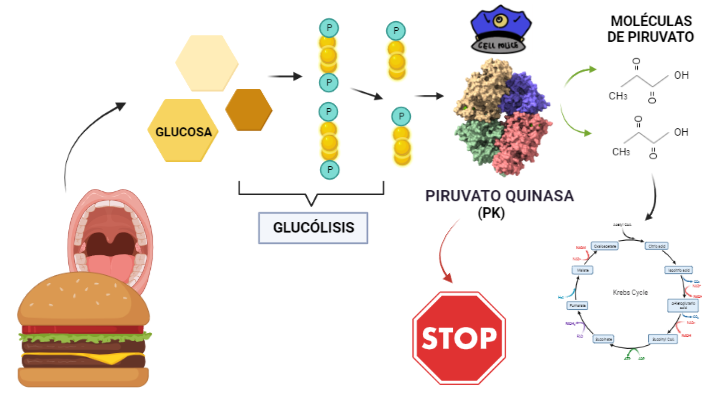
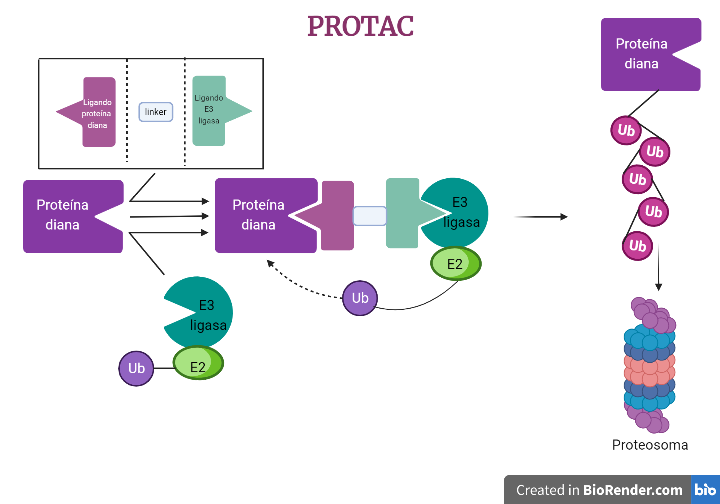
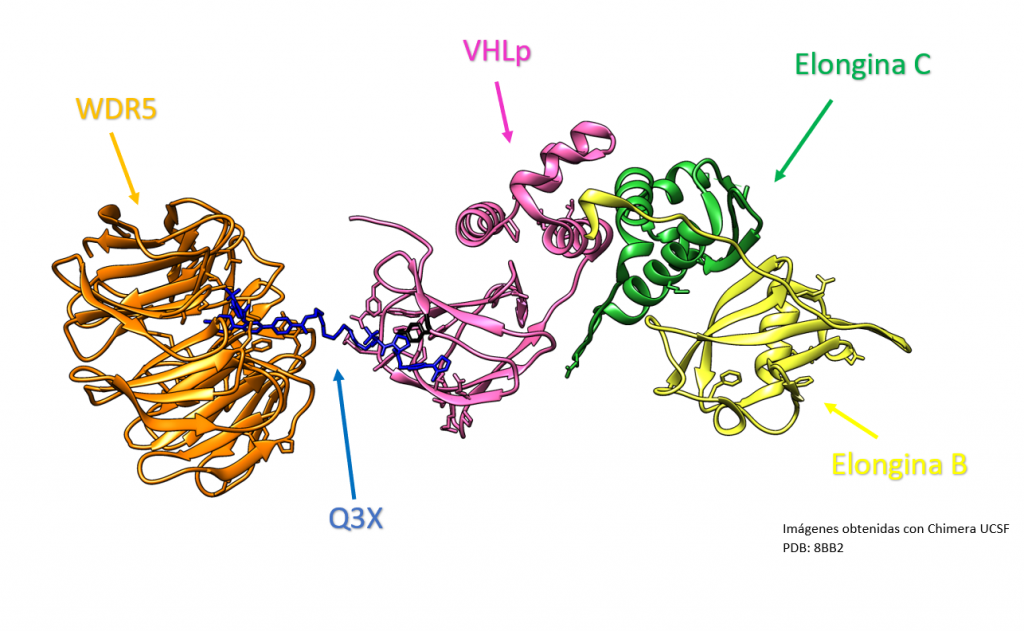
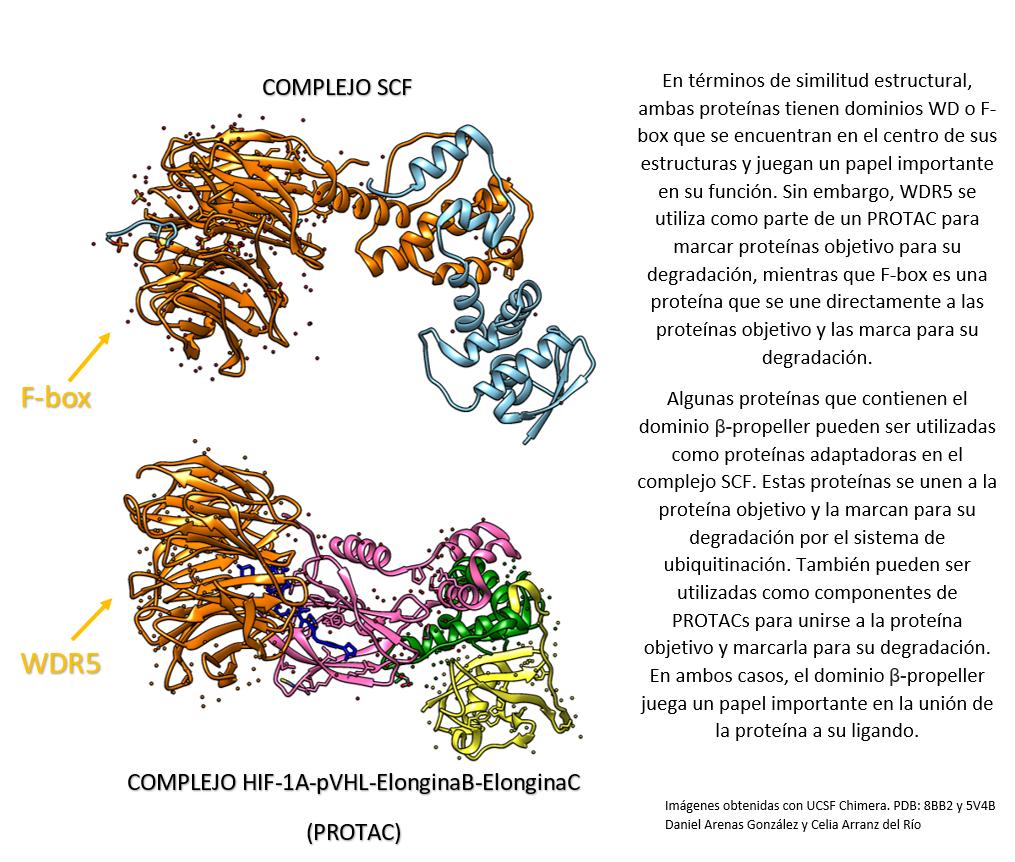
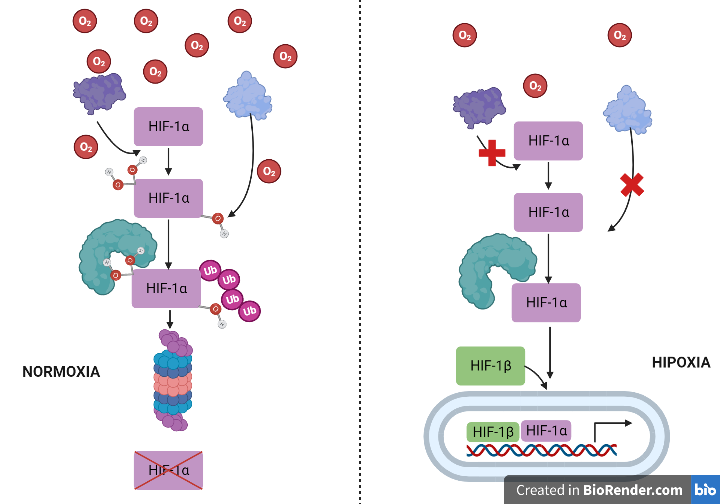
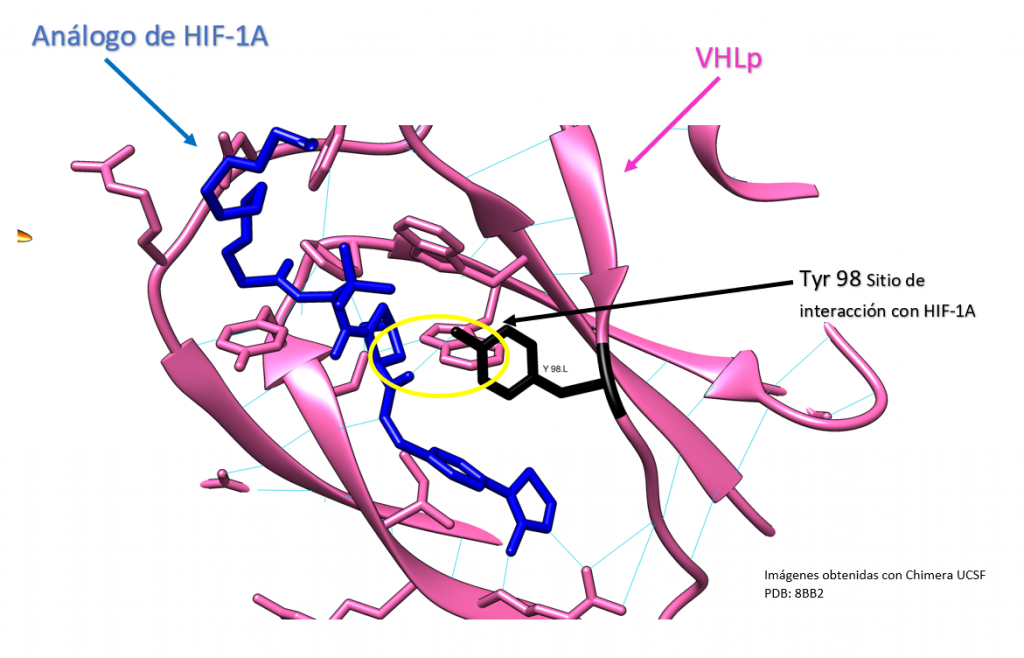
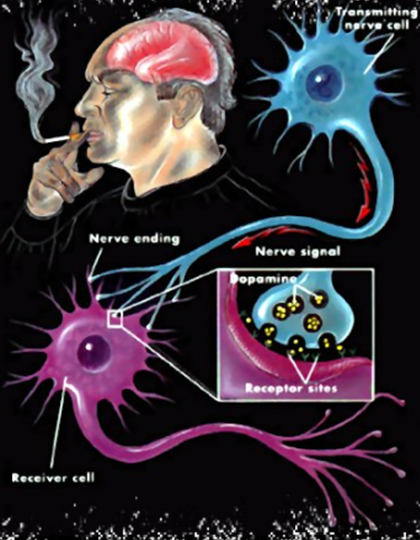
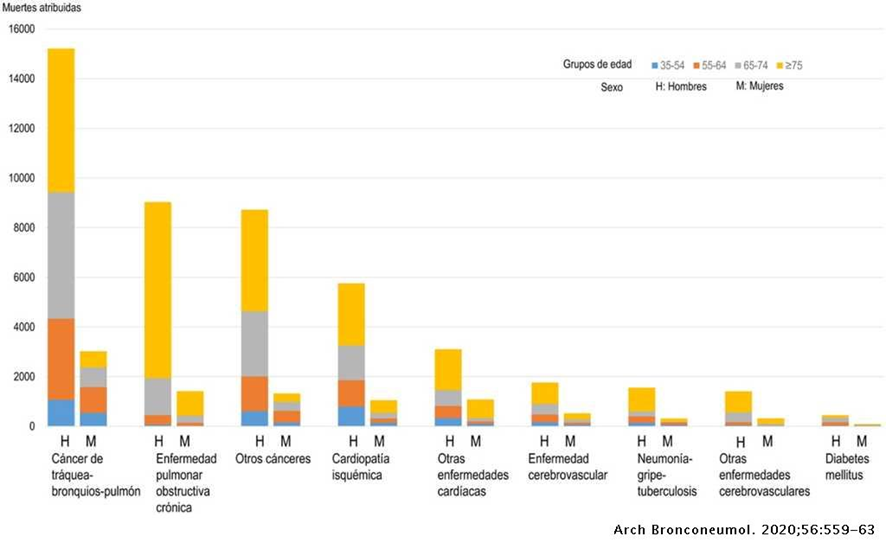
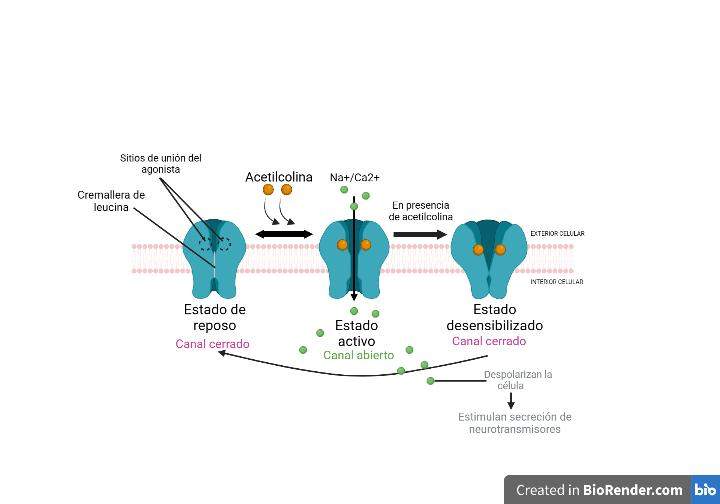
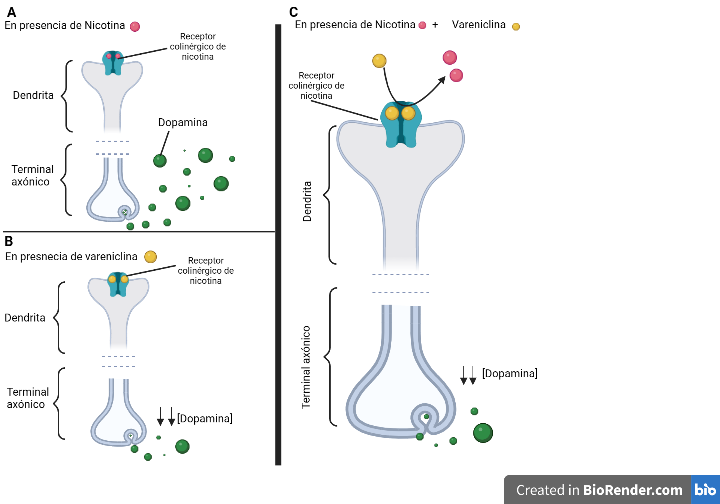
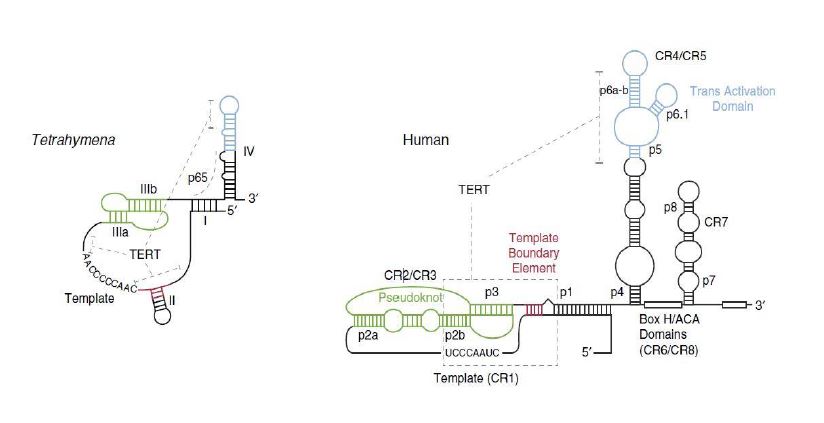
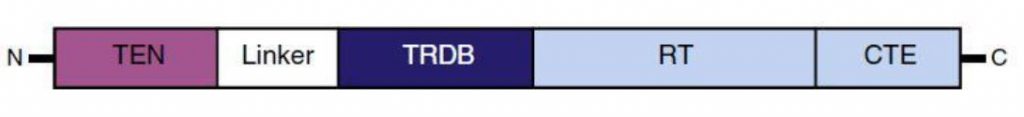
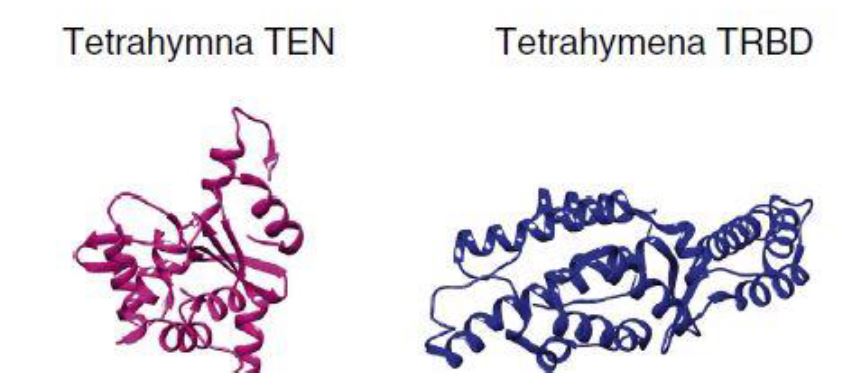
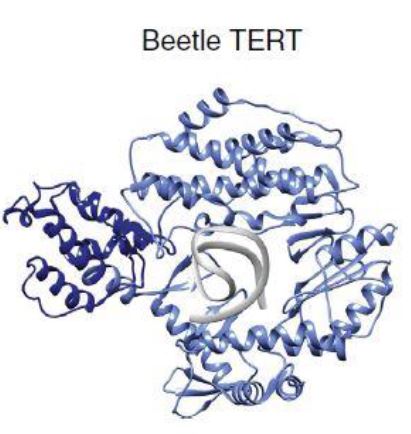
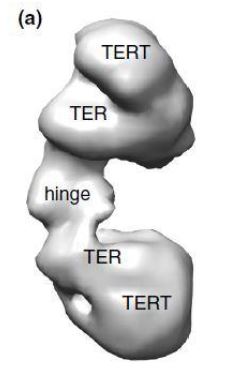
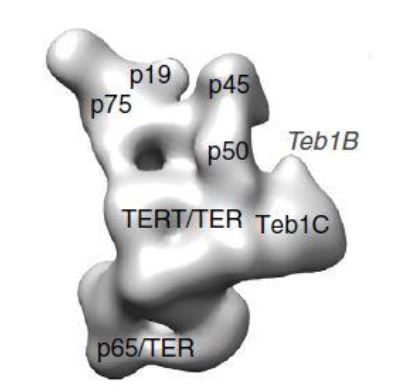
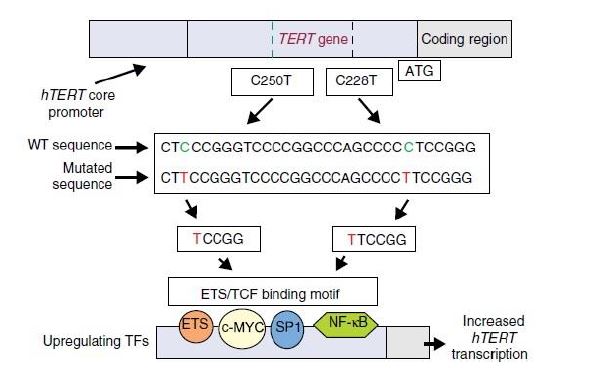
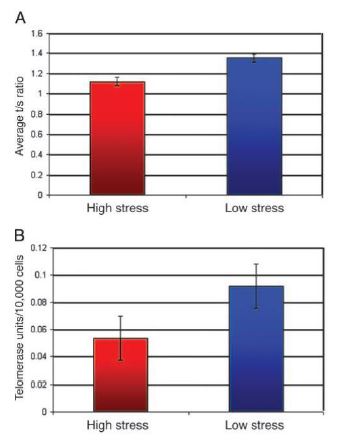
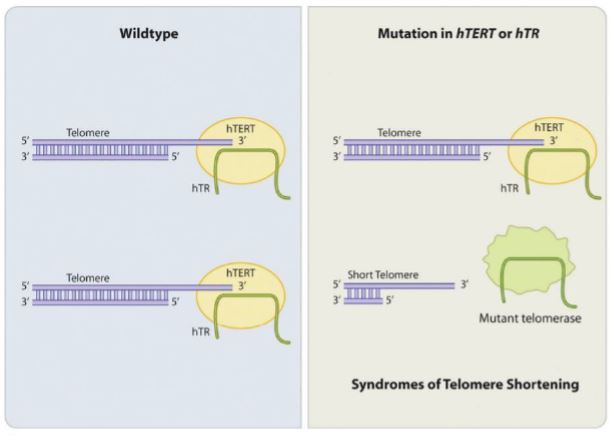
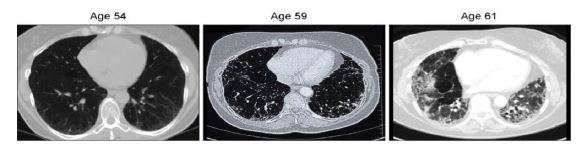
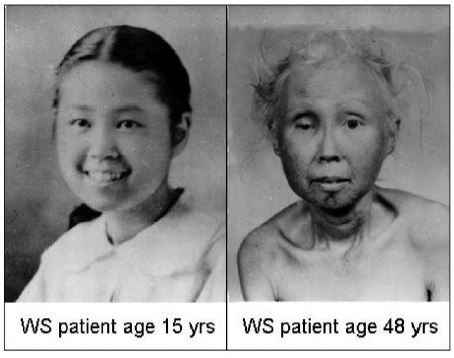
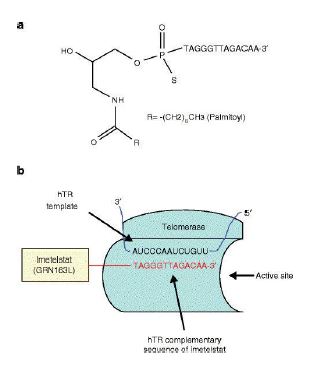
La Piruvato quinasa (PK) es una enzima que se encuentra al final de la ruta metabólica conocida como glucólisis y su función es permitir la salida del piruvato a la siguiente vía metabólica con el fin de obtener energía. Para entender la relevancia de esta molécula, quizás es conveniente explicar de forma concisa el recientemente popular término, conocido como “metabolismo”.
