora». Pero no queremos que nadie desatasque la fotocopiadora para que pueda seguir funcionando, ¿verdad?. Como es lógico, la Naturaleza tiene sistemas que «desatascan» el problema, rompiendo el nucleótido fake y reemplazandolo por uno correcto. Para evitar ésto, los químicos se dieron cuenta de que haciendo otra modificación, que era usar el C-nucleótido, en vez del N-nucleótido natural, el sistema de copia del virus no podía «desatascar» la polimerasa. A ésta solución no se llega de urgencia. El desarrollo de los fármacos basados en «nucleótidos fake» que se usan como antivirales y quimioterápicos, comenzó en los años 1940. Mi tesis doctoral versaba sobre un nucleótido «fake»: las tiopurinas, así que es un tema que me resulta particularmente interesante.
No obstante, aunque antivirales como el remdesivir puedan resultar útiles, al menos en casos hospitalizados y graves, el futuro del tratamiento se va a encontrar en la biología molecular e inmunología: vacuna, anticuerpos… Esta pandemia está mostrando al público que los científicos tienen dudas y tienen que aprender mucho y sobre la marcha, que cambian de idea y parecer según aparecen nuevas evidencias, que el desarrollo de las investigaciones es largo y costoso, que la burocracia y la política son lastres en el desarrollo científico. Esperemos que, junto con la solución al problema, tengamos un gran aprendizaje todos.
Referencias consultadas
Campillo-Balderas-2020-¿-Qué-fue-primero-los-virus-o-las-células-Which-came-first-viruses-or-the-cellsDownload
Andersen, K. G. et al. (2020) ‘The proximal origin of SARS-CoV-2’, Nature Medicine, 26(4), pp. 450–452. doi: 10.1038/s41591-020-0820-9.
Gao, Y. et al. (2020) ‘Structure of the RNA-dependent RNA polymerase from COVID-19 virus.’, Science (New York, N.Y.), 7498(April), pp. 1–9. doi: 10.1126/science.abb7498.
Hackbart, M., Deng, X. and Baker, S. C. (2020) ‘Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors’, Proceedings of the National Academy of Sciences, 117(14), pp. 8094–8103. doi: 10.1073/pnas.1921485117.
Heald-Sargent, T. and Gallagher, T. (2012) ‘Ready, set, fuse! the coronavirus spike protein and acquisition of fusion competence’, Viruses, 4(4), pp. 557–580. doi: 10.3390/v4040557.
Henderson, R. et al. (2020) ‘Controlling the SARS-CoV-2 spike glycoprotein conformation’, Nature Structural and Molecular Biology. Springer US. doi: 10.1038/s41594-020-0479-4.
Hoffmann, M. et al. (2020) ‘SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor’, Cell, 181(2), pp. 271-280.e8. doi: 10.1016/j.cell.2020.02.052.
Kaushik, N. et al. (2017) ‘Zinc Salts Block Hepatitis E Virus Replication by Inhibiting the Activity of Viral RNA-Dependent RNA Polymerase’, Journal of Virology. Edited by J.-H. J. Ou, 91(21), pp. e00754-17. doi: 10.1128/JVI.00754-17.
Kirchdoerfer, R. N. and Ward, A. B. (2019) ‘Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors’, Nature Communications. Springer US, 10(1), pp. 1–9. doi: 10.1038/s41467-019-10280-3.
Koonin, E. V. (2014) ‘The origins of cellular life’, Antonie van Leeuwenhoek, International Journal of General and Molecular Microbiology, 106(1), pp. 27–41. doi: 10.1007/s10482-014-0169-5.
Korber, B. et al. (2020) ‘Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus’, Cell, pp. 1–16. doi: 10.1016/j.cell.2020.06.043.
Matheson, N. J. and Lehner, P. J. (2020) ‘How does SARS-CoV-2 cause COVID-19?’, Science, 369(6503), pp. 510–511. doi: 10.1126/science.abc6156.
Ogando, N. S. et al. (2020) ‘SARS-coronavirus-2 replication in Vero E6 cells: replication kinetics, rapid adaptation and cytopathology’, Journal of General Virology, (March). doi: 10.1099/jgv.0.001453.
Wang, Qihui et al. (2020) ‘Structural and functional basis of SARS-CoV-2 entry by using human ACE2’, Cell, pp. 1–11. doi: 10.1016/j.cell.2020.03.045.
Shang, J. et al. (2020) ‘Structural basis of receptor recognition by SARS-CoV-2.’, Nature. Springer US, pp. 1–8. doi: 10.1038/s41586-020-2179-y.
Te Velthuis, A. J. W., Van Den Worm, S. H. E. and Snijder, E. J. (2012) ‘The SARS-coronavirus nsp7+nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension’, Nucleic Acids Research, 40(4), pp. 1737–1747. doi: 10.1093/nar/gkr893.
te Velthuis, A. J. W. et al. (2010) ‘Zn2+ Inhibits Coronavirus and Arterivirus RNA Polymerase Activity In Vitro and Zinc Ionophores Block the Replication of These Viruses in Cell Culture’, PLoS Pathogens. Edited by R. Andino, 6(11), p. e1001176. doi: 10.1371/journal.ppat.1001176.
Turoňová, B. et al. (2020) ‘In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges’, 5223(August), pp. 1–12. doi: 10.1101/2020.06.26.173476.
Walls, A. C. et al. (2020) ‘Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein’, Cell, 181(2), pp. 281-292.e6. doi: 10.1016/j.cell.2020.02.058.
Wang, Qihui et al. (2020) ‘Structural and functional basis of SARS-CoV-2 entry by using human ACE2’, Cell, pp. 1–11. doi: 10.1016/j.cell.2020.03.045.
Weissleder, R. et al. (2020) ‘COVID-19 diagnostics in context’, Science Translational Medicine, 12(546), pp. 1–7. doi: 10.1126/scitranslmed.abc1931.
Weissman, D. et al. (2020) ‘D614G Spike Mutation Increases SARS CoV-2 Susceptibility to Neutralization.’, medRxiv, (573), p. 2020.07.22.20159905. doi: 10.1101/2020.07.22.20159905.
White, J. M. et al. (2008) ‘Structures and Mechanisms of Viral Membrane Fusion Proteins’, Critical Reviews in Biochemistry and Molecular Biology, 43(3), pp. 189–219. doi: 10.1080/10409230802058320.Structures.
Wu, Y. et al. (2020) ‘A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2’, Science, p. eabc2241. doi: 10.1126/science.abc2241.
Xia, S. et al. (2020) ‘The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin’, Signal Transduction and Targeted Therapy, 5(1). doi: 10.1038/s41392-020-0184-0.
Xu, Y. et al. (2004) ‘Structural basis for coronavirus-mediated membrane fusion: Crystal structure of mouse hepatitis virus spike protein fusion core’, Journal of Biological Chemistry, 279(29), pp. 30514–30522. doi: 10.1074/jbc.M403760200.
Yin, W. et al. (2020) ‘Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir’, Science, 1560(May), p. eabc1560. doi: 10.1126/science.abc1560.
Zhang, L. et al. (2020) ‘The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity.’, bioRxiv : the preprint server for biology. doi: 10.1101/2020.06.12.148726.