Anticuerpos monoclonales como terapia para el Alzheimer
Sofía Pérez Rubio y Celia Navas González
Generalidades de los anticuerpos monoclonales
Los anticuerpos (Ac) son moléculas lipoproteicas que forman parte del sistema inmunitario humoral, y los cuales reconocen de manera específica antígenos (Ag). Dicha unión Ac-Ag es reversible y su fuerza es lo que conocemos como afinidad. En el caso concreto de los anticuerpos monoclonales, estos son producidos por un solo clon activado de células B, por lo cual son activos frente a un determinante antigénico único (1,2).
A nivel general, los anticuerpos poseen una forma espacial de Y, y constan de dos cadenas ligeras y dos pesadas, unidas por puentes disulfuro (Figura 1). A su vez, cada una de las cadenas consta de una región constante (CL y CH respectivamente) que se mantiene, y una región variable (VL y VH) en los extremos, que es la que genera especificidad. También es de importancia su extremo carboxiterminal, del cual dependerá la respuesta efectora que ocasiona el Ac al determinar las distintas uniones a receptores de membrana, y los extremos aminoterminales, los cuales reconocen y se unen a los antígenos (1).
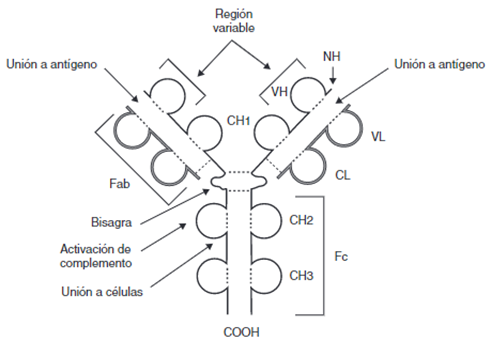
CH: dominios de la región constante de la cadena pesada; CL: dominio constante de la cadena ligera; COOH: extremo carboxiterminal; Fab y Fc: fragmentos resultantes de proteólisis; NH: extremo aminoterminal; VH: dominio variable de la cadena pesada; VL: dominio variable de la cadena ligera; – – -: puentes disulfuro. Extraído de (1)
Los anticuerpos monoclonales en los que nos basaremos fueron descubiertos por los científicos Milstein y Köhler en la década de los 70, por lo cual serían galardonados con un premio Nobel posteriormente (1). Como decimos, estos anticuerpos son los que provienen de una misma célula B y poseen la misma especificidad, por lo que para conseguirlos fusionaron células de mieloma de ratón con células de bazo inmunizado con el Ag de interés. Lo que se consigue de esta manera es un hibridoma, el cual es altamente ventajoso, ya que los linfocitos B producen anticuerpos deseados y aportan la memoria inmune, mientras que las células neoplásicas aportan una capacidad de multiplicación indefinida (Figura 2). En resumen, se consigue una fuente ilimitada de anticuerpos monoclonales específicos que derivan de un único linfocito B (1,2,3).
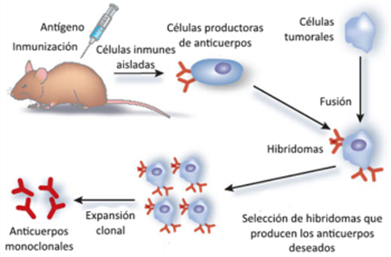
Estos anticuerpos monoclonales obtenidos fueron usados como tratamiento, pero se observó que el hecho de que fueran de origen murino provocaba un problema de tolerancia. Para solventarlo, se emplean Ac quiméricos en los que solo las regiones variables son de origen murino, o humanizados en los que solo lo son las regiones hipervariables. También existe la alternativa de anticuerpos monoclonales humanos que se producen en animales transgénicos (1,2).