El metabolismo, al que hacen referencia desde dietistas y entrenadores hasta profesores de bioquímica, es quizás un proceso que para el ciudadano de a pie resulta abstracto en su relación con la ingesta de comida. Brevemente, se trata de un conjunto de reacciones que consisten en tomar una molécula grande (glucosa) con alto potencial energético y dividirla en paquetes más pequeños (piruvatos, en el caso de la glucólisis) con el fin de distribuir la energía por los distintos sistemas celulares y permitir que los organismos vivos se sigan considerando como tales. Esa glucosa, llega a las células por medio de la alimentación y se convierte en energía química útil mediante las distintas rutas metabólicas. De modo, que el correcto funcionamiento de la PK, es fundamental para que estos pequeños paquetes se introduzcan en la siguiente ruta metabólica (el ciclo de Krebs) que se encargará de distribuir la energía. De hecho, modificaciones de esta molécula podrían degenerar en condiciones tan serias como el Cáncer o el Alzheimer, dado que impediría que la energía alcanzase los destinos pertinentes.
PAPEL BIOLÓGICO
El rol que asume la PK a nivel biológico se traduce a la función que tiene por ejemplo el personal de seguridad a la entrada de un museo. Éste, en caso de que el turista no presente ninguna irregularidad (posesión de armas, comida, etc), procede a concederle la entrada al establecimiento; del mismo modo la PK permite el acceso al piruvato a la siguiente ruta, siempre que no suponga una amenaza contra el correcto funcionamiento de la célula. Al tratarse la PK de una quinasa, esto la convierte en una molécula con la capacidad de fosforilar (añadir grupos fosfato a otras moléculas). De modo que para concederle el acceso a la siguiente ruta, la PK retira los fosfatos de las triosas procedentes de la glucosa y los añade a moléculas de ADP, dando lugar a ATP (que es una molécula que transporta energía). Como resultado de este proceso, se sintetizan dos piruvatos, a los que finalmente les será concedido el acceso al ciclo de Krebs.
La PK en humanos, aparte de la función biológica general descrita anteriormente, también tiene una serie de isoformas cuya función e importancia biomédica es más específica. Las cuatro isoformas de la PK (PKM, PKK, PKR, PKL) fueron descubiertas en 1965 y reciben su nombre a partir del nombre tejido donde se encuentra cada una. La PKM (actualmente denotada como PKM1) se encuentra en los tejidos musculares (muscle, M) del corazón y en el cerebro, la PKK (actualmente denotada como PKM2) se encuentra en los tejidos de los riñones (kidneys, K), el intestino y las células cancerosas; la PKR en el tejido sanguíneo (red blood cells, R) y la PKL en el tejido del hígado (liver, L) y en los riñones también. La diferenciación (que ocurre tras la fase fetal, ya que todas provienen de la isoforma PKM2) favorece la aparición de patologías mucho más especializadas debido a ligeros cambios en la conformación proteica.
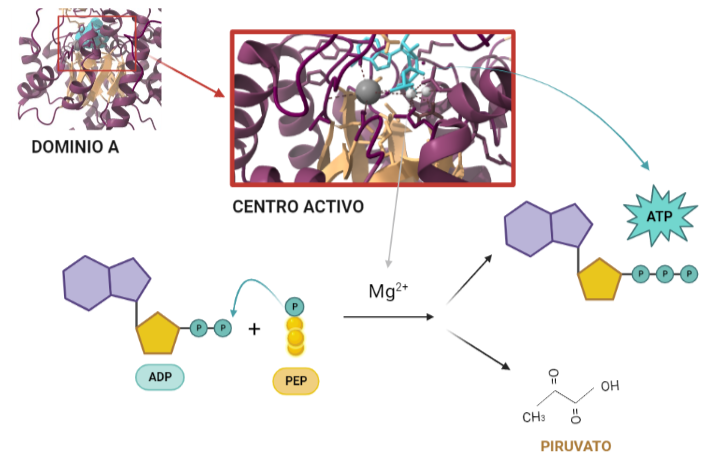
Ilustración que refleja el papel de la Piruvato quinasa en el metabolismo de la glucosa. La molécula de la que partimos es la glucosa, que tiene 6 carbonos, ésta, mediante una concatenación de reacciones da lugar a 1,3-bifosfoglicerato (las bolas amarillas representan los carbonos y las P los grupos fosfato) que tras otra serie de reacciones, dan lugar a fosfoenolpiruvato (PEP) que es el precursor del piruvato. La PK es la encargada de catalizar la reacción de desfosforilación que da lugar al paso de PEP a piruvato cuyo fosfato restante se utiliza para fosforilar ATP. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
ESTRUCTURA Y CÓMO FUNCIONA
Desglose estructural
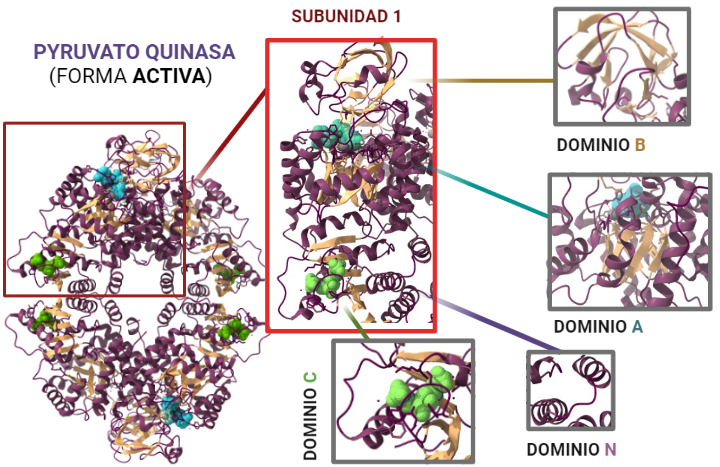
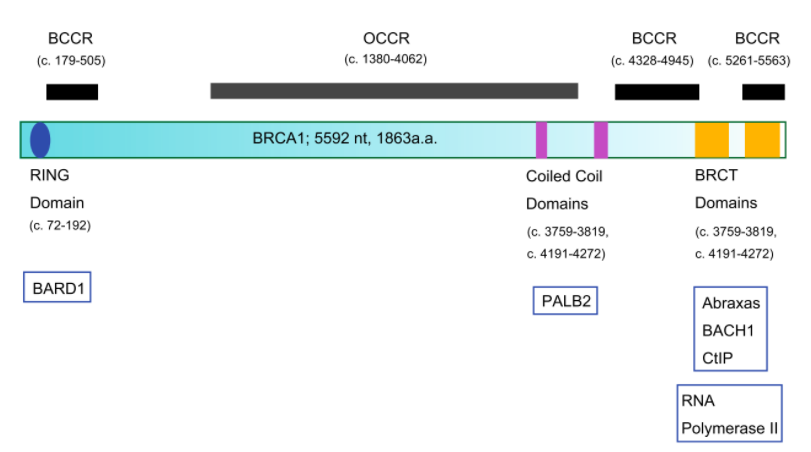
Para entender el papel que desempeña la PK es fundamental comprender su estructura, dado que de ésta dependerá el funcionamiento de la misma. ¿Qué constituye a la PK? La piruvato quinasa es una proteína conformada por 531 aminoácidos que dan lugar a un tetrámero, cuyas cuatro subunidades son iguales. Éstos están organizados en motivos de hélices alfa, láminas beta y bucles.
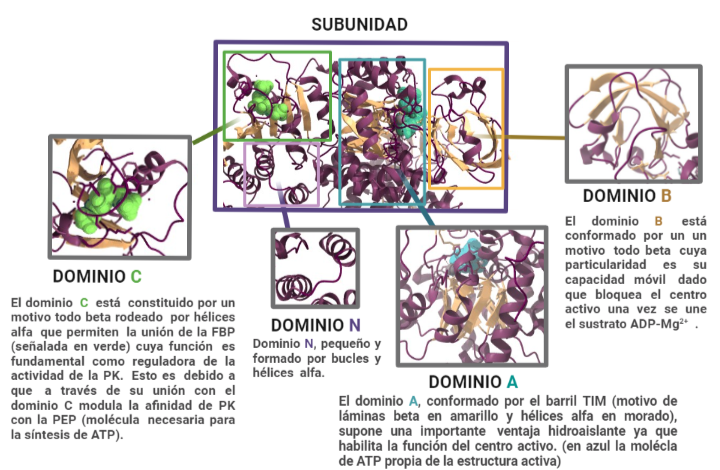
Estas subunidades se organizan a su vez en tres grandes dominios rotulados como A, B y C junto con un dominio N-terminal. El dominio A está conformado por un barril TIM α8/β8 cuyo centro activo se ubica entre el dominio A y el B, éste es además el dominio más grande de la subunidad. El dominio B sin embargo es móvil y bloquea el centro activo una vez que se le une el sustrato ADP-Mg2+. Finalmente, el dominio C contiene la fructosa-1,6-bifosfato (FBP) que es un potente activador alostérico.
Descripción gráfica del desglose estructural que presenta la Piruvato quinasa en estado activo. Los motivos de láminas beta quedan indicados en amarillo, las hélices alfa en púrpura, los bucles en violeta y los ligandos: ATP en azul y FBP en verde. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
Esto significa, que a través de la unión de la FBP al dominio C, se facilitará la unión del fosfoenolpiruvato (PEP) que es fundamental para la regulación de la actividad de la PK, ya que ésta depende de la afinidad con el PEP. En ausencia de activadores alostéricos como la FBP, la PK tiene poca afinidad con el PEP. O sea, si la PK fuera un niño pequeño y su voluntad para realizar los deberes fuera análoga a la actividad de la quinasa, éste necesitaría una motivación para realizarlos. Si se impone la condición de recibir un caramelo a cambio de la tarea, éste cumplirá. De igual forma si la PK presenta la FBP unida al dominio C, ésta aumentará su afinidad a la PEP alterando su actividad. Además, la unión de FBP estabiliza la molécula en estado activo y promueve la tetramerización. Cabe destacar, que todas las isoformas de la PK se unen con la FBP exceptuando la PKM1 que debido a una discrepancia estructural es suficientemente estable por si sola (siendo además insensible a los moduladores alostéricos) y no presenta ni la región de unión a la FBP ni el interfaz dímero-dímero debido a éstas se expresan en los exones específicos de las isoformas.
Desglose de la funcionalidad de las distintas estructuras que conforman cada subunidad de la Piruvato quinasa en estado activo. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
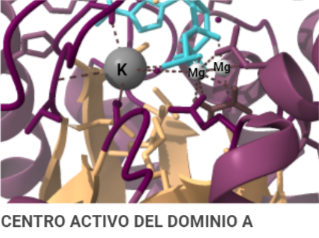
Otro detalle que cabe mencionar de la estructura de la PK es su capacidad de unión a cofactores (K+ y Mg 2+) cuya intervención en la activación de la molécula es esencial. En el caso del Mg 2+, se ha mencionado que está involucrado en bloquear el acceso al centro de activo (gracias al cambio conformacional que desplaza al dominio B) cuando éste forma un complejo de sustrato al unirse al ADP (formando el sustrato ADP-Mg2+). En el caso del K+ sin embargo, se ha observado que ante su presencia, el mecanismo cinético de la PK se mantiene desordenado (forma natural), esto supone que favorece la forma activa de la PK y permite que se unan el PEP o el complejo ADP-Mg2+ de forma independiente (mecanismo aleatorio). En ausencia de K+, por el contrario, el ADP no se pude unir al centro activo hasta que el PEP no haya terminado de formar un centro activo completamente funcional. De forma, que se deduce que el K+ es el encargado de inducir el cierre del centro activo y de que los residuos encargados de la unión al nucleótido adopten la conformación correspondiente.
Ilustración que señala la presencia de cofactores ( K+, Mg2+) en el Dominio A, fundamentales para la adopción de la conformación activa de la PK. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
Función ¿Qué hace?
Entonces, ¿Qué función tiene? Una vez conocida la estructura, es posible dilucidar qué función lleva a cabo, y cómo. La función catalítica de la PK consiste en fosforilar moléculas, debido a su condición de quinasa; en concreto, moléculas de ADP a partir de moléculas de PEP de la etapa anterior de la glucólisis. Todo ello para dar lugar a dos productos. Por un lado, piruvatos estables a partir de sus precursores PEP y por otro, obtener energía en forma de moléculas de ATP a través de la fosforilación del ADP. De modo, que el nombre surge del producto (piruvato) + tipo de enzima (quinasa). Para ello, al centro activo del dominio A se unen el PEP y el complejo ADP-Mg2+ dado que los cationes de Mg2+ median y facilitan la transferencia del grupo fosfato del PEP al ADP dando lugar al ATP y a los piruvatos. Todo ello es posible debido a la alta energía que libera PEP al ser hidrolizada. Al perder el fosfato, el PEP pasa a su forma de enolpiruvato que es menos estable, de modo que se llevará a cabo un proceso de tautomerización que consiste en que el enolpiruvato acepte un protón procedente de una molécula de agua convirtiéndose así en un piruvato estable y favoreciendo la fosforilación del ADP.
Representación gráfica de la reacción que tiene lugar cuando la PK funciona de forma natural.El complejo ADP-Mg2+ se une al centro activo junto con el PEP. Éste le aporta el fosfato que le hace inestable con el fin de utilizarlo para fosforilar al ADP. Como productos se obtienen ATP y piruvato. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
En cuanto a la capacidad alostérica de la PK, aparte de la FBP, que favorece la unión del sustrato PEP, hay otras moléculas que alteran la actividad de la enzima. Por ejemplo, un inhibidor alostérico de esta enzima (PKM1, PKM2) sería la fenilalanina (Phe) cuya unión supone la disminución de afinidad con la PEP mediante la estabilización de la estructura inactiva de la PK. El lugar de unión de Phe también puede albergar a la alanina que actúa como inhibidor, pero solo ante la isoforma PKM2, y esto lo lleva a cabo favoreciendo la conformación dimérica, contraria a la tetramérica a la que se une FBP. A pesar de que en presencia de concentraciones normales de FBP la inhibición de la alanina queda mitigada. La serina sin embargo, también puede ocupar este centro de unión, pero con función activadora no inhibidora, en la PKM2. Dejando a un lado los aminoácidos, hormonas ,como la hormona tiroidea triyodo-L-tironina (T3), actúan también como inhibidor alostérico favoreciendo la conformación monomérica inactiva de la PK. Mientras que el oxalato puede actuar como activador de la PK mediante su interacción con el centro activo por ser análogo al enolpiruvato, en caso de que la concentración de PEP sea baja.
¿PORQUÉ PODRÍA ACABAR CONTIGO?
EL PELIGRO RESIDE EN LA ISOFORMA
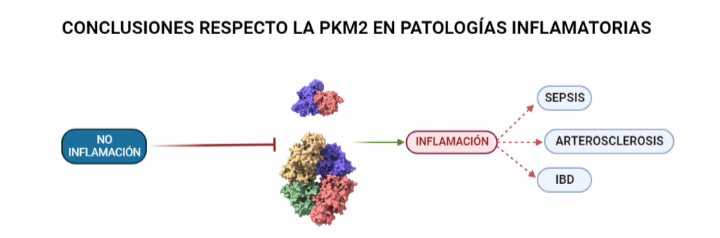
Las isoformas de una proteína son proteínas que provienen del mismo gen que la proteína original, dicho gen se duplica y comienza a acumular mutaciones para dar lugar a las distintas isoformas. En el caso de la PK, tras este proceso de duplicación y modificación por mutaciones se han obtenido 4 isoformas distintas: PKM1, PKM2, PKR y PKL. La importancia de las isoformas recae en que a pesar de realizar la misma función que la proteína inicial, cada una presenta ligeramente distintas: propiedades cinéticas, estructurales, de regulación o de localización en la célula. Estas ligeras diferencias atienden a las necesidades metabólicas del tejido al que pertenecen. O sea, las modificaciones que sufra PKL (L hace referencia a liver, hígado en inglés) afectarán en principio al hígado dado que la estructura de la PKL ha resultado ser la más eficaz a la hora de catalizar las reacciones que precisa este órgano. A pesar de esto, existe una isoforma que destaca en su implicación en numerosas patologías inflamatorias (como la Sepsis, IBD o Arterosclerosis) o enfermedades como el Cáncer o el Alzheimer. Ésta es la PKM2.
