PROTEÍNA P53: UNA MUERTE NECESARIA
Realizado por Leyre Navajo Gómez y Alejandro Muñoz Benito
INTRODUCCIÓN
La proteína p53 es conocida como el “guardián del genoma” por su papel en la regulación del ciclo celular, pero también participa en el mantenimiento de la homeostasis o en la respuesta al estrés. P53 es un factor de transcripción que en su forma activa va a unirse a secuencias del ADN para inducir o reprimir la expresión de genes, produciendo la inhibición del ciclo celular y la activación de la apoptosis.
De forma fisiológica, p53 solo se activa cuando se ha producido un daño en el ADN que los sistemas de reparación de ADN no pueden solucionar y conduce a la célula a procesos de muerte celular para evitar que los daños sean heredados por las células hija, de esta manera está involucrado en el mantenimiento de la integridad génica. Cuando las células no presentan daño en el ADN, p53 va a ser degradada por el proteosoma para impedir que células sanas entren en apoptosis y puedan continuar el ciclo celular.
P53 desarrolla funciones muy importantes dentro de las células, por eso cualquier fallo puede derivar en enfermedades graves como el cáncer. Se ha visto que el 50% de los tumores presentan mutada esta proteína lo que evidencia su importancia y el gran interés que hay en su estudio. Las mutaciones en p53 hacen que pierda su actividad supresora y comience a actuar como una oncoproteína cuya acción conduce a la aparición de células tumorales que escapan de la muerte celular y pierden la regulación del ciclo celular. Generalmente, los tumores con mutación en p53 tienen peor pronóstico, porque aunque se trate al paciente con radioterapia o quimioterapia la vía que conduce a la apoptosis está inhibida y las células pese a la acumulación de tumores seguirán proliferando. Por ello, existen muchos estudios centrados en esta proteína con el fin de entender su estructura, regulación y función para poder desarrollar mecanismos que puedan impedir el progreso de los tumores.
ESTRUCTURA
El gen TP53 se encuentra en el brazo corto del cromosoma 17 (17 p13) y codifica para la proteína nuclear p53, se trata de un homotetrámero donde cada monómero lo constituyen 393 aminoácidos con distintos dominios funcionales. En los tejidos humanos se encuentran 9 isoformas distintas de esta proteína siendo la expresión de cada una de ellas dependiente del tejido en el que se encuentra.