Enfermedad del Alzheimer
Por otra parte, la enfermedad de Alzheimer o EA se caracteriza por la presencia de marañas neurofibrilares, placas seniles y pérdida de neuronas y sinapsis, dando lugar todo ello a una disminución de las habilidades mentales y cognitivas. No obstante, los dos rasgos más característicos de dicha enfermedad son las marañas neurofibrilares de la proteína Tau hiperfosforilada en forma de ovillos neurofibrilares, así como los depósitos del péptido β-amiloide (Aβ) de manera más característica (4,5).
El péptido Aβ, tanto de forma fisiológica como patológica, surge de la degradación proteolítica de la proteína precursora del amiloide (APP) de la membrana plasmática, la cual es cortada por las enzimas β- y γ-secretasas mediante la vía amiloidogénica (Figura 3). Por este motivo, dichas endoproteasas son consideradas posibles blancos terapéuticos de importancia. De forma paralela, en estudios más recientes también se presta atención a la degradación del péptido Aβ además de su formación, en la cual participan neprilisina (NEP) y la enzima insulina degradante (IDE) (4).
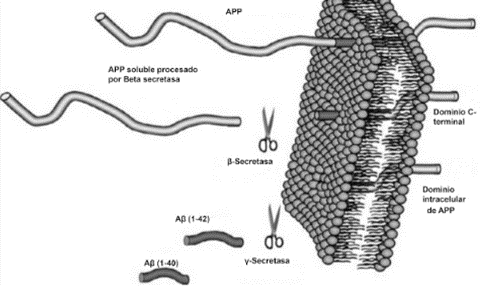
Como se ha descrito, la agregación del péptido es una de las características de EA más importantes, pero la mera presencia del mismo no es la causante de la neurodegeneración ya que se le adjudica un papel fisiológico. Para ser dañino, el péptido Aβ, que de forma normal existe como una estructura α-hélice o “random coil”, debe sufrir un plegamiento incorrecto en estructuras β plegadas que genere la formación de agregados (4).
Sumado a su plegamiento incorrecto, es clave el ensamblaje del péptido para su efecto biológico, siendo el extremo C-terminal el fragmento de mayor importancia para la formación de oligómeros (4). El depósito del péptido β42 produce el entramado de las sustancias conocidas como placas seniles (5), y esto es fundamental para el desarrollo de la enfermedad, ya que estos agregados son los que se depositan posteriormente en el cerebro y causan neurotoxicidad.
Tratamientos inmunoterápicos
A pesar de los grandes avances científicos, los tratamientos disponibles actualmente para la EA son solo sintomáticos, es decir, pueden lograr una mejoría en la calidad de vida, pero no consiguen revertir, frenar o curar la progresión de la enfermedad. Las estrategias farmacológicas están sobre todo enfocadas a inhibir la agregación del péptido Aβ amiloide. (6)
Una de las líneas de investigación más prometedoras contra la EA es la inmunoterapia anti-beta amiloide (Aβ), ya que ha demostrado provocar una respuesta inmune frente a los depósitos de péptidos patógenos y reducirlos (7). En este caso nos centraremos en una inmunoterapia pasiva mediante anticuerpos monoclonales, ya que se ha determinado que los efectos positivos de la inmunización parecen estar mediados por Ac (8). Esta inmunoterapia consiste en la administración por vía intravenosa de anticuerpos anti-βA en el paciente. De este modo, se consigue una respuesta inmunitaria anti-βA sin necesidad de una reacción proinflamatoria mediada por células T. (9). Los estudios en animales transgénicos han demostrado que la inmunización pasiva, además de reducir la carga amiloidogénica neuronal, mejora los déficits cognitivos, incluso antes de eliminar las placas amiloides neuronales. (9)
La inmunización pasiva con anticuerpos monoclonales humanizados comenzó cuando en 1996 Solomon y colaboradores demostraron que el uso de estos anticuerpos monoclonales dirigidos contra el péptido Aβ42 amiloide inhibían su agregación “in vivo” y podían solubilizar algunos precipitados fibrilares. Estos hallazgos pueden ser explicados mediante la activación de la microglía por el complejo Ag-Ac, la movilización de los depósitos de Aβ42 hacia la circulación sistémica o la disolución pasiva del complejo Ag-Ac. (6)