PKM2 Y UN COMPENDIO DE LO QUE PUEDE SALIR MAL
PKM2 en el Cáncer
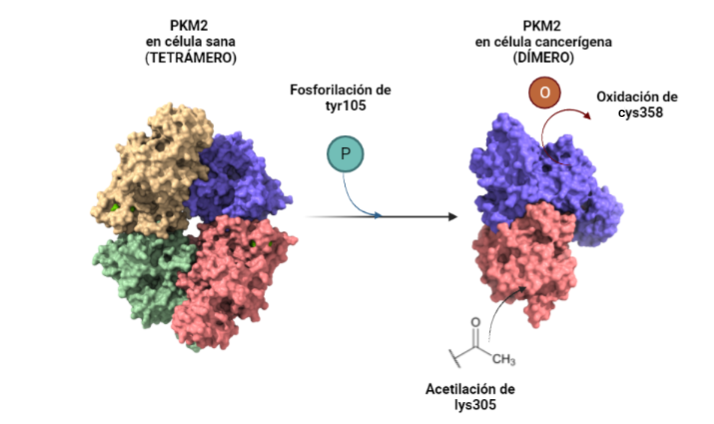
Ante el desarrollo de células neoplásicas, la PKM2 tiene un comportamiento que podría clasificarse como moonlighting. Esto se debe a que en condiciones normales la PK transforma PEP en piruvato y este sigue la ruta metabólica normal hacia el ciclo de Krebs, mientras que, ante una situación de estrés, como puede ser el desarrollo de células cancerígenas, ésta altera su forma tetramérica natural y pasa a su forma dimérica. Al dimerizarse mediante la fosforilación de su tirosina 105 la proteína deja de realizar su función natural y divierte el proceso de la glucólisis hacia la síntesis de metabolitos necesarios para la síntesis de serina. Esto se debe a que dicho aminácido regula a mTORC1 ( mammalian target of rapamacyn complex 1), que es fundamental para favorecer la proliferación celular, característica de las células cancerígenas. Otra de las modificaciones que sufre, es la acetilación de su lisina 305, junto con la oxidación de su cisteína 358 que provoca una alteración en la ruta de la glucólisis haca la PPP (pentose phosphate pathway, vía de la pentosa fosfato) que favorece la síntesis de nucleótidos para sufragar los efectos de la interrupción de la ruta glucolítica.
Descripción gráfica de la dimerización y modificación de la piruvato quinasa en presencia de cáncer. Este proceso consiste en dimerizar la proteína mediante la fosforilación de su tirosina 105 y alterar sus propiedades mediante la oxidación de su cisteína 358 y la acetilación de su lisina 305. A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
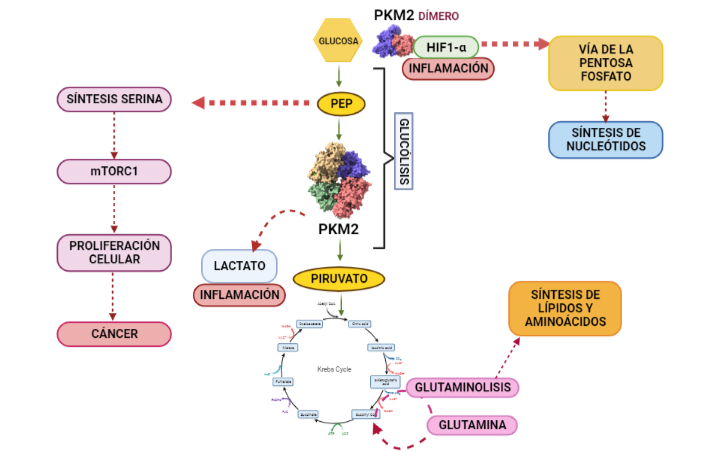
Estas modificaciones de la PK se conocen como el efecto Warburg o Glucólisis Aeróbica. El efecto Warburg supone que las células cancerígenas conviertan el piruvato en lactato, disminuyendo su producción de ATP. La disminución de síntesis de ATP se soluciona mediante los procesos mencionados anteriormente, mientras que el aumento de lactato supone la aparición de un ambiente tumorigénico dado que es excretado, reduciendo así el pH extracelular (esto favorece las condiciones para la proliferación celular) y provocando inflamación. El aumento de lactato también se utiliza para acceder a un recurso de energía alternativo, como es la glutamina. Esto es posible, dado que se disminuye la entrada de piruvato en el ciclo de Krebs, por lo que comienzan a introducirse metabolitos de glutamina en su lugar y aumenta por lo tanto la síntesis de lípidos y aminoácidos. Finalmente, la PKM2 es translocada al núcleo donde se une a HIF1-α (hypoxia-inducible factor-1 alfa) promoviendo la transactivación de HIF1 que favorece la aparición de un ambiente tumorigénico y la desviación de la glucólisis hacia la vía de la pentosa fosfato.
Diagrama que dilucida la modificación de la vía metabólica regulada por la piruvato quinasa en presencia de cáncer (efecto Warburg). Las flechas rosas indican los procesos que surgen bajo condiciones de dicho efecto, mientras que las verdes indican el curso habitual de la vía. A partir de PDB 1a3w y 6wp3(en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
PKM2 en el Alzheimer
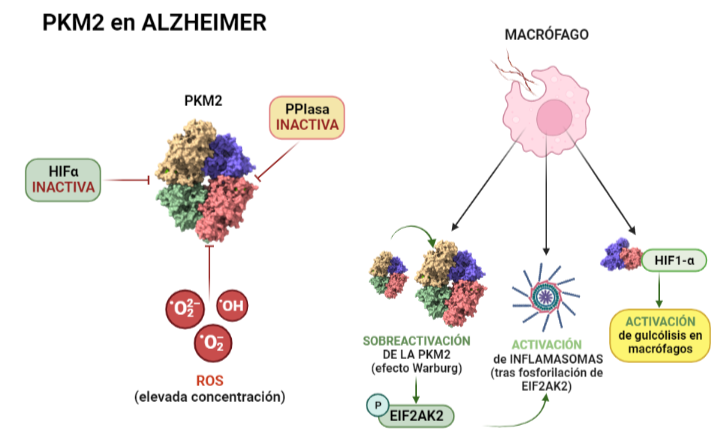
Debido a que el origen y funcionamiento del Alzheimer todavía no se ha esclarecido, se están explorando nuevas vías de investigación. Entre ellas, destacan factores como el estrés oxidativo, fallos en el transporte y metabolismo de la glucosa, y la inflamación. Dado que la piruvato quinasa juega un papel central en el metabolismo de la glucosa, su función se ve afectada por la enfermedad. Como consecuencia de la alteración de la ruta metabólica de la glucosa (debida a la acumulación de β-amiloides), se da la inactivación HIF1-α lo que supone que la PKM2 en dímero no puede unirse a ella, quedando inhibida. Otra forma en la que la PKM2 queda inhibida (en presencia de esta enfermedad) es mediante la inactivación o la regulación insuficiente de PPIasa (peptidil-prolil cis-trans-isomerasa) o la elevada concentración de ROS (especies reactivas de oxígeno), ya que ambas impiden la translocación de la PKM2 al núcleo, y consecuentemente esta no se une a HIF1-α, dando lugar en este caso a un desequilibrio en el metabolismo mediante una cascada de PEP. Respecto al papel de la PKM2 en la respuesta inflamatoria, la PKM2 regula la respuesta de los macrófagos, que son esenciales a la hora de retirar los amiloides para evitar la formación de placas. Esto se debe a que ante la inflamación, los macrófagos adoptan un fenotipo que favorece la síntesis de ATP y se ha observado que algunos incluso activan la Glucólisis Aeróbica (efecto Warburg), que se da cuando hay una sobreactivación de la PKM2. La Glucólisis Aeróbica mediada por PKM2 también promueve la activación de inflamasomas mediante la fosforilación de EIF2AK2 (factor 2 de iniciación de traducción de la α quinasa 2). Por otro lado, mediante la interacción con HIF1-α se promueve la transcripción de genes que activan la glucólisis en macrófagos. Finalmente, la PKM2 también es responsable de la glucólisis de la α-sinucleína que es una proteína presináptica asociada a la fisiopatología del Alzheimer.
Esquema que ilustra las consecuencias de la enfermedad del Alzheimer sobre la actividad de la piruvato quinasa. A la izquierda se indican las principales formas mediante las que se inhibe a la PK en condiciones patológicas (inactivación de las moléculas HIF1-α, PPIasa y la elevada concentración de radicales libres). A la derecha sin embrago, se exponen los procesos que se activan como consecuencia de la patología. A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
PKM2 en diversas enfermedades inflamatorias
La respuesta inflamatoria, como se ha expuesto en los dos casos anteriores, requiere de un alto consumo de energía. Células del sistema inmune, como los macrófagos, activan mecanismos similares al denominado efecto Warburg en el cáncer para aumentar la síntesis de energía y poder responder de manera eficaz. Esto está íntimamente relacionado con la glucólisis por lo que la implicación de la PK es crucial. En el caso de la Sepsis la implicación de la PKM2 en la glucólisis anaeróbica que adoptan las células bajo la patología supone un perjuicio para la supervivencia del individuo. Esto se ha observado tras inhibir su actividad comprobando que de este modo también quedaban inhibidas moléculas como el inflamasoma NLRP3 y HMGB1 (proteína de alta movilidad del grupo 1) cuya represión favorece la supervivencia del individuo. En el caso de la Arterosclerosis se ha observado que la inhibición de PKM2 en linfocitos B y T favorece la atenuación de la inflamación. Esto está unido a que parte de los productos que se sintetizan durante la glucólisis aeróbica (adoptada por las células del sistema inmune bajo condiciones de estrés) producen inflamación como se expone en la segunda imagen de la PKM2 en el cáncer. Incluso en la IBD (enfermedad inflamatoria de Bowel) la PKM2 se está valorando como posible bioindicador de la enfermedad según estudios recientes. Una vez más, la supresión de la PKM2 favoreció el control sobre la inflamación ya que en su versión nuclear dimérica contribuye a la adaptación de las necesidades metabólicas de los linfocitos. Esto significa que la presencia (en muestras) de esta proteína en estado dimérico podría ayudar a identificar la enfermedad. También cabe mencionar, que se ha observado que la PKM2 regula a Bcl-xL impidiendo la apoptosis de las células del epitelio intestinal.
Ilustración que refleja concisamente la influencia que tiene la PK en enfermedades inflamatorias como son: la Sepsis, la Arterosclerosis y la IBD. A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
Israelsen, W. J., & Vander Heiden, M. G. (2015). Pyruvate kinase: Function, regulation and role in cancer. Seminars in cell & developmental biology, 43, 43–51. https://doi.org/10.1016/j.semcdb.2015.08.004
June 2022, Faiza Ahmed, Jonathan Ash, Thirth Patel, Auriel Sanders, David Goodsell, Shuchismita Dutta
Oria-Hernández, J., Cabrera, N., Pérez-Montfort, R., & Ramírez-Silva, L. (2005). Pyruvate kinase revisited: the activating effect of K+. The Journal of biological chemistry, 280(45), 37924–37929. https://doi.org/10.1074/jbc.M508490200
Patel, S., Das, A., Meshram, P., Sharma, A., Chowdhury, A., Jariyal, H., … & Shard, A. (2021). Pyruvate kinase M2 in chronic inflammations: a potpourri of crucial protein–protein interactions. Cell Biology and Toxicology, 37(5), 653-678.
The RCSB PDB «Molecule of the Month»: Inspiring a Molecular View of Biology D.S. Goodsell, S. Dutta, C. Zardecki, M. Voigt, H.M. Berman, S.K. Burley (2015) PLoS Biol13(5): e1002140. doi: 10.1371/journal.pbio.1002140
Yang, L., Venneti, S., & Nagrath, D. (2017). Glutaminolysis: a hallmark of cancer metabolism. Annual review of biomedical engineering, 19, 163-194.
Vivir así es morir de amor: Síndrome de Takotsubo
escrito por sofialvan_3B | 28 enero, 2023
Realizado por Sofía Alván Benito
«Morir de amor»… Esa frase que hemos escuchado y leído miles de veces en poesías, libros o en la letra de famosas canciones como la de Camilo Sesto. Suena dramático e irreal, pero a veces la realidad supera a la ficción.
Introducción
El síndrome de Takotsubo (ST) es una patología que afecta al corazón. Suele aparecer en pacientes que han estado sometidos a un gran estrés físico o emocional (Lyon et al., 2016). Estos detonantes pueden ser, por ejemplo, la pérdida de un ser querido, un gran cambio inesperado en nuestra vida o incluso una ruptura con nuestra pareja.
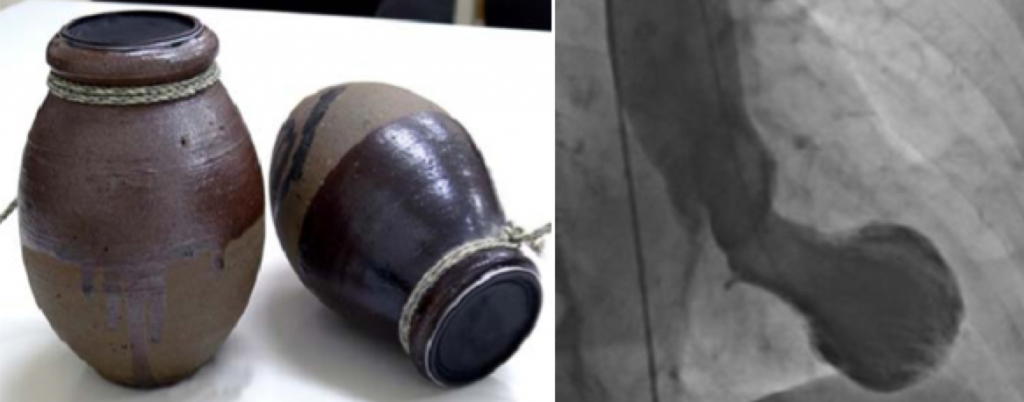
Fue diagnosticado por primera vez por un médico japonés, que bautizó a esta enfermedad con el nombre que tiene actualmente. La palabra «Takotsubo», en japonés, es el nombre de una vasija que se utiliza para cazar pulpos y que, curiosamente, tiene una forma muy similar a la que adquiere el corazón al sufrir esta enfermedad (Sato et al., 1990). (Figura 1)
Figura 1: Comparación entre una vasija japonesa Takotsubo y un corazón de un paciente con ST. (Foto adaptada de la original tomada por: Profesor Christian Templin, Hospital Universitario de Zurich)
Síndrome de Takotsubo vs Infarto de Miocardio
Este síndrome, también llamado síndrome del corazón roto, se asemeja bastante a un infarto de miocardio (IM). Sin embargo, ambas patologías tienen grandes diferencias. (Tabla 1) (Falola, Fonbah, & McGwin, 2013; Gupta & Gupta, 2018)
Síndrome de Takotsubo
Infarto de Miocardio
Antecedentes cardiovasculares
No
Sí
Obstrucción arterial
No
Sí
Factores de riesgo
Estrés emocional o físico
Tabaco, obesidad, hipertensión, diabetes
Partes afectadas
Ventrículo izquierdo
Corazón (general)
Tabla 1: Diferencias entre el ST y el IM. (De elaboración propia)
Epidemiología
Realmente, el Síndrome de Takotsubo es una enfermedad rara. Afecta únicamente, más o menos, a un 2% de todos los pacientes que fueron inicialmente diagnosticados con Síndrome Agudo del Miocardio. (Deshmukh et al., 2012)
Suele aparecer con mayor frecuencia en mujeres mayores de 50 años. La mayoría de casos se producen por un estímulo, que lleva a la aparición de la enfermedad. (Lyon et al., 2016). Sin embargo, en un 30% de los casos no hay detonante físico ni emocional. (Khera, Light-Mcgroary, Zahr, Horwitz, & Girotra, 2016). Dentro de los casos que sí se deben a un evento desencadenante, un 90% corresponden a eventos negativos, son los casos del síndrome del corazón roto propiamente dicho. (Templin et al., 2015). El 10% restante se debe a eventos positivos, como por ejemplo ganar la lotería, y constituyen una variante de esta patología que recibe el nombre de síndrome del corazón feliz. (Ghadri et al., 2016)
La tasa de mortalidad es muy baja, de un 4.5%. (Singh et al., 2014). En general, esta patología tiene un buen pronóstico y la mayoría de pacientes se recuperan en unos meses. (Elesber et al., 2007). Suele ser una enfermedad transitoria, aunque en ocasiones puede llegar a ser recurrente.
En hombres, aunque la enfermedad es menos frecuente, la mortalidad es superior a la de las mujeres. (Khera, Light-Mcgroary, Zahr, Horwitz, & Girotra, 2016)
Síntomas y manifestaciones clínicas
Como ya ha sido mencionado anteriormente, los síntomas de un paciente con Síndrome de Takotsubo son muy parecidos a los de un paciente que sufre un infarto de miocardio.