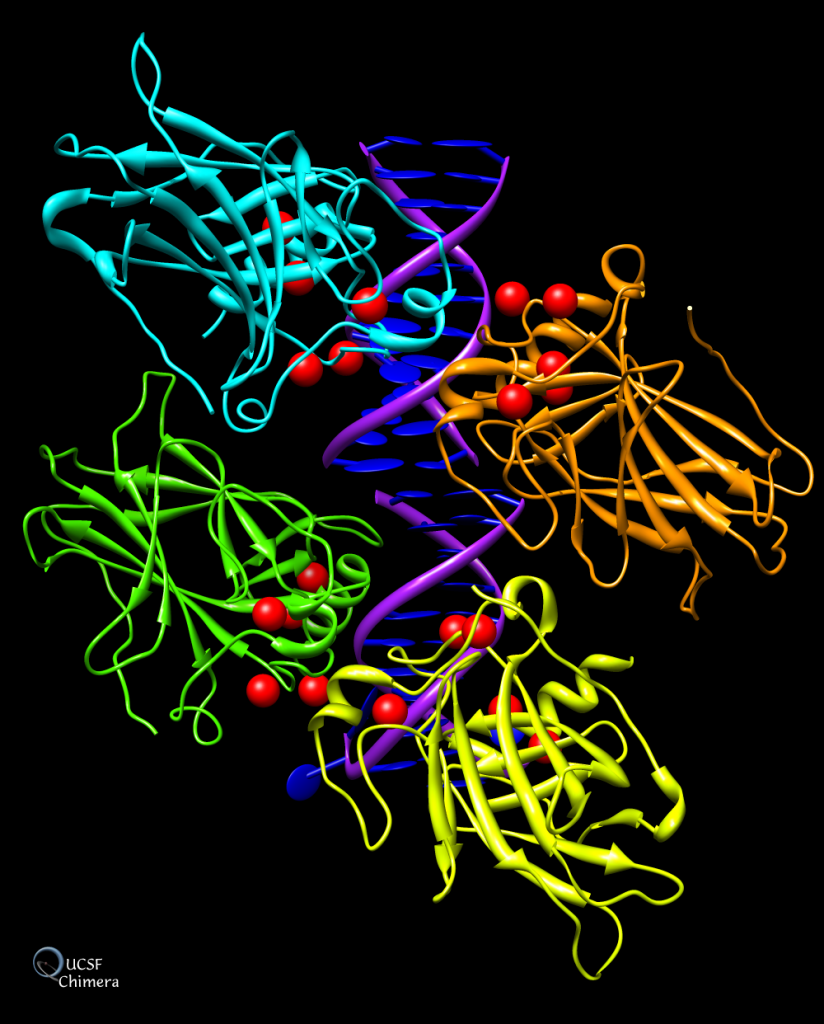
Las cuatro regiones que constituyen cada monómero son:
- Dominio aminoterminal. p53 contienen dos dominios de transactivación (AD1, AD2) que son necesarios para activar la transcripción de los genes diana, una región rica en prolinas que participa en la regulación de la apoptosis Y una señal de exportación nuclear (NES). Ante diferentes señales, la proteína sufre modificaciones postraduccionales (fosforilaciones) que conducen a su separación de la proteína MDM2 permitiendo su activación. (2)
- Dominio de unión de ADN (Core). Localizado entre los aminoácidos 94-294, es un dominio resistente a proteasas y con una secuencia conservadas en otros miembros de su familia (p63 y p73) Este dominio se encarga de reconocer y unirse de manera específica a secuencias promotoras del ADN para regular la transcripción de los genes diana. Su estructura la constituyen dos láminas β antiparalelas, dos grandes “loops” (L2 y L3) que se estabilizan con Zinc y un motivo loop lámina-hélice al final del dominio. Este dominio tiende a ser rico en aminoácidos básicos que permiten la unión al ADN. (3)
- Dominio de tetramerización (Tet). Se encuentra entre los aminoácidos 320-360 y participa en la formación del tetrámero. Lo forma un dímero de dímeros, con dos laminas-β y dos α-hélices entrecruzadas (4). Consta de una región flexible de 30 aminoácidos aproximadamente, que conecta este dominio con cada uno de los dominios centrales.
- Dominio carboxiloterminal. Localizado entre los aminoácidos 363-393, presenta muchos residuos básicos (arginina y lisina) siendo capaz de unirse al ADN de forma inespecífica por interacciones electroestáticas. Ante diversas señales de estrés sufre modificaciones postraduccionales (glicosilación, fosforilación o ubiquitinación). (1)

REGULACIÓN
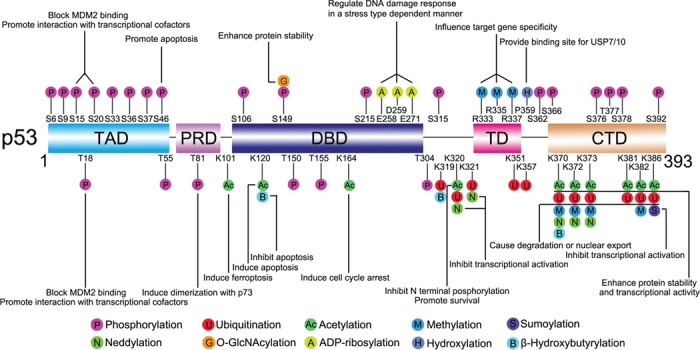
P53 sufre modificaciones postraduccionales que incluyen fosforilación, ubiquitinación, acetilación y metilación; todas ellas están implicadas en la estabilidad, conformación, localización e interacciones de p53. La desregulación de alguna de estas rutas aparecen entre las principales causas de enfermedades del desarrollo asociados con p53, especialmente en cánceres.