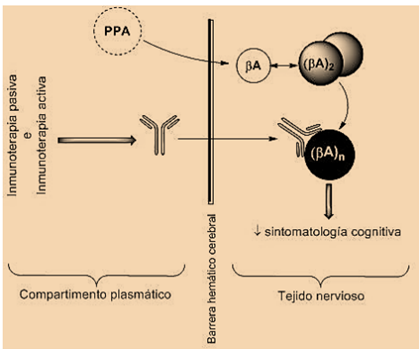
Debido a la localización de EA como enfermedad cerebral, es de importancia para su tratamiento la barrera hematoencefálica (BHE) que aísla el Sistema Nervioso Central. Varias investigaciones demuestran que el Aβ soluble se desplaza a través de la BHE en un equilibrio dinámico bidireccional (4,5,7). Por otra parte, existen diferentes conclusiones en cuanto a los anticuerpos monoclonales anti-Aβ: ciertos artículos defienden que estos provocan un incremento en los niveles plasmáticos de Aβ a pesar de que no se unieran a los acúmulos cerebrales, mientras que otros demostraron que sí atravesaban la BHE uniéndose a las placas amiloides, siendo la única diferencia el método de administración (intravenosa o intraperitoneal) (5,7).
En estas técnicas de inmunización pasiva caben destacar dos aproximaciones distintas en cuanto al mecanismo de acción: aclaramiento periférico (plasmático) y central (tisular) del Aβ. Dicha inmunoterapia requiere la administración repetida de Ac anti-Aβ humanizados. Estos pueden estar dirigidos a la región N-terminal del péptido actuando por la vía de aclaramiento central, o bien a la región central en cuyo caso no se detectan en el cerebro, es decir, actúan mediante aclaramiento periférico (7).
Fármacos
En la actualidad existen cuatro fármacos en el mercado que han sido aprobados por la FDA (Food and Drug Association) para el tratamiento de la EA. Sus mecanismos de acción están basados en modular los circuitos corticales involucrados en procesos cognitivos, así como otros de respuesta celular que se activan ante estímulos de toxicidad cerebral por aumento de la excitabilidad neuronal. Estos tratamientos se clasifican en dos grupos: inhibidores de la acetilcolinesterasa (AChEI), cuyo mecanismo de acción consiste en aumentar la transmisión colinérgica mediante la inhibición de la acetilcolinesterasa en la hendidura sináptica, y antagonistas de los receptores de ácido N-metil-D-aspártico (NMDAR), que reducen la excitotoxicidad por el bloqueo de este receptor inotrópico. Se ha demostrado que estos fármacos tienen un simple efecto paliativo y que su eficacia disminuye con el tiempo (5,6,9).
Bapineuzumab y Solanezumab son los dos anticuerpos monoclonales que actualmente han llegado a las fases más avanzadas del desarrollo experimental (pero fracasaron en los ensayos en fase III en pacientes con EA leve-moderada, pues su objetivo era conseguir la inmunización de los pacientes que sufrían ese grado de EA sin producir meningoencefalitis) (5,6,9). Ambos son anticuerpos monoclonales humanizados contra la proteína A pero actuando en diferentes regiones de la misma. Es de destacar que Bapineuzumab, a pesar de haber reducido la concentración de biomarcadores clave como la placa amiloide y proteína Tau fosforilada medida en líquido cefalorraquídeo, falló en producir mejoras cognitivas significativas (6,9,10,11). Cabe agregar que no fue posible mantener dosis máximas de Bapineuzumab debido a la aparición de efusión y edema cerebral, siendo los principales efectos adversos de esta terapia. El uso de Solanezumab no logró reducir la carga de amiloide en cerebro, y por ende tampoco se asoció a efusión ni edema relacionado con amiloide. Sin embargo, a pesar de que como se ha dicho anteriormente fracasó en los ensayos en fase III con pacientes con enfermedad leve-moderada, posteriormente se observó que en los estadios iniciales de la enfermedad había un deterioro cognitivo menor, habiendo también una mejora de las capacidades funcionales (5,10,11). Se ha observado que otro fármaco, el Crenezumab, produce efectos muy similares al Solanezumab en fase II de estudio (10,11).