Los principales síntomas son dolor de pecho, disnea o dificultad para respirar, palpitaciones, insuficiencia cardíaca, paro cardíaco… (Templin et al., 2015)
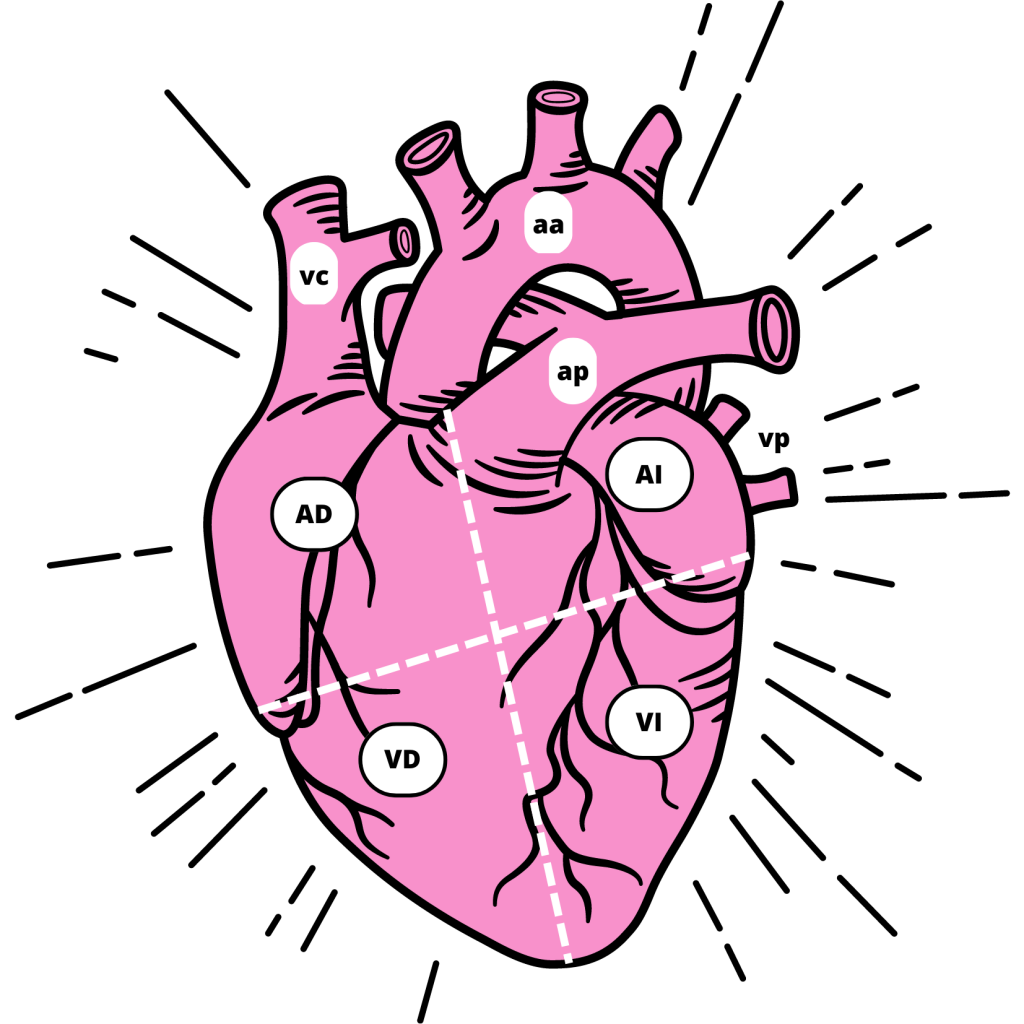
Los pacientes con este síndrome no presentan ningún otro problema cardiovascular, ni obstrucción en las arterias. A pesar de ello, se ve una pérdida de la función del ventrículo izquierdo del corazón, que es la parte más afectada. (Figura 2)
Figura 2: Partes del corazón y vasos sanguíneos principales. AD: Aurícula Derecha; AI: Aurícula Izquierda; VD: Ventrículo Derecho; VI: Ventrículo Izquierdo; vc: vena cava; aa: arteria aorta; ap: arteria pulmonar; vp: vena pulmonar (De elaboración propia)
En condiciones normales, el corazón utiliza como fuente principal de energía la que procede del metabolismo de ácidos grasos en vez de la glucosa. Cuando se sufre este síndrome, el corazón cambia su metabolismo a uno en el que utiliza más glucosa y menos ácidos grasos. (Gupta & Gupta, 2018)
Además, se puede ver elevación de biomarcadores cardíacos como la troponina o el péptido natriurético. Esto puede servir para el diagnóstico de la enfermedad. (Budnik et al., 2016)
Puede haber alteraciones del electrocardiograma, en algunos casos. (Migliore, Zorzi, Perazzolo Marra, Iliceto, & Corrado, 2015)
Este síndrome también recibe el nombre de síndrome de balonamiento apical transitorio, ya que se puede observar una especie de abultamiento en forma de «balón» en la región apical. (Lyon et al., 2016)
Causas
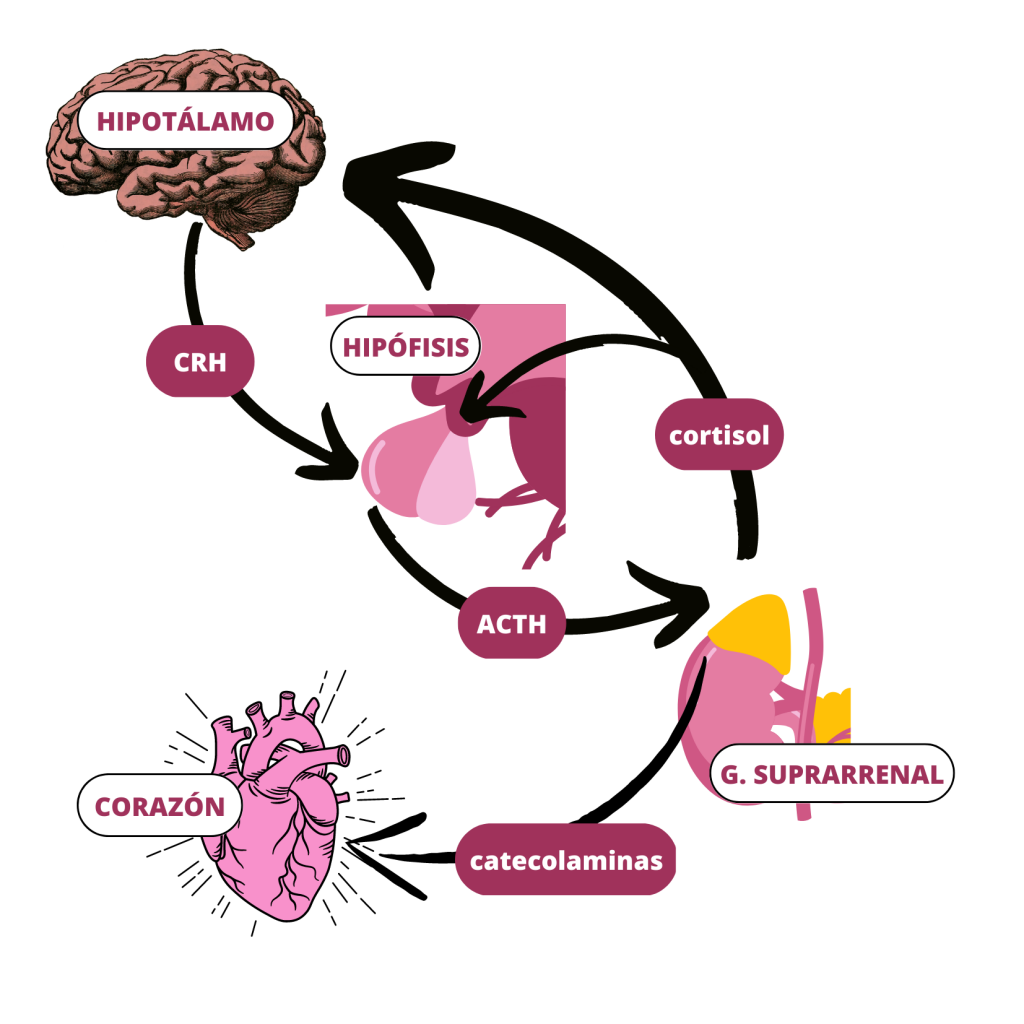
Una de las posibles explicaciones a esta patología es que se produzca debido al exceso de estimulación del eje hipotálamo-hipófisis-adrenal. (Figura 3). En situaciones de estrés, se activa esta vía, para producir hormonas como los glucocorticoides (cortisol) y las catecolaminas (epinefrina y norepinefrina). (Figura 4)
Figura 3: Eje hipotálamo-hipófisis-adrenal y su papel en el corazón. CRH: Hormona Liberadora de Corticotropina ; ACTH: Hormona Adenocorticotropa o Corticotropina (De elaboración propia)Figura 4: Estructura química del cortisol, norepinefrina y epinefrina (de izquierda a derecha)
Cuando se produce esta patología, los niveles plasmáticos de epinefrina y norepinefrina son altos, lo que indica que pueden estar ejerciendo un efecto desmesurado sobre el corazón, lo que derivaría en este síndrome. (Wittstein et al., 2005)
La adrenalina y la noradrenalina actúan sobre receptores adrenérgicos para ejercer su función. En el corazón, el ventrículo izquierdo es el lugar en el que se encuentran el mayor número de receptores adrenérgicos. Esto explica que el ventrículo izquierdo del corazón sea la zona que se ve más afectada en el Síndrome de Takotsubo. (Gupta & Gupta, 2018)
Se producen cambios en el metabolismo celular, que llevan a una disminución de la contractibilidad del corazón, principalmente en el ventrículo izquierdo. (Gupta & Gupta, 2018)
Como curiosidad, algunas publicaciones científicas apuntan a una posible relación entre esta patología cardíaca y el cáncer. Se dice que es posible que las catecolaminas que se liberan en esta enfermedad actúen sobre células tumorales, induciendo su crecimiento. (Sattler et al., 2017)
Además, se ha postulado que el hecho de que afecte más frecuentemente a mujeres puede deberse a la influencia de ciertas hormonas sexuales. (Alkhoury et al., 2016)
Otros posibles factores que podría jugar algún papel en el desarrollo de esta enfermedad son los aspectos genéticos. (Ikutomi et al., 2014)
Tratamiento
No hay un solo tratamiento claro para esta condición.
Dependiendo de las circunstancias personales de cada paciente o de otras enfermedades coexistentes, el tratamiento podría incluir beta-bloqueadores (para frenar el efecto de las catecolaminas sobre sus receptores), ventilación mecánica, levosimendan (para la insuficiencia cardíaca)… (Santoro et al., 2013; Templin et al., 2015)
Conclusión
El Síndrome de Takotsubo es un gran ejemplo de que existe una conexión entre el cerebro y diferentes órganos de nuestro cuerpo, en este caso, entre el cerebro y el corazón. No solo influyen las cuestiones físicas sobre la salud del cuerpo, sino que las cuestiones emocionales a veces pueden ser tan influyentes que no solo afectan a nuestro estado de ánimo, también pueden llegar a afectar al funcionamiento de órganos tan importantes como lo es el corazón.
En definitiva, si solías ser de los que dicen «de amor nadie se muere», apuesto a que ahora habrás cambiado de opinión y tendrás que decir «de amor sí se puede morir».
Referencias
Alkhoury, J., Lundgren, J., Ali, A., Mesinovic, D., Redfors, B., & Omerovic, E. (2016). Updates on publication trends in takotsubo syndrome doi:10.1016/j.ijcard.2016.07.059
Budnik, M., Kochanowski, J., Piatkowski, R., Wojtera, K., Peller, M., Gaska, M., . . . Opolski, G. (2016). Simple markers can distinguish takotsubo cardiomyopathy from ST segment elevation myocardial infarction. International Journal of Cardiology, 219 doi:10.1016/j.ijcard.2016.06.015
Deshmukh, A., Kumar, G., Pant, S., Rihal, C., Murugiah, K., & Mehta, J. L. (2012). Prevalence of takotsubo cardiomyopathy in the united states. American Heart Journal, 164(1) doi:10.1016/j.ahj.2012.03.020
Elesber, A. A., Prasad, A., Lennon, R. J., Wright, R. S., Lerman, A., & Rihal, C. S. (2007). Four-year recurrence rate and prognosis of the apical ballooning syndrome. Journal of the American College of Cardiology, 50(5) doi:10.1016/j.jacc.2007.03.050
Falola, M., Fonbah, W., & McGwin, G. (2013). Takotsubo cardiomyopathy versus ST-elevation myocardial infarction in a large case-control study: Proposing a new mechanism. International Journal of Cardiology, 167(3) doi:10.1016/j.ijcard.2012.10.059
Ghadri, J. R., Sarcon, A., Diekmann, J., Bataiosu, D. R., Cammann, V. L., Jurisic, S., . . . Prasad, A. (2016). Happy heart syndrome: Role of positive emotional stress in takotsubo syndrome. European Heart Journal, 37(37) doi:10.1093/eurheartj/ehv757
Gupta, S., & Gupta, M. M. (2018). Takotsubo syndrome doi:10.1016/j.ihj.2017.09.005
Ikutomi, M., Yamasaki, M., Matsusita, M., Watari, Y., Arashi, H., Endo, G., . . . Ohnishi, S. (2014). Takotsubo cardiomyopathy in siblings. Heart and Vessels, 29(1) doi:10.1007/s00380-013-0345-y
Khera, R., Light-Mcgroary, K., Zahr, F., Horwitz, P. A., & Girotra, S. (2016). Trends in hospitalization for takotsubo cardiomyopathy in the united states. American Heart Journal, 172 doi:10.1016/j.ahj.2015.10.022
Lyon, A. R., Bossone, E., Schneider, B., Sechtem, U., Citro, R., Underwood, S. R., . . . Omerovic, E. (2016). Current state of knowledge on takotsubo syndrome: A position statement from the taskforce on takotsubo syndrome of the heart failure association of the european society of cardiology doi:10.1002/ejhf.424
Migliore, F., Zorzi, A., Perazzolo Marra, M., Iliceto, S., & Corrado, D. (2015). Myocardial edema as a substrate of electrocardiographic abnormalities and life-threatening arrhythmias in reversible ventricular dysfunction of takotsubo cardiomyopathy: Imaging evidence, presumed mechanisms, and implications for therapy. Heart Rhythm, 12(8) doi:10.1016/j.hrthm.2015.04.041
Santoro, F., Ieva, R., Ferraretti, A., Ienco, V., Carpagnano, G., Lodispoto, M., . . . Brunetti, N. D. (2013). Safety and feasibility of levosimendan administration in takotsubo cardiomyopathy: A case series. Cardiovascular Therapeutics, 31(6) doi:10.1111/1755-5922.12047
Sato, H., Tateishi, H., Uchida, T., Dote, K., Ishihara, M., Kodama, K., … & Hori, M. (1990). Clinical aspect of myocardial injury: from ischemia to heart failure. Kagaku Hyoronsha, 2, 55-64.
Sattler, K., El-Battrawy, I., Lang, S., Zhou, X., Schramm, K., Tülümen, E., . . . Akin, I. (2017). Prevalence of cancer in takotsubo cardiomyopathy: Short and long-term outcome. International Journal of Cardiology, 238 doi:10.1016/j.ijcard.2017.02.093
Singh, K., Carson, K., Shah, R., Sawhney, G., Singh, B., Parsaik, A., . . . Horowitz, J. (2014). Meta-analysis of clinical correlates of acute mortality in takotsubo cardiomyopathy doi:10.1016/j.amjcard.2014.01.419
Templin, C., Ghadri, J. R., Diekmann, J., Napp, L. C., Bataiosu, D. R., Jaguszewski, M., . . . Lüscher, T. F. (2015). Clinical features and outcomes of takotsubo (stress) cardiomyopathy. New England Journal of Medicine, 373(10) doi:10.1056/nejmoa1406761
Wittstein, I. S., Thiemann, D. R., Lima, J. A. C., Baughman, K. L., Schulman, S. P., Gerstenblith, G., . . . Champion, H. C. (2005). Neurohumoral features of myocardial stunning due to sudden emotional stress. New England Journal of Medicine, 352(6) doi:10.1056/nejmoa043046
ADN G-cuadruplexos, diana farmacológica frente al cáncer
escrito por Anaycristina_3C | 28 enero, 2023
Realizado por Ana Jiménez y Cristina Iruela – 3º de Biología Sanitaria, UAH
Los G-cuadruplexos son unas estructuras químicas que llevan años en el punto de mira por su característica estructra y localización. Cada vez se apuesta más por ellos como terapia frente al cáncer dada su interacción con estrucutras y moléculas íntimamente relacionadas con la enfermedad. A continuación se expondrá una breve revisión sobre el tema.
Estructura y función de los ADN-G cuadruplexos
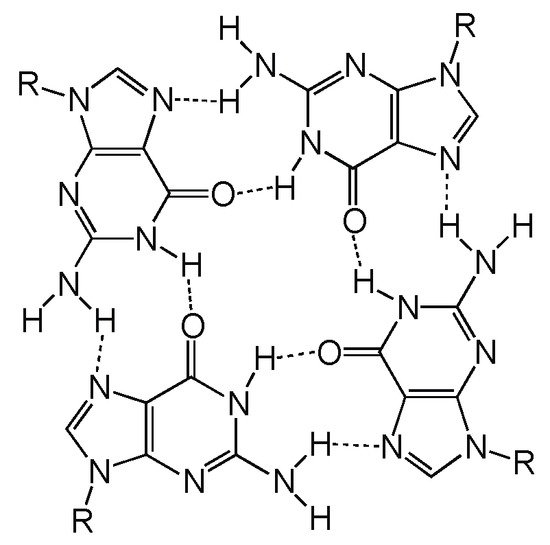
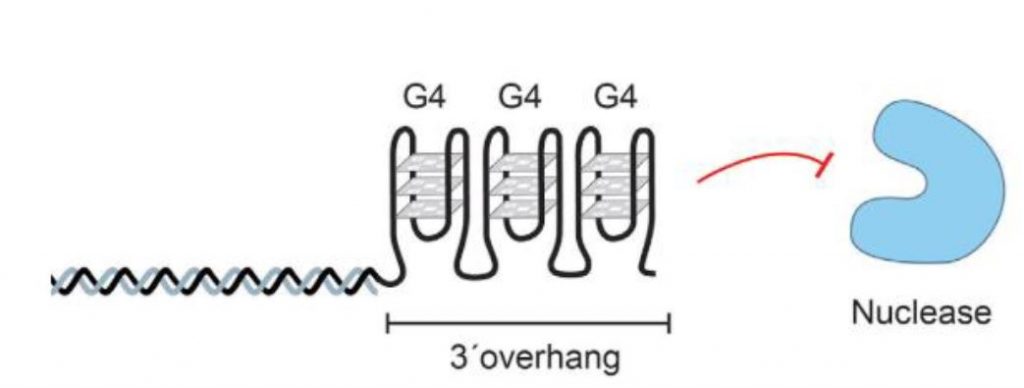
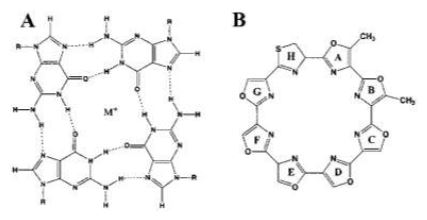
Las secuencias de ADN ricas en guanina pueden plegarse en estructuras secundarias no canónicas de cuatro cadenas denominadas G-cuadruplexos (G4). Estas estructuras secundarias se forman tanto en el ADN como en el ARN. Consiste en 4 guaninas unidas por puentes de hidrógeno de tipo Hoogsteen, en los que cada guanina puede actuar como donante y aceptor de dos puentes de hidrógeno formando una estructura plana denominada tétrada G [1].
Dos o más tétradas G se pueden apilar una encima de otra para formar un G-cuadruplexo, siendo esta su unidad estructural. Esta se forman conectando 4 guaninas a través de 8 puentes de hidrógeno. En la tétrada G, se forman dos de estos puentes que emparejan guaninas adyacentes, en los que están involucrados los nitrógenos número 1,7, 2 y el oxígeno 6 de cada nucleótido de guanina [2].
Figura 1 Estructura química de una tétrada G Tomada de Kolesnikova, S., & Curtis, E. A. (2019). Structure and Function of Multimeric G-Quadruplexes. Molecules (Basel, Switzerland), 24(17), 3074. https://doi.org/10.3390/molecules24173074
Además, es necesaria la presencia de un catión metálico (Na+, K+) para estabilizar la estructura [3].
En el ARN, los G4 formados en la región 5’UTR del ARNm inhiben la traducción dependiente de cap y mejoran la traducción independiente de caperuza mediada por IRES. También influyen en otros mecanismos moleculares que tienen lugar en el ARN, como el empalme, cambios en el marco de lectura, localización del ARNm o la maduración de los miARN [3].
En base a experimentos in vitro, se predijo que los G-cuadruplexos se forman en regiones que albergan un motivo G4 específico. Sin embargo, estudios actuales muestran que también pueden formarse dentro de regiones con bucles formados por 3 o más guaninas por repetición, así como en regiones que no siguen este motivo G4 estricto [1].
No se distribuyen al azar en todo el genoma, sino que abundan en ciertas regiones, como promotores, telómeros, sitios de unión de factores de transcripción u orígenes de replicación. La estabilidad de esta estructura depende, entre otros factores, del número de guaninas por repetición y de la longitud de los bucles [1].