La estructura multimodular de p53 hace que presente múltiples sitios donde pueden realizarse modificaciones que influirán en su función, estas modificaciones pueden ser reversibles por la acción de otras enzimas.
https://pubmed.ncbi.nlm.nih.gov/31282934/
Algunos ejemplos de modificación postraduccional son (5):
- Ubiquitinación. La ubiquitina es una pequeña proteína que se une a las proteínas que van a ser degradadas, se trata de una cascada de reacciones enzimáticas donde participan ligasas (E1: activación, E2: ligación, y E3: conjugación) cuyo objetivo es generar proteínas marcadas con una cola de ubiquitinas que se dirigirán al proteosoma para ser degradadas. Mdm2 es la principal ubiquitina ligasa E3, puede modificar p53 en seis residuos de lisina dentro de CTD. Cuando en la célula hay niveles altos de mdm2 se conduce a la poliubiquitinación y degradación nuclear de p53; mientras que si los niveles son bajos se promueve la monoubiquitinación y exportación nuclear de p53, en el citoplasma p53 realiza funciones diferentes a las de un factor de transcripción. Por tanto, mdm2 es un regulador negativo de p53.
- Fosforilación. En p53 aparecen muchos sitios ricos en treonina y serina que pueden fosforilarse por acción de Ser-Thr quinasas, la mayoría se acumulan en el extremo N-terminal y se modifican como respuesta al estrés celular para iniciar la reacción de p53. Algunos sitios (como T55, S376 y S378) se fosforilan constitutivamente en células no estresadas, en condiciones normales de crecimiento celular el factor TAF1 fosforila p53 en T55 promoviendo su degradación a través del proteosoma. En respuesta al daño del ADN, la fosfatasa 2A puede desfosforilar T55, mejorando la estabilidad de p53 para promover la detención del ciclo celular. Cuándo ocurre la fosforilación (antes o después de la activación de p53) y qué efectos tiene la fosforilación en p53 (activación o represión) son muy variables en diferentes condiciones. Hay fosforilaciones que impiden la unión a mdm2 a p53 para mejorar su estabilidad, otras modificaciones como fosforilaciones en el dominio C-terminal que promueven la unión de p53 al ADN
- Acetilación. CBP/p300 podría acetilar p53 en varias lisinas C-terminales conduciendo a la unión de p53 con el gen diana para activar vías posteriores como detención del ciclo celular, senescencia o apoptosis. Además, la acetilación puede impedir la ubiquitinación de p53 y aumentar su estabilidad. Dos sitios modificados por CBP/p300 son K164 y K101, la acetilación de K164 promueve la inducción de la detención del ciclo celular mediante la expresión de p21 y la acetilación de K101 es fundamental para la regulación de la ferroptosis. En respuesta al daño del ADN, también se puede acetilar p53 en K320 para impedir la fosforilación de serinas en el extremo N-terminal de p53 y permitir la expresión de genes específicos que promueven la supervivencia celular, no la apoptosis . La p53 acetilada puede ser desacetilada por varias desacetilasas como la histona desacetilasa-1 (HDAC1), el cual puede ser reclutado por mdm2.
- Metilación. Al igual que la acetilación, es una marca epigenética importante en la lisina de las colas de histonas, también destaca la metilación de arginina. Esta modificación puede estabilizar p53 y restringirlo en el núcleo. También se asocia con una transcripción mejorada de algunos genes diana como p21. Destaca la proteína arginina metiltransferasa 5 (PRMT5) que se encarga de la metilación de p53 en R333, R335 y R337, esta reacción afecta a la especificidad del gen diana de p53 haciéndola más sensible a ciertos promotores o potenciadores.