Otro fármaco que se ha desarrollado, el Aducanumab, se ha evaluado en estudios fase I y fase II y ha demostrado una disminución de la concentración de proteína anormal Αβ en cerebro, es decir, actúa a nivel de β-amiloide, reduciendo su acúmulo en las placas seniles interneurales en personas en estadio inicial de la enfermedad (5). En el caso de este fármaco, se ha observado, además de la reducción de la carga amiloide cerebral, una mejora en las funciones cognitivas, lo cual no ocurría en los dos fármacos anteriores (6).
Otro anticuerpo monoclonal totalmente humano diseñado para unirse con una elevada afinidad a un epítopo conformacional en las fibra de βA, el Gantenerumab, se está ensayando con el objetivo de evaluar su potencial modificador en personas con riesgo de desarrollar la EA presenil, por un mutación genética de carácter autosómico dominante. El fundamento terapéutico es que actúa degradando las placas amiloides mediante un proceso de reclutamiento de la microglía y activación de la fagocitosis. Los estudios experimentales con ratones transgénicos apoyan esta hipótesis (5,9).
Hay otro fármaco llamado Azeliragon, cuya acción está orientada a la desagregación de las placas β-amiloide. Se está ensayando en pacientes con estadios avanzados de la enfermedad (5).
Cabe destacar que todos los fármacos son anticuerpos monoclonales de origen humanizado, excepto el Gantenerumab y el Aducanumab, que son de origen humano (11).
Conclusiones
Finalmente, se puede concluir que la inmunoterapia a base de anticuerpos monoclonales es un tratamiento esperanzador en la enfermedad de Alzheimer, y que podría contribuir a sanar a personas de esta demencia para la cual hasta ahora sólo existían cuidados paliativos.
Referencias
(1) Merino, A. G. (2011). Anticuerpos monoclonales. Aspectos básicos. Neurología, 26(5), 301-306.
(2) Machado, N. P., Tèllez, G. A., & Castaño, J. C. (2006). Anticuerpos monoclonales: desarrollo físico y perspectivas terapéuticas. Infectio, 10(3), 186-197.
(3) Castillo, L. V. (2015). Producción de anticuerpos monoclonales. Univ Alcalá, 1-12.
(4) Rodríguez, A. E. E., & Signoret, V. C. Z. (2017). Papel de la agregación del péptido Beta amiloide en la enfermedad de Alzheimer. Revista de Educación Bioquímica, 36(1), 2-11.
(5) Costa Vera, E. (2017). Avances en el tratamiento del Alzheimer.
(6) Acosta, G. T., Delgado, K. R., & Nassar, J. S. (2021). Enfermedad de Alzheimer e Inmunoterapia: revisión de tres anticuerpos monoclonales humanizados dirigidos contra el Aβ amiloide (bapineuzumab, solanezumab y aducanumab). Revista Médica de Costa Rica y Centroamérica, 85(627), 2-7.
(7) González, M. M., Piñera, P. P., Calatayud, M. T., & Ménez, B. B. (2005). Inmunoterapia para la enfermedad de Alzheimer. Archivos de Medicina, 1(4).
(8) Janus, C., Pearson, J., McLaurin, J. et al. Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 408, 979–982 (2000).
(9) J. Folch, M. Ettcheto, D. Petrov, S. Abad, I. Pedrós, M. Marin, J. Olloquequi, A. Camins, Una revisión de los avances en la terapéutica de la enfermedad de Alzheimer: estrategia frente a la proteína β-amiloide, Neurología, Volume 33, Issue 1,2018, Pages 47-58.
(10) Selkoe, D. J., & Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO molecular medicine, 8(6), 595–608.
(11) van Dyck C. H. (2018). Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biological psychiatry, 83(4), 311–319.