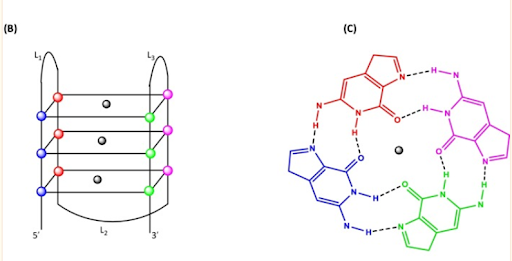
Figura 2 Estructura de los G-cuadruplexos Nota: B) Una representación 2D de un pliegue G4 típico que muestra los tres cuartetos planos. Las esferas en los vértices de los cuartetos representan una guanina de cada uno de los cuatro G-tripletes. La esfera negra en el centro denota el catión metálico central (Na + , K + ) necesario para estabilizar la estructura G4. (C) Una vista superior de un cuarteto G plano que muestra los enlaces Hoogsteen (líneas discontinuas), los átomos de los mismos y un catión en la cavidad central. Las figuras no están dibujadas a escala. Fragmento tomado de: Saranathan, N., & Vivekanandan, P. (2019). G-Quadruplexes: More Than Just a Kink in Microbial Genomes. Trends in microbiology, 27(2), 148–163. https://doi.org/10.1016/j.tim.2018.08.011
La relevancia fisiológica de estas estructuras se debe a la existencia de proteínas que pueden unirse a ellas o desplegarlas. Existen 3 clases de proteínas que interactúan con los G-cuadruplexos descritas en la literatura: proteínas de unión a G-cuadruplexos, estabilizadoras de G-cuadruplexos y desarrolladoras de G-cuadruplexos (como helicasas). Se ha descrito qué mutaciones y/o delecciones en estas proteínas conducen a cambios en la formación de estas estructuras. Lo que, a su vez, puede dar lugar a cambios en las vías biológicas (cambios transcripcionales) y aumentar la inestabilidad del genoma [1].
La formación transitoria de G4 en condiciones termodinámicamente favorables tiene funciones reguladoras importantes dictadas por su ubicación en el genoma [3]. Entre ellas se encuentran la regulación de la transcripción, traducción, replicación del ADN y localización del ARN [4]. Destaca la función de los G-cuadruplexos en relación a la inhibición de la actividad de la telomerasa [3].
Relación con los telómeros + telomerasa + cáncer
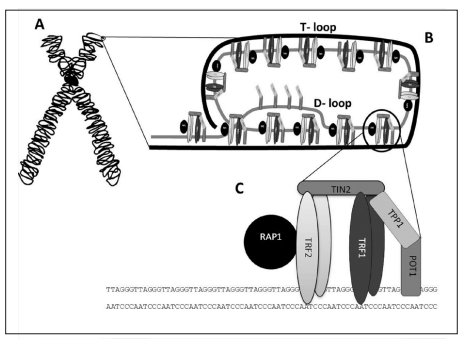
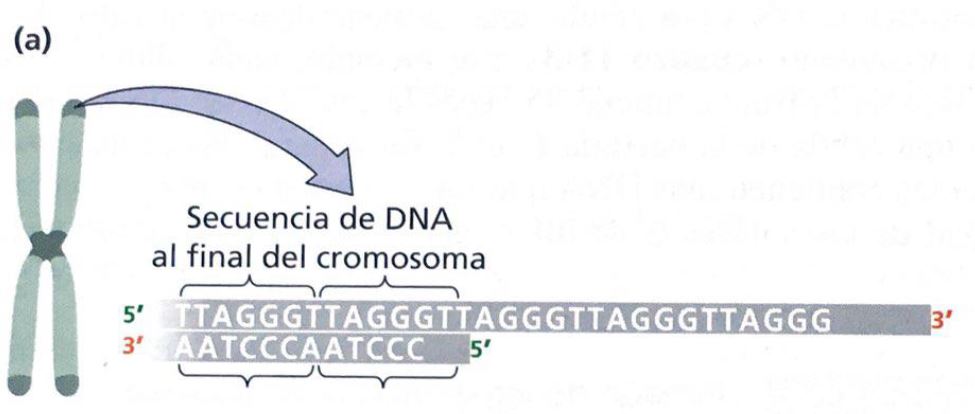
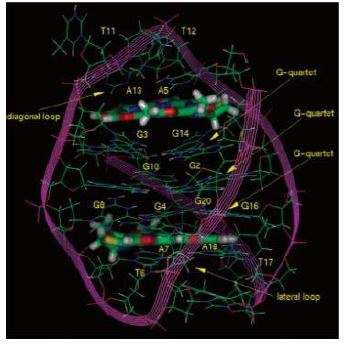
Como ya se ha mencionado, los telómeros son un ejemplo de la presencia de G4 cuadruplexos en el genoma de los vertebrados, basándose en la secuencia consenso: (5’-TTAGGG-3’) [5] que evidencia la presencia repetitiva de las guaninas (dicha secuencia es específica para los mamíferos y cambia según la especie de los mismos).
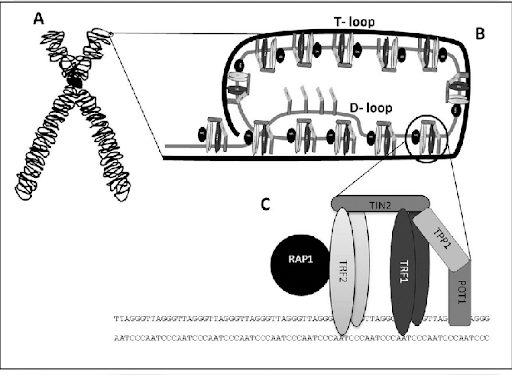
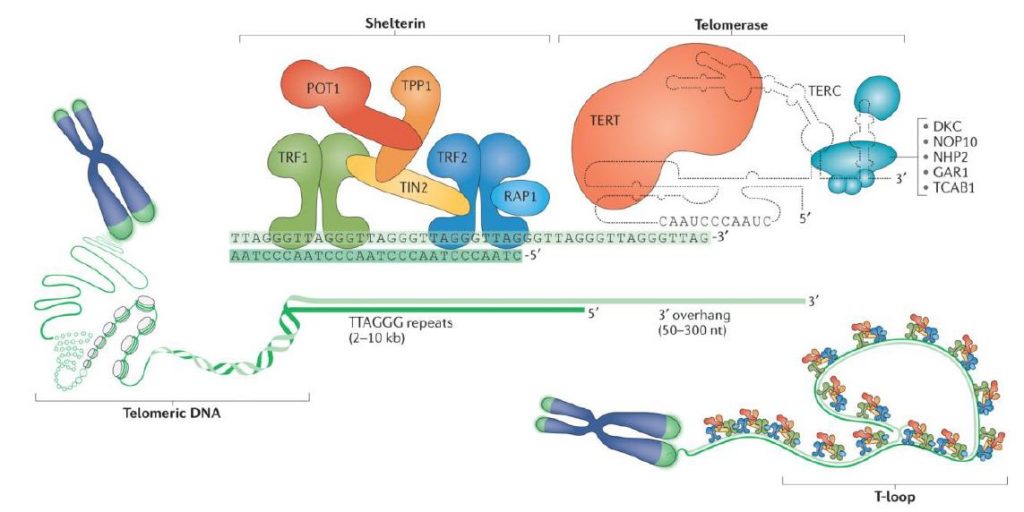
Para comprender la importancia de los telómeros, es clave entender la estructura de los mismos, cuya formación es una respuesta evolutiva al problema encontrado en los extremos 3’ cuando la maquinaria de replicación de nuestras células no puede rellenar el hueco al no tener un extremo 5’ anterior que le sirva de molde para la síntesis de la nueva cadena. Esto tiene como resultado la formación de un T-loop y un D-loop originados por la invasión de un extremo 3’ que sobresalía respecto al extremo 5’ complementario [6]. Además, se encontrará el complejo de Shelterina, el cual poseerá diferentes proteínas que regularán la actividad de la telomerasa, enzima encargada de la elongación de los telómeros por medio de la adición de unidades (TTAGGG).
Esta respuesta evita la pérdida de información en cada ronda de replicación y evitan que la célula reconozca esta región sobrante como un daño en el ADN y lo elimine. De todas maneras, estos telómeros se irán acortando igualmente con el tiempo: acortamiento telomérico de Hayflick, resultando en un punto crítico de longitud activando la llamada senescencia replicativa, siendo este proceso la base del envejecimiento celular que resulta en poner fin a su división [7]. La regulación de dicha senescencia es clave para el organismo para evitar su envejecimiento y como supresor de tumores [8].
Figura 3 Representación de un cromosoma y terminación telomérica Nota: A) Esquema de un cromosoma indicando la ubicación de un telómero. B) Estructura del telómero: T-loop secuestrando el extremo terminal del cromosoma, y D-loop donde se observa la triple hebra de ADN. C) Complejo Shelterina de proteínas asociadas a los telómeros. Tomado de Mengual Gomez, Diego & Armando, Romina & Farina, Hernán & Gomez, Daniel. (2014). Telomerasa y telómero: su estructura y dinámica en salud y enfermedad. Medicina. 74. 69-76.
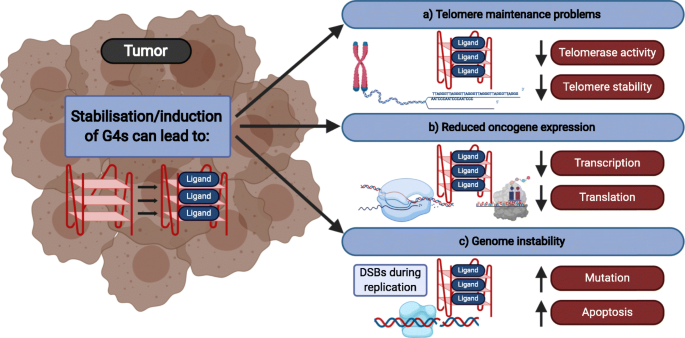
Los G4 tienen un papel de represión de determinados genes en células sanas impidiendo la entrada de la maquinaria necesaria para la replicación y transcripción. En células sanas, estos evitan la expresión de oncogenes como: MYC, sufriendo así un proceso de regulación negativa [5].
Figura 4 Resumen esquemático de los efectos de los ligandos de G4 en las células cancerosas Tomado de: Kosiol, N., Juranek, S., Brossart, P., Heine, A., & Paeschke, K. (2021). G-quadruplexes: a promising target for cancer therapy. Molecular cancer, 20(1), 40. https://doi.org/10.1186/s12943-021-01328-4
Lo que ocurre en enfermedades como el cáncer es que el acortamiento de los telómeros se evita hasta tal punto que las células se inmortalizan y escapan al proceso de muerte celular. La base patológica de esto es la activación de la telomerasa la cual está además sobre expresada en los tejidos cancerosos [9], cuya activación será siempre el reflejo de una respuesta anómala. Los G4 presentes en los telómeros de sus células no tendrán la misma eficacia que en las células sanas, puesto que la telomerasa se introduce y favorece la elongación de dicho telómero. Este suceso tendrá como consecuencia el desarrollo del fenotipo inmortal que adoptarán las células del tejido afectado y que se volverán cancerosas [5]. Es importante destacar que la alteración de la unión de los G4 con la telomerasa se ha observado tanto in vivo como in vitro [1].
Cabe mencionar las regiones TERRA, región telomérica de RNA no codificante [5]. Esta, es el transcrito resultante del telómero llevado a cabo por la enzima RNA polimerasa II la cual puede aparecer como ARN nucleoplásmico libre o en forma de un nuevo loop en la estructura de los telómeros: R-loop (correspondiente a un híbrido entre ADN y ARN) [8].
Cuando el telómero se acorta hasta dicho punto crítico anteriormente mencionado, este R-loop se asocia con el resto de TERRA promoviendo la reparación dirigida por homología (denominada HDR-mediated). Este proceso va a permitir la recombinación del telómero con su propia secuencia perpetuando así su vida celular y evitando la senescencia prematura. Además, este mismo mecanismo será utilizado por algunas células cancerosas para la elongación de los telómeros en caso de no poseer telomerasa funcionando como mecanismo de alargamiento alternativo, siendo la base de los tumores ALT [8].
Paradójicamente, algunos estudios han dado evidencia de la longitud reducida de los telómeros de las células cancerígenas respecto a las células de tejidos libres de cáncer, así como un aumento del número de los G4 en las mismas [5][9]. Para esto se siguen formulando diferentes hipótesis.
Otras utilidades bioquímicas
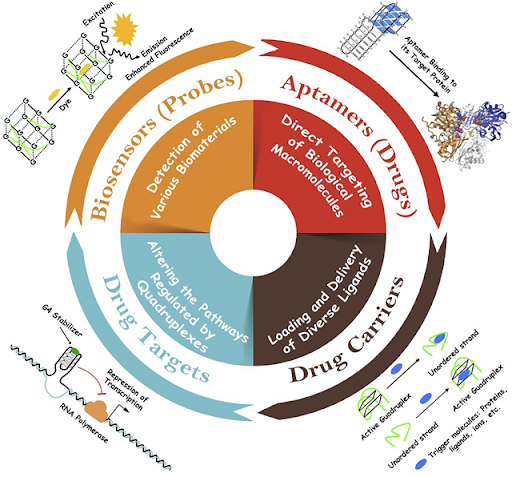
Además de la función anteriormente mencionada, se han estudiado cada vez más aplicaciones:
Son utilizados como sondas, solas o en complejo con hemina, una estructura de porfirina que contiene hierro para detectar la presencia de diferentes ligandos [10].
También como transportadores, gracias a su capacidad para secuestrar ligandos, actuando como agentes de administración de fármacos [10].
En los últimos años, se ha extendido su uso como fármacos, en concreto como aptámeros (ácidos nucleicos de cadena sencilla aislados de genotecas de oligonucleótidos por selección in vitro), interactuando con biomoléculas, como proteínas e interfiriendo con sus funciones [10].
O como dianas farmacológicas explotando su capacidad para interactuar con ligandos específicos, lo que puede alterar funciones importantes si el G-cuadruplexo se encuentra en regiones esenciales en el genoma del virus o de la célula huésped [10].
Figura 5 Aplicaciones de los G-cuadruplexos Nota: Representación gráfica de las principales aplicaciones de los G-cuadruplexos. Tomado de: Abiri, A., Lavigne, M., Rezaei, M., Nikzad, S., Zare, P., Mergny, J. L., & Rahimi, H. R. (2021). Unlocking G-Quadruplexes as Antiviral Targets. Pharmacological reviews, 73(3), 897–923. https://doi.org/10.1124/pharmrev.120.000230
Telomestatina
En múltiples estudios, se ha propuesto que las mejores dianas farmacológicas serían aquellas que solo se expresasen en las células cancerosas o aquellas que fuesen esenciales para mantener el fenotipo maligno de las mismas. La telomerasa, es una diana clave [6][7][9].
Se trata de un producto natural aislado de Streptomyces anulatus que es un ligando de los G4 teniendo una afinidad muy alta por la secuencia concreta de los telómeros: (5’-TTAGGG-3’). Al interaccionar, inhibe de manera eficaz la actuación de la telomerasa, por lo que se detiene la elongación de los telómeros de las células cancerígenas y como consecuencia suprime su proliferación. Esta actividad anticancerígena provoca que algunos de los factores claves encontrados en el complejo de Shelterina del telómero, como TRF2 y POT1, se liberen de dicho telómero, evitando así que lleven a cabo su función de retrasar la senescencia [6].
Además, la telomestatina es un ligando que tiene una mayor afinidad por los G4intramoleculares, tanto si han sido formados a partir de un ADN telomérico dúplex, como de uno monocatenario, teniendo la función anteriormente mencionada. Esto supone una ventaja frente a otros compuestos como TMPyP4, el cual posee afinidad por los G4 intermoleculares y teniendo un efecto totalmente diferente el cual no se ha observado en la telomestatina: formación de puentes de anafase en erizos de mar [6].
A pesar de sus ventajas estabilizado los G4 cuadruplexos, arrastra algunas características que resultan contraproducentes así como sus solubilidad o inestabilidad, por lo que se empezaron a utilizar algunos compuestos análogos sintéticos [5].
Búsqueda de otros fármacos
En definitiva, la existencia de análogos sintéticos de G4s es lo que ha permitido contemplar una nueva forma de terapia para el cáncer [5][11], dado que reprime el correcto funcionamiento de las células cancerosas, llegando a conseguir la destrucción de la misma; así como análogos de la telomestatina [11], aunque estas terapias siguen en constante estudio y desarrollo.
El silvestrol es un compuesto obtenido de la corteza de los árboles de la familia flavaglina cuya estructura permite inhibir el factor de transcripción: eIF4A, tratándose de una análogo sintético. El factor posee una actividad helicasa clave para el proceso fisiológico de la transcripción al permitir deshacer las estructuras secundarias que pueden aparecer en la cadena de ADN y que impedirían la continuación del proceso. Al mismo tiempo tiene un papel clave en la carcinogénesis al facilitar la leucemia linfoblástica aguda de las células T al promover la transcripción de oncogenes como MYC, CDK6 o MDM2 al desenrollar los G4 de la región 5’ UTR de sus mRNAs. Este compuesto lo que hará, será inhibir al eIF4A [5], interfiriendo indirectamente en el mantenimiento de la estructura de los ADN G cuadruplexos.
Otro análogo que también afecta al gen MYC es: TMPyP4, anteriormente mencionado. Este se basa en la represión de proto-oncogenes de dicho gen por medio de la estabilización de los G4 cuadruplexos [5].
Los análogos “pirodistatina” y CX-3542 provocan daño en células cancerosas también. El primero, induce la formación de un nuevo loop en la estructura del telómero: “R-loop”, siendo un híbrido de DNA y RNA transcrito causando un daño en el ADN canceroso. El segundo causa daño y muerte celular con mayor eficacia en 2 tipos celulares cancerosos concretamente: células ATRX deficientes y células BRCA1/2 deficientes [5].
En relación a la función de estas estructuras como fármacos, existen secuencias cortas en los ácidos nucleicos derivadas del motivo hexanucleotido TGGGAG, denominadas “secuencias de Hotoda” que son potentes inhibidores anti-VIH. Estas secuencias cortas también se encuentran activas en otros virus como en los que aparecen secuencias de 6 nucleótidos con la siguiente estructura GGGGGT, la cual, da lugar a G-cuadruplexos. Este se une al dominio C-terminal de la proteasa del virus de la hepatitis A y es un fuerte inhibidor de la proteasa 3C de este virus [10]. Al inhibirla, impide que el virus descomponga sus proteínas para poder multiplicarse. Por lo tanto, deja de propagarse.
Un argumento notable es que estas secuencias cortas son demasiado cortas para ser específicas. Además, pueden actuar sobre otros componentes celulares del huésped, que se unen a estructuras secundarias de ADN no canónicas [10].
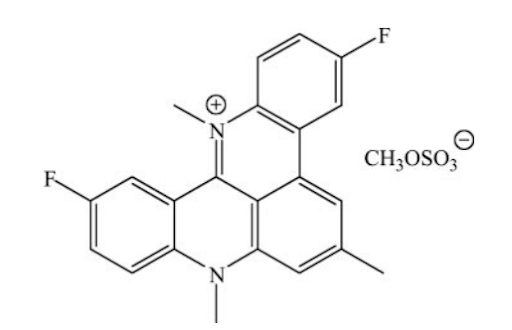
Otro fármaco que ha resultado ser un potente inhibidor de la telomerasa es RHPS4, tratándose de un mutante de la subunidad de la telomerasa denominada hTERT. La expresión de dicha subunidad mutante ha dado evidencias de inhibir el proceso de la telomerasa al unirse y competir por el sitio de unión. Tras estudiar su efecto en células tumorales, se concluyó que la línea celular MCF-7 de las células pertenecientes al cáncer de mama sufren una detención del crecimiento similar a la senescencia [7].