FUNCIÓN
La proteína p53 en condiciones normales está presente en su forma inactiva a muy baja concentración, mientras que en situaciones de estrés celular, falta de nutrientes o de daño en el ADN, se produce su activación y estabilización. Si el daño es leve detiene la progresión del ciclo celular para permitir su reparación y posteriormente reiniciarlo. Ante un daño grave o irreparable, activa el proceso de apoptosis, reduciendo de esta manera la posibilidad de que células con mutaciones puedan sobrevivir y contribuir al proceso de carcinogénesis. (6)(7)
Por ello, dentro de las funciones más importantes de p53 encontramos la detención del ciclo celular y activación de la apoptosis además de otras funciones secundarias como regular ciertos procesos metabólicos, la respuesta antioxidante y la reparación del ADN. Todas estas funciones confluyen en la idea de que p53 tiene como misión principal ser un supresor de tumores. (6)(7)
DETENCIÓN DEL CICLO CELULAR
La detención del ciclo celular mediada por p53 (7)
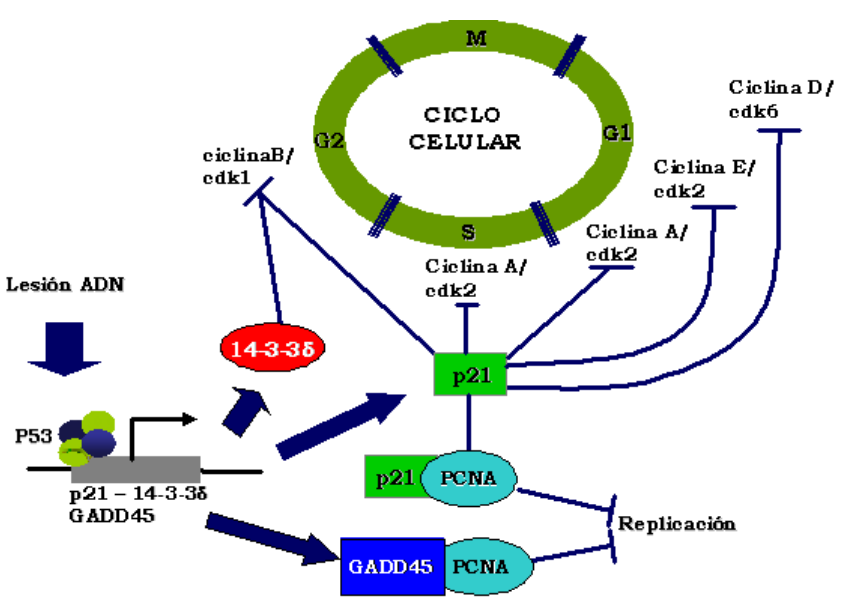
Está mediada principalmente por la activación transcripcional de p21/WAF1. P53 se une a dos sitios aguas arriba del promotor de P21. El sitio 5′ en el promotor p21 es uno de los sitios de unión a p53 más fuertes analizados, con una constante de disociación muy baja (nM).
El ARNm de p21 es altamente inducido después de la activación de p53. p21 se une a los complejos ciclina E/Cdk2 y ciclina D/Cdk4 para causar la detención de G1 en el ciclo celular. La inhibición de Cdk2 y Cdk4 por p21 bloquea la fosforilación de pRb, promueve la unión de pRb a E2F1 y promueve el silenciamiento de la transcripción de los objetivos E2F1 imprescindibles para la replicación del ADN y la progresión del ciclo celular. p21 también interactúa con el antígeno nuclear celular proliferante (PCNA) e inhibe la replicación del ADN, lo que puede contribuir a su actividad de detención del ciclo celular. Se ha demostrado que la ausencia del factor p21 causa deficiencias en la detención del ciclo celular después del lo que indica que es un mediador importante por p53.