Figura 6 Estructura de RHPS4 Tomada de: Cookson, J. C., Dai, F., Smith, V., Heald, R. A., Laughton, C. A., Stevens, M. F., & Burger, A. M. (2005). Pharmacodynamics of the G-quadruplex-stabilizing telomerase inhibitor 3,11-difluoro-6,8,13-trimethyl-8H-quino[4,3,2-kl]acridinium methosulfate (RHPS4) in vitro: activity in human tumor cells correlates with telomere length and can be enhanced, or antagonized, with cytotoxic agents. Molecular pharmacology, 68(6), 1551–1558. https://doi.org/10.1124/mol.105.013300
Referencias consultadas
Kosiol, N., Juranek, S., Brossart, P., Heine, A., & Paeschke, K. (2021). G-quadruplexes: a promising target for cancer therapy. Molecular cancer, 20(1), 40. https://doi.org/10.1186/s12943-021-01328-4
Yuan, W. F., Wan, L. Y., Peng, H., Zhong, Y. M., Cai, W. L., Zhang, Y. Q., Ai, W. B., & Wu, J. F. (2020). The influencing factors and functions of DNA G-quadruplexes. Cell biochemistry and function, 38(5), 524–532. https://doi.org/10.1002/cbf.3505
Saranathan, N., & Vivekanandan, P. (2019). G-Quadruplexes: More Than Just a Kink in Microbial Genomes. Trends in microbiology, 27(2), 148–163. https://doi.org/10.1016/j.tim.2018.08.011
Kolesnikova, S., & Curtis, E. A. (2019). Structure and Function of Multimeric G-Quadruplexes. Molecules (Basel, Switzerland), 24(17), 3074. https://doi.org/10.3390/molecules24173074
Nakanishi, C., & Seimiya, H. (2020). G-quadruplex in cancer biology and drug discovery. Biochemical and biophysical research communications, 531(1), 45–50. https://doi.org/10.1016/j.bbrc.2020.03.178
Kim, M. Y., Gleason-Guzman, M., Izbicka, E., Nishioka, D., & Hurley, L. H. (2003). The different biological effects of telomestatin and TMPyP4 can be attributed to their selectivity for interaction with intramolecular or intermolecular G-quadruplex structures. Cancer research, 63(12), 3247–3256.
Cookson, J. C., Dai, F., Smith, V., Heald, R. A., Laughton, C. A., Stevens, M. F., & Burger, A. M. (2005). Pharmacodynamics of the G-quadruplex-stabilizing telomerase inhibitor 3,11-difluoro-6,8,13-trimethyl-8H-quino[4,3,2-kl]acridinium methosulfate (RHPS4) in vitro: activity in human tumor cells correlates with telomere length and can be enhanced, or antagonized, with cytotoxic agents. Molecular pharmacology, 68(6), 1551–1558. https://doi.org/10.1124/mol.105.013300
Pérez-Martínez, L., Wagner, T., & Luke, B. (2022). Telomere Interacting Proteins and TERRA Regulation. Frontiers in genetics, 13, 872636. https://doi.org/10.3389/fgene.2022.872636
Kelland L. R. (2005). Overcoming the immortality of tumour cells by telomere and telomerase based cancer therapeutics–current status and future prospects. European journal of cancer (Oxford, England : 1990), 41(7), 971–979. https://doi.org/10.1016/j.ejca.2004.11.024
Abiri, A., Lavigne, M., Rezaei, M., Nikzad, S., Zare, P., Mergny, J. L., & Rahimi, H. R. (2021). Unlocking G-Quadruplexes as Antiviral Targets. Pharmacological reviews, 73(3), 897–923. https://doi.org/10.1124/pharmrev.120.000230
Teng, F. Y., Jiang, Z. Z., Guo, M., Tan, X. Z., Chen, F., Xi, X. G., & Xu, Y. (2021). G-quadruplex DNA: a novel target for drug design. Cellular and molecular life sciences : CMLS, 78(19-20), 6557–6583. https://doi.org/10.1007/s00018-021-03921-8
El precio de la inmortalidad
escrito por adrarranz2_3B | 28 enero, 2023
Andrea Adrados Santa Elena, Alba Arranz Benayas y Laura Arranz Ortega, 3º Biología Sanitaria UAH
La muerte nos asusta a todos pero, ¿merecería la pena alcanzar la inmortalidad?
La esperanza de vida cada vez es mayor con el paso de los años, gracias a los avances médicos, y la mejora de las condiciones de vida; pero nuestras células siguen teniendo una fecha de caducidad.
Existen varias estrategias para frenar ese envejecimiento celular y así prolongar su duración. Esto se podría conseguir gracias a una enzima, la telomerasa, que permitiría alargar los telómeros acortados, que son los responsables de la muerte celular, lo que supondría un “rejuvenecimiento” de la célula. De esta manera se conseguiría incrementar nuestros años de vida.
No obstante, la telomerasa tiene su lado negativo, puesto que está presente en la mayoría de cánceres en los que las células tienen un crecimiento ilimitado. Por lo tanto, conseguir una activación constante de las células de nuestro cuerpo puede ser un gran peligro, ya que si no se controla, en vez de darnos más años de vida, nos los estaría quitando.
Telómeros
¿Qué son los telómeros?
Los telómeros son complejos nucleo-proteicos que constituyen las estructuras de los extremos de los cromosomas lineales permitiéndoles mantener su integridad ya que otorgan estabilidad y protección.
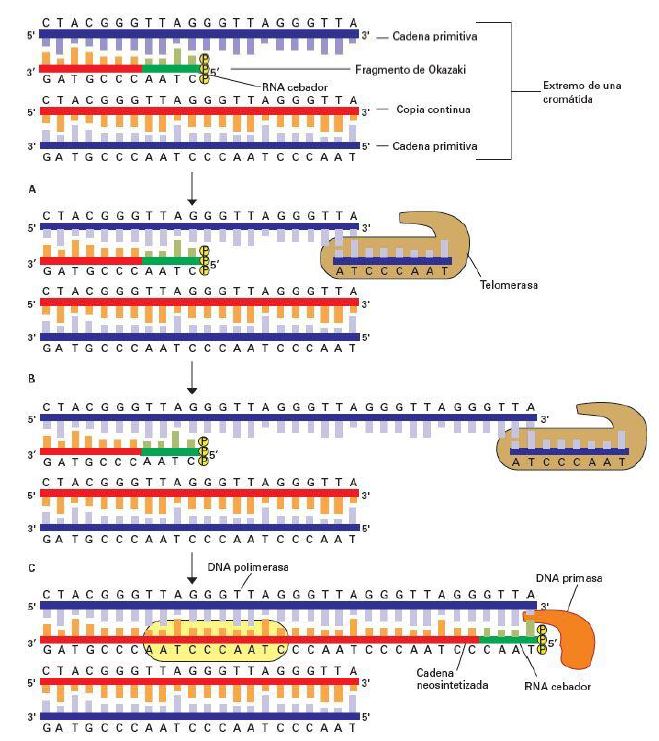
Las ADN polimerasas, enzimas encargadas de replicar el ADN, necesitan un extremo OH 3’ libre sobre el que ir añadiendo nucleótidos y rellenar el hueco que queda tras eliminar el cebador. Sin embargo, al tratarse de un cromosoma lineal dicho extremo no está presente y en cada ronda de replicación se pierde un determinado número de bases. Los telómeros desempeñan por tanto una función clave ya que evitan que se pierda información vital, en su lugar se perderán bases de su estructura.
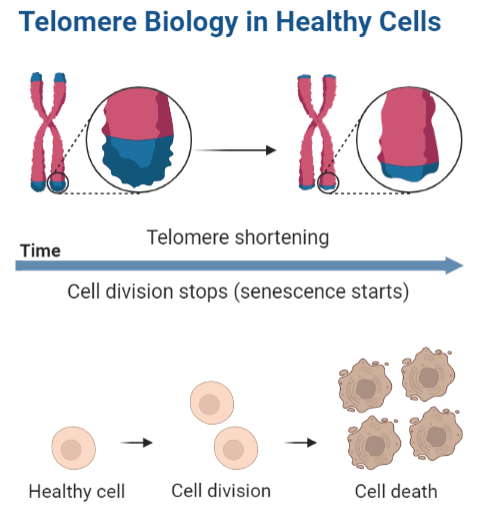
Además, están a cargo del reloj mitótico y por consiguiente la senescencia celular, es decir, determinan el número de divisiones que tendrá la célula y la proliferación celular se frenará cuando la longitud de los telómeros sea crítica. Llegados a ese punto se dirige la célula a la muerte celular, que es lo que desemboca en degeneración tisular y se traduce, en lo que cotidianamente vemos como envejecimiento
Imagen 1: Esquema del acortamiento de telómeros dirige a la muerte celular. Plantilla elaborada con BioRender.
Estructura de los telómeros
Su estructura difiere del resto de la cromatina, de manera que no se llevan a cabo procesos de degradación, recombinación o fusión, es decir, les permiten no ser reconocidos por los sistemas de reparación del ADN.
Podemos encontrar tres regiones:
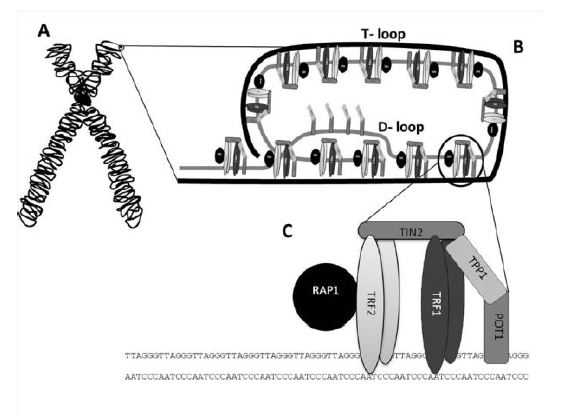
Extremo saliente (extremo 3’ overhang): son las secuencias no replicadas que quedan libres en forma de cadena sencilla. Posibilita la formación de unas estructuras secundarias en forma de bucle (T-loop y D-loop) por inserción de dicho extremo 3’ overhang en la región de doble cadena y posterior hibridación por complementariedad. Esto evita que los extremos de los cromosomas sean confundidos con ADN dañado.
Repeticiones teloméricas: se trata de secuencias cortas repetidas y conservadas entre las especies. Son ricas en nucleótidos G y T (en el caso de los humanos las secuencias teloméricas son TTAGGG) y pueden formar los G-cuadruplexos, estructuras complejas donde 4 guaninas quedan unidas por puentes de hidrógeno de Hoogsten formando planos cuadrados. Estos están implicados en el mantenimiento de los telómeros, pero hay que regular su apertura para permitir la replicación del ADN.
Áreas que están entre la primera secuencia de un gen y las repeticiones.
En los seres humanos, los telómeros interactúan con el complejo de la shelterina, formado por una serie de proteínas que incluyen a TRF1 y TRF2, las cuales interactúan con RAP1, TIN1, TPP1 y POT1. La función del complejo es impedir la activación del mecanismo de reparación del ADN en los extremos, protegiendo frente a la degradación, y regular la actividad de la telomerasa.
TRF1: es la secuencia C-terminal. Reconoce específicamente el fragmento de ADN telomérico y actúa como regulador negativo de la longitud telomérica (represor de la telomerasa)
TRF2: regulador negativo de la longitud telomérica, estabiliza la secuencia G repetitiva y previene de fusiones entre extremos teloméricos de distintos cromosomas
Por tanto, ambos restringen la actividad de la telomerasa, impidiendo la elongación telomérica
Reloj mitótico
A medida que los telómeros de las células se van acortando estas se vuelven senescentes, siendo la senescencia una situación en la que las células a pesar de ser viables y activas metabólicamente ya no proliferan, es irreversible, y conduce hacia la muerte celular. Cuando se detecta una longitud crítica de los telómeros se pone en marcha un mecanismo que bloquea el avance del ciclo celular y promueve la entrada en apoptosis gracias a la activación de las proteínas p53 y Rb (proteínas inhibidoras del ciclo celular).
Los telómeros pueden presentarse de dos formas: protegidos (capped) si están formando el T-loop, y desprotegidos (uncapped) si están de manera lineal, pudiendo haber transiciones entre ambas estructuras. El último caso se da cuando los telómeros son tan cortos que ya no son capaces de formar estructuras secundarias lo que hace que sean susceptibles de sufrir el ataque de nucleasas y fusión de extremos. Esto conduce a la inestabilidad cromosómica y senescencia, con la consecuente entrada en apoptosis ya mencionada.
Existe una teoría que sugiere que los telómeros del cromosoma 17 que es donde está codificada la proteína p53 sea el sitio donde se activa este mecanismo.
Telomerasa
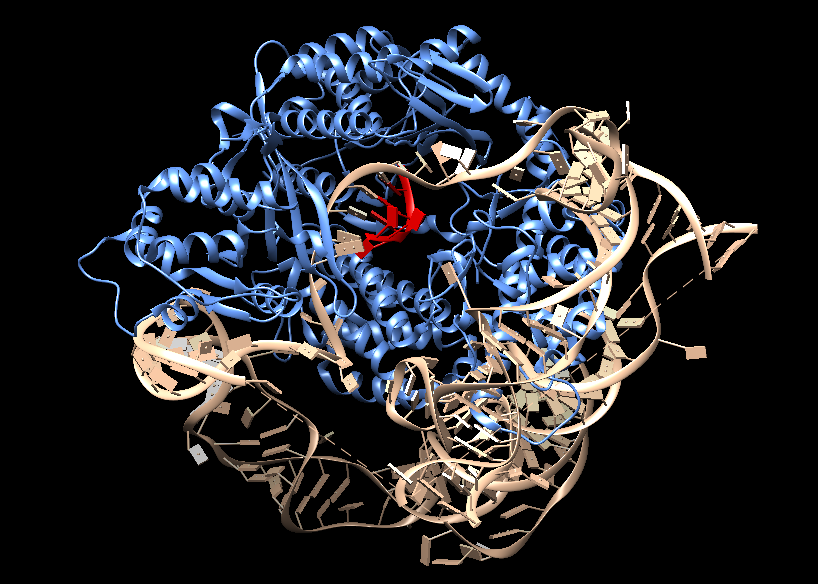
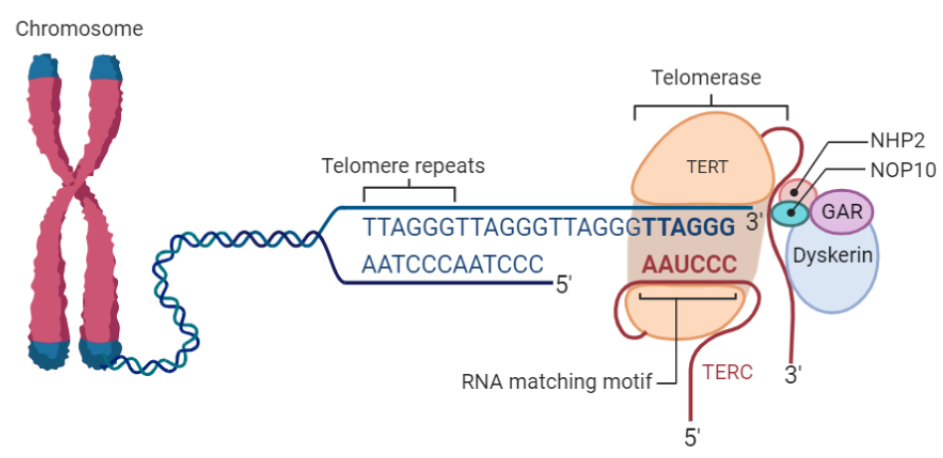
Estructura de la telomerasa
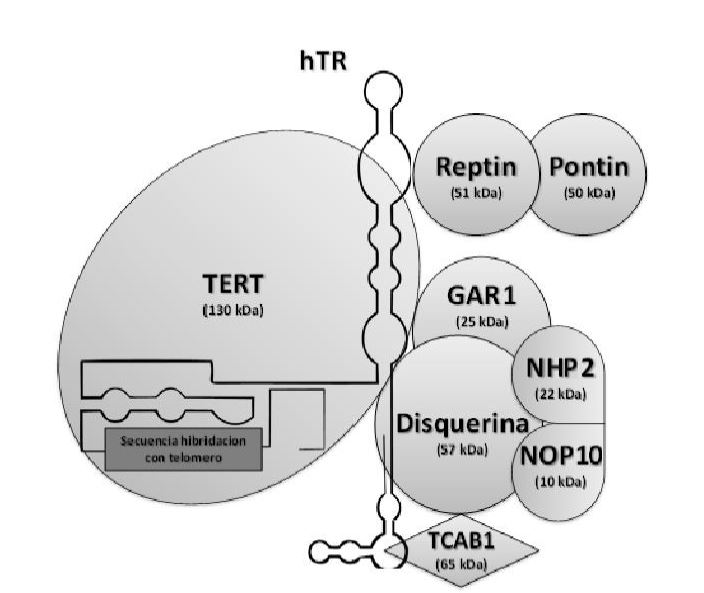
La telomerasas es una ribonucleoproteína formada por una subunidad catalítica llamada TERT, que es una retrotranscriptasa y un componente de ARN llamado TERC, que actúa como molde para la adición de secuencias teloméricas repetitivas en el extremo 3’.
Imagen 3: Telomerasa humana: subunidad catalítica TERT (azul), componente ribonucleico TERC (beige) y unidad telomérica de DNA correspondiente a secuencia corta de repetición TTAGGG (rojo). Fuente: imagen elaborada con Chimera, código PDB 7TRD
Función de la telomerasa
La telomerasa es la encargada de alargar los telómeros, se encuentra activa en todos los tejidos durante la embriogénesis y tras esta únicamente permanece en células de líneas germinales productoras de gametos y en células cancerosas. Su función está regulada tanto por proteínas quinasas (añaden un grupo fosfato) que aumentan su actividad, como por fosfatasas (eliminan un grupo fosfato) que reducen su actividad, por ello el balance entre ambas juega un papel esencial en la tumorigénesis.
Esta enzima tiene una gran afinidad por las secuencias ricas en G, reconoce y se une al extremo 3’ overhang del telómero, al ser reclutada por complejo de la shelterina, y lo alarga mediante la adición de nucleótidos en sentido 5’ → 3’ usando como molde su propia fracción de RNA (TERC). Dicha unión es posible gracias a que TERC presenta una serie de bases complementarias al ADN telomérico de tal modo que se produce el apareamiento entre ellas.
Imagen 4: inicio del mecanismo de acción de la telomerasa. Plantilla sacada de BioRender
A continuación, se recluta la primasa y la ADN polimerasa α que sintetizan el primer, un pequeño fragmento mixto de ARN y ADN que proporciona el extremo 3’ OH necesario para que intervenga la ADN polimerasa δ y complete el fragmento. Finalmente se da la eliminación del cebador y el ligado de los extremos, quedando nuevamente un extremo 3’ overhang.
También hay que destacar que TERT es el componente limitante de la telomerasa. Mientras que TERC tiene una expresión constitutiva en la mayoría de los tejidos, será la expresión de TERT la que conduzca hacia una activación de la función de la telomerasa por lo que su transcripción estará reprimida en células somáticas.