Sin embargo, estas células nulas de p21 no son completamente defectuosas para la detención de G1, lo que se sugiere que otros genes diana de p53 también contribuyen a la detención del ciclo celular.
La activación de p53 también detiene las células en las fases G2/M. Aunque p21 también puede inhibir la ciclina B/Cdc2 para inhibir la progresión del ciclo celular a través de la mitosis, otros genes diana de p53, como el 14-3-3σ, pueden participar en el bloqueo de la transición G2/M.
También se ha demostrado que la represión p53 del promotor cdc25C promueve la detención de G2 / M después del daño del ADN. Estudios recientes ex vivo sugieren que la inducción de micro ARN mir34a contribuye a la detención del crecimiento por p53. Sin embargo, la eliminación de la familia de genes mir34 en ratones no afectó la detención y la apoptosis mediadas por p53, lo que sugiere que la función de mir34 in vivo es limitada o redundante.
En las células de mamíferos, la detención del ciclo celular mediada por p53 proporciona importantes funciones como punto de control:
- Las roturas de doble cadena de ADN que se producen por acción de la radiación ionizante activa la (ATM) quinasa que inhibe la actividad de la ligasa MDM2 E3 a través de la fosforilación, causando una rápida acumulación de p53 e inducción de p21. Al detener las células en la fase G1 se da tiempo a la reparación de roturas de doble cadena potencialmente letales.
- P53 mantiene la integridad cromosómica y mejora la supervivencia de las células dañadas ya que además de forzar un punto de control del ciclo celular, p53 también regula un grupo de genes implicados en la recombinación y reparación del ADN.
- P53 regula los genes implicados en la formación de heterocromatina para facilitar la reparación oportuna de las roturas de la cadena de ADN.
PROMOVER APOPTOSIS
La apoptosis se define como una forma de muerte celular programada en la que las células «se suicidan» fragmentándose en trozos empaquetados de membrana denominados cuerpos apoptóticos. (9)
En la activación de p53, ciertos tipos de células (leucemia o fibroblastos transformados) sufren predominantemente apoptosis en lugar de detención del ciclo celular. (7)
Las limitadas anomalías del desarrollo asociadas con la mayoría de los ratones p53-nulos indican que la proteína p53 puede ser prescindible, en gran medida, para los procesos apoptóticos que ocurren durante el desarrollo normal. (9)
Sin embargo, en condiciones de estrés celular que ponen en peligro la integridad del genoma, p53 se vuelve crucial para restringir la proliferación celular inapropiada. De esta manera, p53 evita la expansión clonal de las células que poseen defectos genéticos peligrosos, restringiendo así la neoplasia. Las situaciones de estrés celular que pueden inducir la apoptosis mediada por p53 incluyen daño en el ADN, hipoxia, escasez de ribonucleótidos y estrés oxidativo. (9)