El lado oscuro de la telomerasa: cáncer
El cáncer es una enfermedad originada a partir de la transformación maligna de una célula que comienza a dividirse sin control y escapa de la muerte celular programada dando lugar a gran cantidad de células hijas, las cuales presentan también alteraciones en los mecanismos de proliferación, diferenciación y apoptosis.
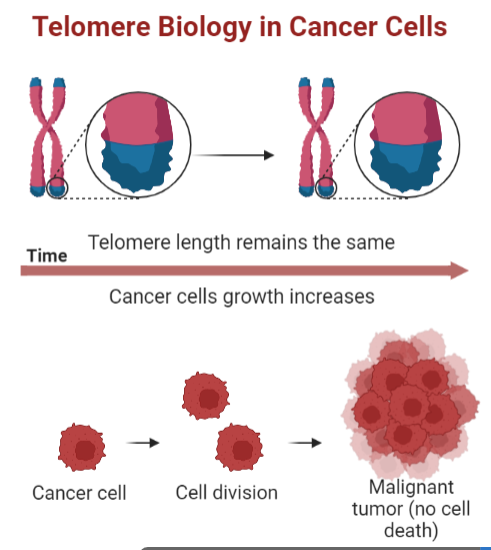
Varios estudios han demostrado la participación de la telomerasa en el proceso de carcinogénesis, puesto que se ha visto que varias líneas celulares cancerosas la presentan y además la inmortalización de las células in vitro ocurre a la vez que la activación de la enzima.
Las células somáticas no presentan telomerasa, por lo que tienen una capacidad limitada para replicarse, siendo esto una barrera de la proliferación. Sin embargo, los tumores malignos tienen una proliferación infinita, gracias a que sus células poseen la telomerasa activa que permite esa replicación sin límites.
Imagen 5: esquema de como la acción de la telomerasa puede conducir a célula tumoral. Plantilla elaborada con BioRender
Un posible tratamiento contra el cáncer
Últimamente ha habido investigaciones enfocadas en el acortamiento de los telómeros de las células cancerosas, sin embargo, esto es algo impreciso ya que tendría que ser específico para la longitud de estos en cada una de las células a tratar y podría demorarse demasiado tiempo. Las estrategias más prometedoras son aquellas que inhiben la protección de los telómeros atacando a la telomerasa, de este modo se podría lograr entrar en apoptosis en pocos días e incluso podría funcionar con telómeros largos.
Los estudios estiman que la telomerasa se detecta en un 80-90% de los tumores cancerosos, por lo que podría ser considerada un marcador de malignidad en tumores. Esto la convierte en un blanco ideal para la terapia contra el cáncer, pero como su regulación es muy compleja se han estudiado diferentes estrategias:
Nivel transcripcional: la clonación de las regiones promotoras de los genes que codifican para las subunidades TERT y TERC han permitido identificar reguladores positivos y negativos, conociendo estos se puede aumentar o inhibir su transcripción
Nivel postranscripcional: se está buscando bloquear el ARNm de las subunidades TERT y TERC mediante el uso de ribozimas con actividad ribonucleasa
Nivel post-traduccional se ha demostrado que para que la enzima funcione necesita el ensamblaje de todos los constituyentes, por lo que bloqueando alguna de las proteínas que forman parte del complejo se podría bloquear su acción
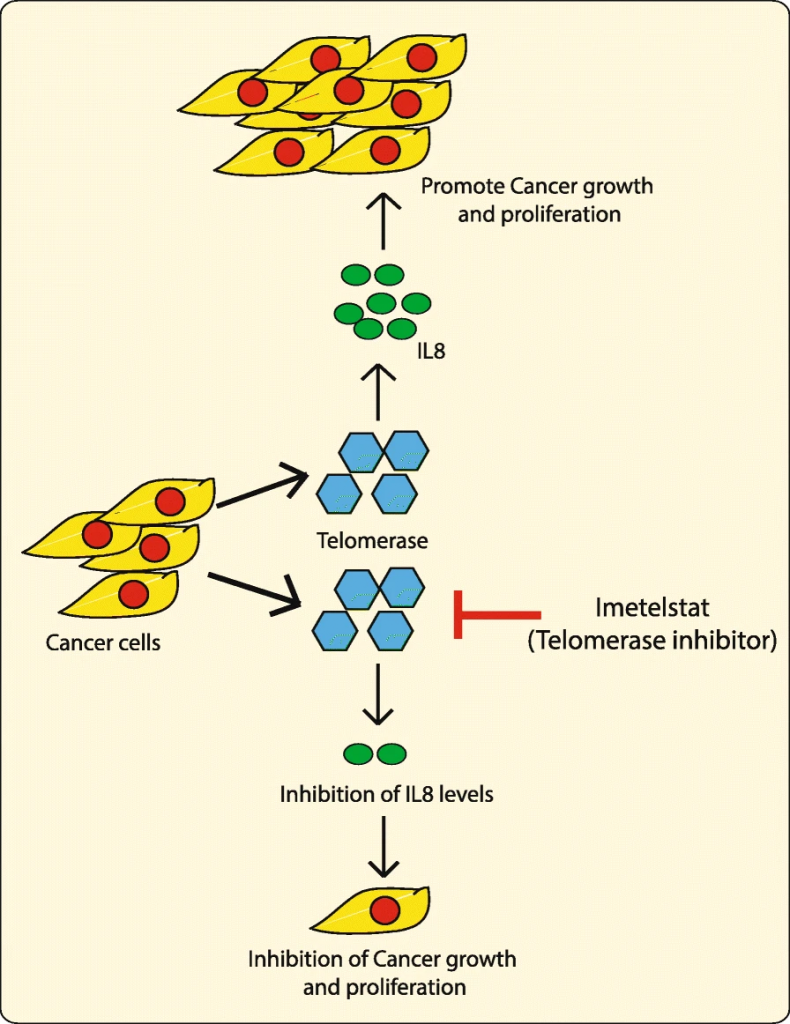
Imagen 6: representación de como la inhibición de la telomerasa detiene la proliferación del cáncer. Fuente: Interleukin 8 is a biomarker of telomerase inhibition in cancer cells
El lado bueno de la telomerasa: la inmortalidad
Como ya hemos mencionado las células germinales expresan la enzima telomerasa mientras que las somáticas no, por lo que estas últimas en cada división van acortando los telómeros entrando así en senescencia.
Por un lado la senescencia, sirve como mecanismo de supresión celular, ya que las células senescentes no son capaces de replicarse, por tanto, no se replicarán cromosomas anormales. Uso que se daría en un posible tratamiento contra células cancerosas.
Sin embargo, se ha propuesto que la reconstitución de la actividad de la telomerasa en distintos tejidos podría ser empleada como terapia para enfermedades asociadas al envejecimiento y que están caracterizadas por una disminución de la capacidad proliferativa y regeneración celular.
Se ha buscado usar la telomerasa como diana terapéutica en medicina regenerativa frente a enfermedades crónicas, por ejemplo, frente a enfermedades de la piel: estimulando células madre para que expresen el componente de ARN de hTERT de tal modo que se active la telomerasa reemplazando la piel perdida. También serviría para enfermedades cardiovasculares y neurodegenerativas asociadas al envejecimiento e incluso el propio envejecimiento.
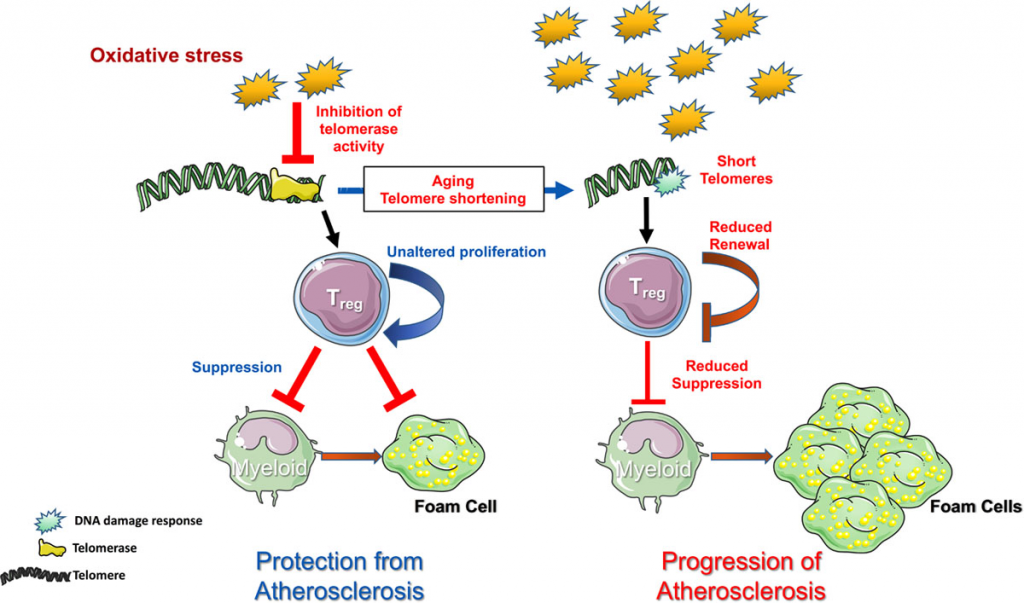
Imagen 7: Ejemplo del papel de la telomerasa en la prevención de enfermedad cardiovascular. Fuente: Telomerase as a Therapeutic Target in Cardiovascular Disease
La última estrategia, desarrollada por un grupo del CNIO en 2012, se basa en una terapia génica que activa el gen de la telomerasa durante unas pocas horas, por lo que la enzima puede ejercer su función reparadora un tiempo limitado, y así se disminuye los riesgos.
Podríamos pensar que una activación continua de la telomerasa implicaría que nuestras células no murieran, lo que se podría considerar conseguir la inmortalidad. Sin embargo, no sería tan fácil porque la actividad constante de esta enzima es muy probable que derive en un cáncer como hemos comentado.
Conclusiones
El acortamiento de los telómeros es el mecanismo fisiológico de nuestro cuerpo que explica el envejecimiento. La naturaleza ha sido capaz de evitar esa muerte celular, mediante la activación de la telomerasa en células somáticas que derivan en células cancerígenas. Sin embargo, lo que encontramos hoy en día es que la ciencia quiere aprovechar esa idea a nuestro favor. Si se consigue activar la telomerasa de una manera regulada, se podrá extender los años de vida; lo que sería un paso más cerca de esa idea ficticia que tenemos de la inmortalidad.
Bibliografía
Mengual Gómez, D. L., Armando, R. G., Farina, H. G., & Gómez, D. E. Telomerasa y telómero: su estructura y dinámica en salud y enfermedad. MEDICINA (Buenos Aires), 74(1), 69-76 (2014).
Cascales Angosto, M., Álvarez Gómez, J. A. Anales de la Real Academia de Doctores de España. Volumen 14, pp. 49-70 (2010).
Isnais Luna Rodríguez(1), Odania Mondeja Ortiz(2), Maritza Roque Tarife(3). Telomerasa. Enzima del futuro. Revista médica electrónica de ciego de Ávila, Vol.11, No. 1 (2005)
Arvelo, F., & Morales, A. Telómero, telomerasa y cáncer. Acta Científica Venezolana, 55, 288-303. (2004).
Greider, C. W., & Blackburn, E. H. Telómeros, telomerasa y cáncer. Investigación y Ciencia, 235, 20-26.(1996).
Figueroa, E. F., & Mayani, H. Cromosomas, control celular y cáncer: una cuestión de telomerasas. Revista Ciencia, julio – septiembre (2003)
Dias, J. Proliferación celular y regulación de la telomerasa en cáncer de mama (Doctoral dissertation, Universidad de Málaga). (2017).
Hernández Fernández, R. A. Telómeros y telomerasas. Revista Cubana de Investigaciones Biomédicas, 18(2), 121-129. (1999).
Saretzki G. Telomerase inhibition as cancer therapy. Cancer Lett;194(2):209-19 (2003)
Sarborit, A. & Muñiz, C. Telomerasa: Salud y Envejecimiento. Morfovirual (2020)
Virus oncolíticos como nueva terapia frente al cáncer
escrito por natpmv_3C | 28 enero, 2023
Realizado por Natalia López Escobar y Pablo Martín Valenzuela.
Biología molecular. 3º Biología Sanitaria. Grupo C.
1. Introducción
Actualmente, existen diversas terapias frente al cáncer, por un lado, las tradicionales, donde encontraríamos la quimioterapia, la cirugía y la radioterapia; y por el otro, las de nueva incorporación, donde nos encontraríamos la terapia dirigida, la inmunoterapia y la terapia hormonal láser entre otras.
Una de las líneas de investigación más recientes frente al cáncer es el uso de virus oncolíticos. Estos virus son modificados genéticamente para reconocer al cáncer e infectarlo. Gracias a esto, no sólo conseguimos que las células del tumor infectadas mueran, si no que permite, además, una activación del sistema inmune del hospedador. Al lisar a las células tumorales, se liberan antígenos que serán reconocidos por células del sistema inmune que activarán la respuesta inmune.
1.1. El cáncer
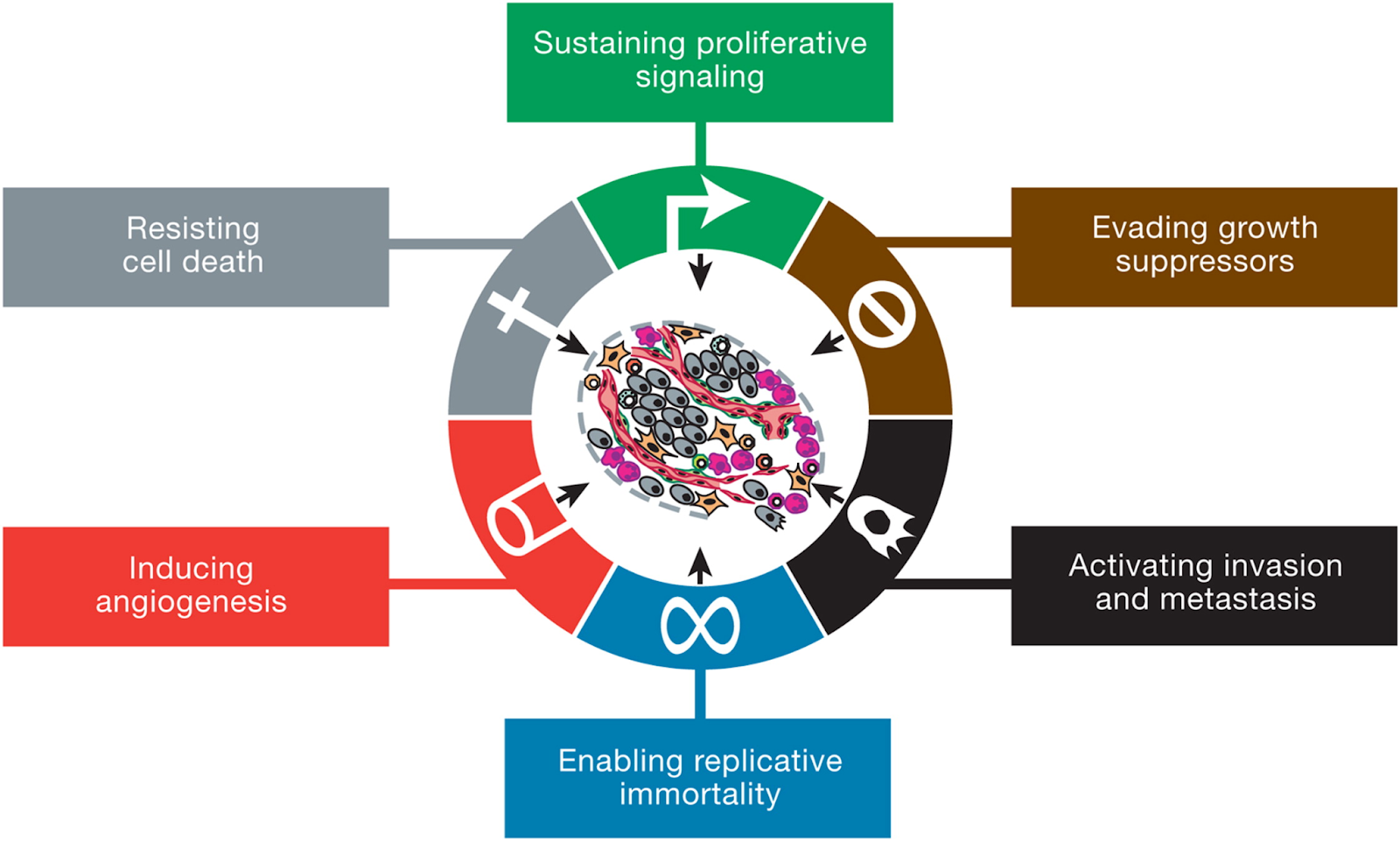
El cáncer es un conjunto de enfermedades que se presentan cuando las células se multiplican sin control y se diseminan a los tejidos que los rodean. Las características que deben cumplir las células para ser consideradas células tumorales fueron descritas en 2011 por Hanahah y Weinberg (1 y 2). Estas características son:
Autosuficiencia de señales de crecimiento, es decir, no necesitan señales externas para crecer.
Insensibilidad a señales antiproliferativas.
Evasión de la apoptosis.
Adaptación metabólica.
Inmortalización, mediante el alargamiento de los telomeros
Capacidad de invasión y angiogénesis, es decir, capacidad de crear nuevos vasos sanguíneos.
Capacidad de colonización de otros tejidos (metástasis).
Evasión de la respuesta inmune
Catherine Sánchez, 2013. Conociendo y comprendiendo la célula cancerosa: Fisiopatología del cáncer
1.2. Los virus oncolíticos
La idea de que los virus pueden ser utilizados contra el cáncer no es novedosa, proviene de mediados del siglo XX, cuando se observaron, en pacientes con linfomas y leucemias, remisiones del tumor, coincidentes con infecciones por virus, como el de la hepatitis o el del sarampión (3). Se empezó entonces a probar la infección de pacientes oncológicos con virus. No tuvo la eficacia esperada y además, se encontraron muchos efectos secundarios causados por los virus, de modo que se detuvo la investigación (4).
Ahora, gracias a los avances de la ingeniería genética, se han podido desarrollar virus oncolíticos más seguros y específicos frente a determinados tipos de tumores.