Además, p53 puede inducir apoptosis sin detención aparente del crecimiento, por ejemplo, en células que tienen expresión desregulada de los protooncogenes c-myc o adenovirus 12S E1A, o el factor de transcripción E2F-1 entre otros, provocan una acumulación y estabilización de p53. (8)
La separación de las vías dependientes de p53 para la detención del crecimiento y la apoptosis puede basarse en la inducción selectiva de productos génicos específicos para cada vía. (8)
Se ha demostrado que p53 regula al alza genes proapoptóticos como Bax y suprime genes antiapoptóticos como BCL-2, mediante la activación transcripcional o represión, alterando así las proporciones relativas de Bax a Bcl-2 y cambiando el equilibrio hacia la apoptosis. (8)
p53 también induce directamente la transcripción de IGF-bp3, una proteína que puede inhibir la actividad antiapoptótica de IGF-1, a través de un motivo de consenso p53 en la región promotora. Esta proteína también puede inducir apoptosis, a través del secuestro de IGF-1. (8)
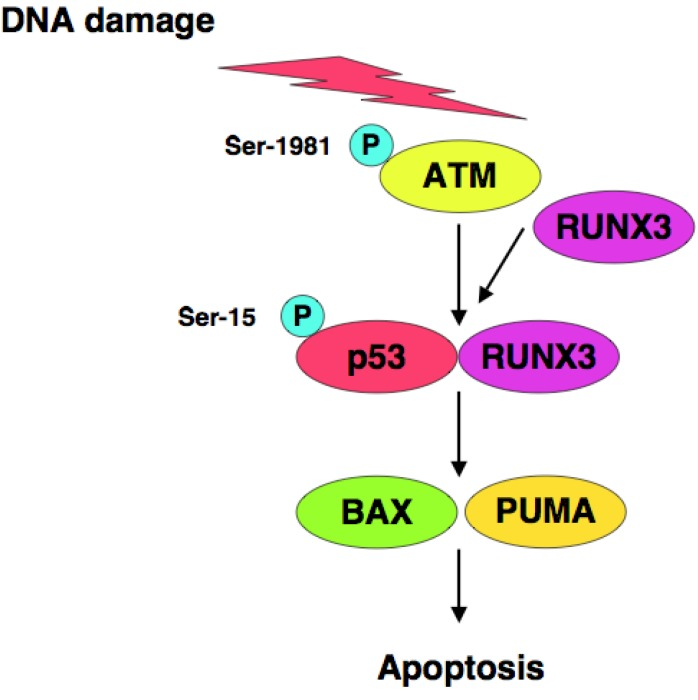
Por otro lado, Se sugiere que podría existir una interacción funcional entre p53 y una proteína llamada RUNX3 en respuesta al daño del ADN, mejorando así la actividad proapoptótica de p53. Cabe destacar que RUNX3 recluta formas fosforiladas de ATM a p53 en SER-15 y por lo tanto activa p53 (10)
La demostración de que la apoptosis mediada por p53 puede ocurrir en ausencia de síntesis de proteínas o ARN implicó que la transactivación específica de secuencias SST no siempre es imprescindible para la apoptosis. Posteriormente, las investigaciones sobre la capacidad apoptótica de los mutantes p53 deficientes en SST dieron lugar a resultados contradictorios. (9)
Se ha propuesto por tanto que la apoptosis mediada por p53 ocurre a través de una combinación de mecanismos dependientes de SST e independientes de SST, actuando de forma sinérgica. En condiciones fisiológicas la apoptosis óptima requiera una combinación de ambos mecanismos. (9)
Es pertinente señalar un importante gen diana de p53 que aparentemente no contribuye a la apoptosis. El gen p21 inducible por p53 codifica un inhibidor de quinasas dependientes de ciclina. Los estudios de fibroblastos deficientes en p21 revelaron la contribución significativa de la inducción de p21 a la detención del ciclo celular en G1. Sin embargo, los mismos estudios también mostraron claramente que p21 no era necesaria para la apoptosis mediada por p53. (9)
Algunos mutantes de p53 pueden transactivar p21 e inducir la detención del ciclo celular, pero no pueden inducir IGF-bp3 o Bax, y por lo tanto muestran una actividad pro-apoptótica deteriorada. Esto indica que la selectividad del promotor de los mutantes p53 puede determinar si p53 induce la detención del crecimiento o la apoptosis. (8)
Se ha sugerido que la inhibición de la apoptosis inducida por p53 está mediada por MDM2, que está regulada al alza por p300, pero que inhibe la función de p53. MDM2 se une al dominio de activación transcripcional de p53, induce una rápida degradación de p53 y bloquea su capacidad para regular genes diana y ejercer efectos antiproliferativos y proapotóticos. (8)
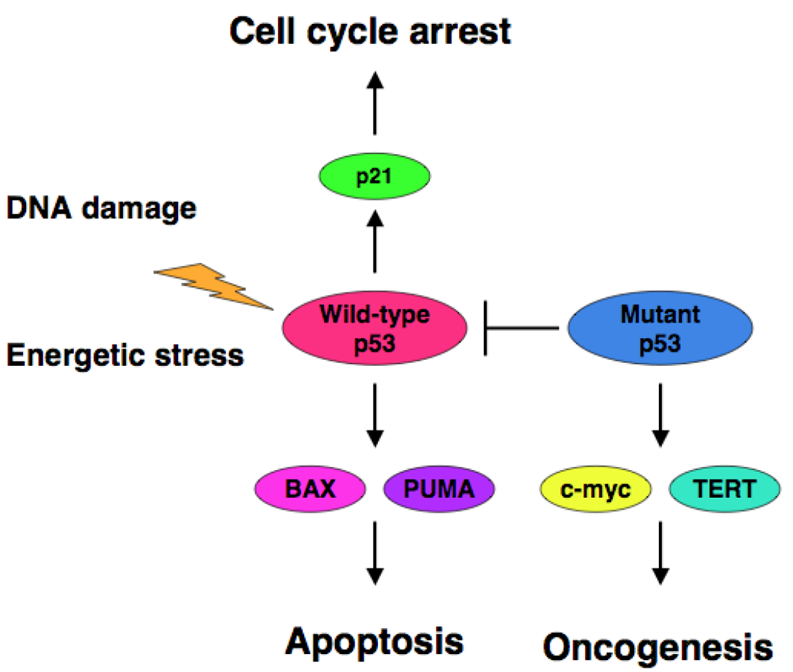
RELACIÓN P53 Y CÁNCER
Ante un daño grave o irreparable en el ADN, p53 activa procesos de apoptosis para disminuir la posibilidad de que las células mutadas sobrevivan y transmitan esos daños a las células hija. Cualquier mutación en p53 u otras proteínas que regulen su función inhiben la actividad de p53 condiciendo a carcinogénesis.
La inactivación de p53 produce un desequilibrio entre la división celular y la muerte celular, que provoca la proliferación continua de la célula. Además, las células con ADN dañado no pueden ser eliminadas y las mutaciones serán heredadas e irán acumulándose en las nuevas células. Como consecuencia, las células tumorales presentan un nuevo fenotipo que las diferencian de las células normales del tejido donde se encuentran, son células que presentan una función y morfología diferentes a las células de las que proceden.
BIBLIOGRAFÍA
1. Appella, E, Anderson, CW (2001.a). Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem., 268, 10:2764-72.
2. Candau, R, Scolnick, DM, Darpino, P, Ying, CY, Halazonetis, TD, Berger, SL
(1997). Two tandem and independent sub-activation domains in the amino terminus
of p53 require the adaptor complex for activity. Oncogene, 15, 7:807-16
3. Cho, Y., Gorina, S., Jeffrey, P. D. & Pavletich, N. P (1994). Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 265, 346-55
4. Jeffrey, PD, Gorina, S, Pavletich, NP (1995). Crystal structure of the tetramerization
domain of the p53 tumor suppressor at 1.7 angstroms. Science, 267, 5203:1498-502
5. Liu, Y., Tavana, O. & Gu, W. (2019). p53 modifications: exquisite decorations of the powerful guardian. Journal of Molecular Cell Biology, 11(7), 564-577.
6. Liu, J., Zhang, C., Hu, W. & Feng, Z. (2018). Tumor suppressor p53 and metabolism. Journal of Molecular Cell Biology, 11(4), 284-292.
7. Chen, J. (2016). The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harbor Perspectives in Medicine, 6(3), a026104.
8. Discussion. (1999b). Biochemical Pharmacology, 58(7), 1089-1095.
9. Gottlieb, T. M. & Oren, M. (1998). p53 and apoptosis. Seminars in Cancer Biology, 8(5), 359-368.
10. Ozaki, T. & Nakagawara, A. (2011). Role of p53 in Cell Death and Human Cancers. Cancers, 3(1), 994-1013