2. Mecanismos moleculares de acción
Los virus oncolíticos son capaces de infectar células anormales a través de dianas celulares específicas: Transcriptasa inversa de telomerasa humana, antígeno específico de próstata, ciclooxigenasa-20, her2/neu…
La Transcriptasa inversa de telomerasa humana o hTERT, es una subunidad catalítica de la enzima Telomerasa. La telomerasa es una polimerasa ribonucleoproteica, que mantiene los extremos de los telomeros. No puede ser considerado un protooncogén, ya que su mutación por sí sola no induce el crecimiento. Si que es importante su papel en la inmortalización de las células tumorales. La mutación en el promotor de hTERT confiere una mayor agresividad al melanoma (5)
HER2/neu: es un tipo de HER (Human EGF Receptor). Es un receptor con actividad Tyr quinasa, que tiene como ligando EGF (Epidermal Growth Factor). HER2 tiene un peculiaridad, ya que presenta la capacidad de activarse sin necesidad de ligando. Se ha visto su sobreexpresión hasta en el 30% de los cánceres de mama. (6 y 7)
Una vez hemos visto ejemplos de algunas dianas que pueden usar los virus para reconocer a las células tumorales, podemos ver los mecanismos que producen la muerte del tumor. La infección viral provoca, en primer lugar, la lisis de células tumorales. Las células dendríticas, reconocen antígenos virales y estimulan la producción de Interferon de tipo I, factor de necrosis tumoral alfa. (TNF-α) y citoquinas como la interleucina 2 (IL-2). El TNF-α regula la expresión del complejo de histocompatibilidad, e influye positivamente en la acción de la enzima caspasa y contribuye a la apoptosis celular en algunos tumores. Además, está molécula está relacionada con la activación de los linfocitos T citotóxicos y las células NK. Por lo tanto, conseguimos la muerte de las células tumorales mediante dos modos: por un lado, la lisis celular provocada por el ciclo de infección del virus. (8)
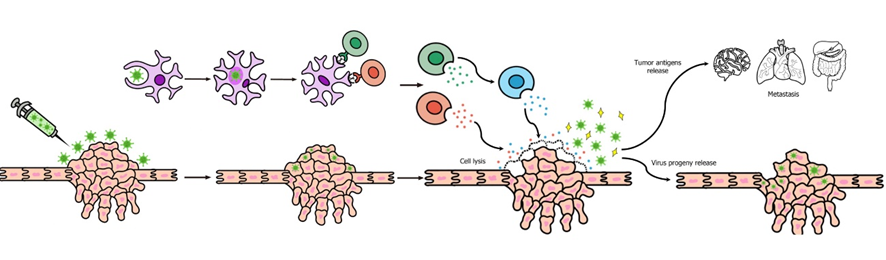
Santos Apolonio et al. Oncolytic virus therapy in cancer.
Una de las principales ventajas que supone el uso de virus oncolíticos es que podría inducir regresión en casos de metástasis (que representan la mayor parte de las muertes por cáncer) ya que, al provocar la lisis celular, salen nuevas partículas virales que pueden viajar hacia zonas lejanas donde haya metástasis. Pero el mecanismo más importante son las nuevas respuestas inflamatorias, que se producen cuando se lisan las células tumorales y salen antígenos al exterior. Estas nuevas respuestas inflamatorias, unidas a la memoria inmune celular, pueden provocar la regresión de las metástasis. (8)
Uno de los virus oncolíticos más prometedores es el CTV-m7, el cual incrementa la acción citotóxica sobre el tumor y es capaz de lisar células metastásicas. Se ha probado su uso en cánceres de próstata y ha demostrado efectividad (9).
Hay un único virus oncolítico aprobado por la FDA, es el T-VEC (Imlygic®), que es el virus del herpes simple (VHS), modificado para atacar a las células cancerígenas del melanoma.
3. Virus de la Enfermedad de Newcastle como nueva aproximación terapéutica para el glioblastoma
3.1. Introducción
Vamos a poner un ejemplo de un estudio que se realizó sobre el virus de la enfermedad de Newcastle, para ver si es adecuado para usarlo como virus oncolítico y como terapia para el glioblastoma.
3.1.1. Glioblastoma (GBM)
El glioblastoma es el tumor cerebral más común en el SNC, siendo muy agresivo debido a su invasividad y alta proliferación. Las personas que lo padecen tienen una esperanza de vida muy corta una vez que se diagnostica, a pesar de la mejora de los tratamientos y establecimiento de terapias.
Este tumor, compuesto por células madre de glioma (GSCs), presenta resistencia a diferentes tratamientos contra el cáncer, como la quimio o la radioterapia, ya que estas células son capaces de autorrenovarse y diferenciarse (10). Las GSCs se cree que también son las causantes de la recurrencia del glioblastoma.
Los rasgos más característicos de este cáncer son la proliferación microvascular y la necrosis, es decir, se agrupan en capas y las células presentan la zona central con necrosis (11).
3.1.2. Virus de la Enfermedad de Newcastle (NDV)
Es un virus aviar, con propiedades oncolíticas e inmunoestimuladoras, por lo que su estudio en viroterapia y ensayos clínicos cada vez es mayor.
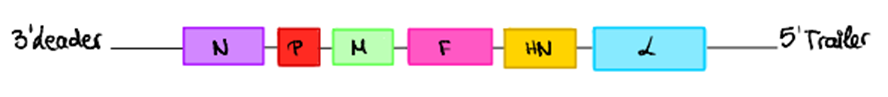
El genoma de este virus consiste en una molécula de ARN monocatenario, con polaridad negativa y formada por dos regiones en los extremos, leader en 3’ y tráiler en 5’, no codificantes; y seis genes que codifican 6 tipos de proteínas diferentes (12).
6 genes para 6 tipos de proteínas
Infecta células y se replica en ellas, destruyéndolas. Esto lo hace más rápido en las células cancerosas humanas, de ahí el interés en su estudio como tratamiento para el cáncer. Presenta dos cepas: las cepas líticas, que dañan la membrana de la célula; y las cepas no líticas que bloquea el metabolismo de la célula. Las cepas líticas son las que se estudian para el cáncer, ya que son capaces de eliminar directamente las células cancerosas; pero las dos cepas se usan en vacunas que ayudan al sistema inmune a combatir el cáncer (13).
Virus de la enfermedad de Newcastle
Los cultivos que se realizaron con GSCs y rNDV muestran como este virus afecta a la viabilidad de las células del tumor, induciendo apoptosis.
3.2. Glioblastoma: aspectos moleculares y patología
Las vías de señalización, moléculas y genes más comúnmente afectadas en el GBM, que hacen que sea resistentes a los tratamientos convencionales (14), son:
Receptores tirosina/quinasa (RTK): se encuentran en la membrana plasmática. Se autofosforilan en presencia de ligando para activarse. Se encargan de activar vías de transducción que continúan con vías de transcripción de genes que regulan el ciclo celular.
Vía de PI3K/AKT/mTOR: PI3K activa a AKT y este activa a mTOR, relacionado con la supervivencia y el ciclo celular.
Señalización de RAS/MAPK: genes transcritos por vías de traducción llevadas a cabo por segundos mensajeros (oncogenes o genes supresores de tumores), que participan en la proliferación celular. RAS es una GTPasa que actúa en la transducción de señal de RTK. Cuando se activan RTK, se activa RAS, que a su vez activa la vía de transducción de las MAPK. Las mutaciones en RAS la activan permanentemente, activando también permanentemente la vía de las MAPK. Esto induce una transcripción activa de genes relacionados con el ciclo celular.
P53 y retinoblastoma (RB): implicadas en regulación del ciclo celular. P53 es un gen supresor que se encarga de inducir apoptosis cuando el ADN está dado. Si p53 está mutado, se sigue con el ciclo celular y el daño en el ADN. También inhibe a mTOR, relacionado con el ciclo celular. El retinoblastoma está relacionado con la mutación de pRb, que hace que no se una a E2F y se siga con el ciclo celular.
Gen EGFR: es el gen del receptor del factor de crecimiento epitelial (GFR). Si está alterado, se hace independiente de EGF, por lo que se activa a muy bajas concentraciones de ligando.
3.3. NDV como agente oncolítico
En 1965 observó por 1ª vez que NDV presentaba un efecto antitumoral y baja neuroafinidad. Este potencial oncolítico que presenta el virus se debe a su propia capacidad de replicarse bastante mejor (unas 104 veces mejor) en las células tumorales que en las células normales, y además, sin afectar a las células sanas. Además, al ser un virus aviar, sus cepas virulentas provocan solo síntomas leves.
3.3.1. Mecanismo de oncólisis de NDV
NDV se asocia principalmente a la inducción de la apoptosis. La apoptosis es un tipo de muerte celular programada que ocurre en todos los tipos celulares. Además, también puede provocar necroptosis, que es un tipo de muerte celular que tiene características tanto de necrosis (por la morfología de las células) como de apoptosis (por lo de programada). También puede inducir la muerte celular por autofagia.
La infección por este virus induce la activación de la respuesta inmune, favoreciendo su efecto oncolítico. Las células tumorales infectadas presentan Ag virales, haciendo que las células de alrededor liberen citoquinas, que activan a macrófagos, NK y o monocitos, provocando la respuesta inmune innata; o haciendo que se activen las células presentadoras de Ag, que activan a los linfocitos T citotóxicos, que activarán la respuesta inmune adaptativa. Todo esto activa el estado de actividad inmunológica antitumoral causando la muerte celular inmunogénica de las células tumorales (15).
El genoma de este virus es muy fácil de modificar, por lo que la técnica de genética inversa es útil para obtener virus recombinantes, teniendo como objetivo aumentar su eficacia antitumoral.
Se han estudiado sus propiedades oncolíticas, dando en algunos casos reducción parcial y en otros total del tumor.
3.4. Resultados
La tesis concluye que el NDV induce cambios en la viabilidad de las GSCs, demostrando la capacidad oncolítica del virus en diferentes tipos de líneas celulares tumorales, incluidas las líneas tumorales de glioma. Además, interfiere en el crecimiento celular de las GSCs, y provoca la inducción de la apoptosis de las diferentes líneas celulares.
En cuanto a os xenotransplantes, también se observó que se reduce el tamaño de los tumores xenotransplantados en ratones Nude. Finalmente se demostró que en los ratones inmunodeprimidos, el virus causa 100% de mortalidad, siendo seguro solo para los ratones inmunocompetentes. Lo que puede suponer una importante limitación en el uso farmacológico del virus de la enfermedad de Newcastle.
4. Conclusión
Nos ha parecido un trabajo interesante y además hemos aprendido muchas cosas que no sabíamos y que nos han gustado mucho. Creemos que la investigación y el estudio de los virus como terapia para el cáncer es algo muy importante y que podría funcionar muy bien para solucionar el problema que provoca esta enfermedad. Si es verdad que aún queda mucho por avanzar, pero creemos firmemente, que de aquí a unos años esta nueva terapia será una opción más para combatir el cáncer.
5. Bibliografía
1. The Hallmarks of CancerHanahan D, Weinberg RCell (2000) 100(1) 57-70
3. Studies in Hodgkin’s syndrome; the association of viral hepatitis and Hodgkin’s disease; a preliminary report. HOSTER H, ZANES R, VON HAAM E. Cancer research (1949) 9(8) 473-80
4. Clinical studies of viruses as antineoplastic agents with particular reference to Egypt 101 virus. SOUTHAM C, MOORE A Cancer (1952) 5(5) 1025-34
5. Telomerase reverse transcriptase promoter mutations in primary cutaneous melanoma Heidenreich B, Nagore E, Rachakonda P, Garcia-Casado Z, Requena C, Traves V, Becker J, Soufir N, Hemminki K, Kumar R. Nature Communications (2014) 5(1) 3401
6. Homeostasis celular: crecimiento celular y cáncer – Bioquímica médica Michie, Alison M.; Paunovic, Verica; Harnett, Margaret M.; Bioquímica médica, Capítulo 28, 397-415
8. Santos Apolonio J, Lima de Souza Gonçalves V, Cordeiro Santos ML, Silva Luz M, Silva Souza JV, Rocha Pinheiro SL, de Souza WR, Sande Loureiro M, de Melo FF. Oncolytic virus therapy in cancer: A current review. World J Virol 2021; 10(5): 229-255
9. Therapy of prostate cancer using a novel cancer terminator virus and a small molecule BH-3 mimetic Sarkar S, Quinn B, Shen X, Dash R, Das S, Emdad L, Klibanov AWang X, Pellecchia M, Sarkar D, Fisher P Fisher POncotarget (2015) 6(13) 10712-10727
10. Piper K, DePledge L, Karsy M, Cobbs C. Glioma Stem Cells as Immunotherapeutic Targets: Advancements and Challenges.
11. Ohgaki and Kleihues, 2013; Perry and Wesseling, 2016; Urbanska et al., 2014
12. Marcos et al., 2005; Triosanti et al., 2018
13. Csatary LK, Eckhardt S, Bukosza I, Czegledi F, Fenyvesi C, Gergely P, Bodey B, Csatary CM. Attenuated veterinary virus vaccine for the treatment of cancer. Cancer Detect Prev. 1993;17(6):619-27.
13. Csatary LK, Moss RW, Beuth J, Töröcsik B, Szeberenyi J, Bakacs T. Beneficial treatment of patients with advanced cancer using a Newcastle disease virus vaccine (MTH-68/H). Anticancer Res. 1999 Jan-Feb;19(1B):635-8.
14. Crespo et al., 2015; Szopa et al., 2017
15. Matveeva et al., 2015; Zamarin and Palese, 2012ª
16. Virus de la Enfermedad de Newcastle como nueva aproximación terapéutica para el Glioblastoma, Rubio S (2018) 24-43
¡Muerte proteica! Descubrimos una quimera destructora de proteínas: Los Protacs
escrito por danielcelia_3b | 28 enero, 2023
Daniel Arenas González y Celia Arranz del Río.
Los PROTACs son moléculas diseñadas para unirse a proteínas diana, ubiquitinizarlas, y conducirlas al sistema de degradación proteosomal. Recientemente, se han utilizado para modular la actividad de la proteína Von Hippel-Lindau (VHL), una proteína capaz de unirse a factores de transcripción y regular la expresión génica.
PROTACs, un sistema de degradación de moléculas.
Los PROTACs (PROteolysis TArgeting Chimera) son moléculas diseñadas para desactivar proteínas específicas en el cuerpo. Es una molécula pequeña diseñada para unirse a una proteína objetivo específica y llevarla a un sistema de degradación proteica llamado sistema ubiquitina-proteasoma. Los PROTACs se componen de dos componentes: un ligando que se une a la proteína objetivo y un ubiquitina ligasa que cataliza la adición de ubiquitina a la proteína objetivo. [1], [2].
Figura1. Representación esquemática del funcionamiento de un PROTAC Imágenes obtenidas con BioRender. Daniel Arenas Gónzalez y Celia Arranz del Río.
Los PROTACs se pueden diseñar para unirse a proteínas específicas que se sabe que están implicadas en enfermedades concretas, y por lo tanto se están considerando como posibles tratamientos para dichas enfermedades. También se utilizan en investigación científica para entender el papel de diferentes proteínas en el cuerpo y cómo pueden ser moduladas para tratar enfermedades.[3]
Figura 2. Estructura de WDR5 humano y pVHL:ElonginC:ElonginB unido a PROTAC con conector PEG. Creado con UCSF Chimera por Daniel Arenas González y Celia Arranz del Río.
Los PROTACs se han utilizado en investigación para modular la actividad de la proteína VHL, de la que hablaremos a continuación. Por ejemplo, se han desarrollado PROTACs que pueden unirse a la proteína VHL y llevarla al sistema de eliminación de proteínas del cuerpo, lo que puede tener efectos terapéuticos en el cáncer causado por pVHL. Esto se logra mediante el mecanismo de acción de los PROTACs descrito anteriormente, es decir, mediante la unión de la proteína objetivo (en este caso, la proteína VHL) a una molécula de deglución, que luego lleva a la proteína objetivo al sistema de eliminación de proteínas del cuerpo.[3],[4]
Video 1. Estructura 360º del PROTAC formado por la pVHL. Creado con UCSF Chimera por Daniel Arenas González y Celia Arranz del Río.
Comparación entre el complejo HIF-1a-pVHL-Elongina B-Elongina C (PROTAC) y el complejo SCF.
Por un lado, WDR5 es una proteína que puede ser utilizada como componente de un PROTAC para unirse a una proteína objetivo y marcarla para su degradación por el sistema de ubiquitinación.
WDR5 tiene un dominio beta propeller. Es frecuente encontrar el dominio beta propeller en proteínas que se utilizan como componentes de PROTACs.
Por otro lado, SCF (complejo de ubiquitina ligasa E3) es un complejo proteico que se encarga de marcar las proteínas para su degradación por el sistema de ubiquitinación. El complejo SCF está compuesto por cuatro proteínas: una proteína E3 ligasa, una proteína adaptadora que se une a la proteína objetivo, una proteína F-box que se une a la proteína adaptadora y una proteína que se une a ubiquitina.[5],[6]
Figura 3. Comparación estructural entre los dominios beta-propeller de F-box y WDR5. Creado con UCSF Chimera por Daniel Arenas González y Celia Arranz del Río.
La proteína Von Hippel-Lindau (VHL).
La proteína VHL (Von Hippel-Lindau) es una proteína codificada por el gen VHL y se encuentra en el núcleo de las células.
Se puede unir a factores de transcripción para regular la expresión génica y a proteínas que tienen un papel en la regulación del ciclo celular y en la reparación del ADN, como la proteína p53. Esto puede ser importante para evitar el crecimiento anormal de las células, que puede llevar a enfermedades como el cáncer. [6]
La proteína VHL es un componente del complejo proteico VCB, que también incluye elongina B, elongina C y cullin-2. Tiene actividad de ubiquitina ligasa E3 y dirige la degradación dependiente del proteasoma de sus proteínas objetivo.
La proteína VHL interactúa con CUL2, y esta interacción depende de la integridad del complejo VBC trimérico.
La proteína VHL interactúa con ADRB2. En condiciones normales de oxígeno esta interacción depende de la hidroxilación de ADRB2 y la subsiguiente ubiquitinación y degradación mediadas por VCB. Sin embargo, bajo hipoxia, la hidroxilación, interacción con VHL, la ubiquitinación y la posterior degradación de ADRB2 disminuyen significativamente.
Además, VHL interactúa con RNF139, USP33 y PHF17, y se encuentra en un complejo compuesto por LIMD1, VHL, EGLN1/PHD2, TCEB2 y CUL2. [7]
Vía VHL-HIF.
La pVHL también interactúa, a través de su dominio beta, con el factor inducible por la hipoxia (HIF1A). Su interacción está regulada mediante la hidroxilación de un residuo de prolina por la enzima HIF-1alfa prolil hidroxilasa dependiente de oxígeno. [8]
HIF es una proteína de expresión constitutiva y ubicua en los tejidos. La regulación de la actividad de este factor de transcripción depende del oxígeno y ocurre a varios niveles de su fisiología molecular (hidroxilación, proteólisis, transporte nuclear, etc.).
En normoxia (niveles de oxígeno normales). En condiciones de normoxia, las subunidades HIF-alfa se unen rápidamente a la prolilhidroxilasa, que las ubiquitiniza, haciendo posible que la enzima E3-ligasa o VHL las reconozca y activando así la proteólisis de HIF mediante la vía del proteosoma. .
En hipoxia o anoxia (niveles de oxígeno mínimos o ausencia de oxígeno). En condiciones hipóxicas, la enzima prolil hidroxilasa no ubiquitiniza al factor HIF-1alfa, impidiendo su reconocimiento por parte de la enzima VHL (E3-ligasa) y, por tanto, su proteólisis. El factor HIF comienza a acumularse en el citoplasma y se transloca al núcleo, donde se transcribirá y originará un ARNm que puede ser traducido. [9],[10].
Figura 4. Vía HIF-VHL en situaciones de normoxia e hipoxia. Imagen creada con Biorender. Daniel Arenas González y Celia Arranz del Río.
Mutación VHL Y98N
Figura 5. Representación del la interacción entre un análogo de HIF-1A con la tirosina del residuo 98 de la VHLp. Creado con UCSF Chimera por Daniel Arenas González y Celia Arranz del Río.
La proteína VHL es una proteína clave en el ciclo celular y en la regulación de la respuesta del cuerpo a los cambios en el oxígeno. En la posición 98 de la proteína VHL hay una tirosina, que es un aminoácido importante en su estructura y función. Sin embargo, cuando hay una mutación que cambia la tirosina por asparragina en esa posición. La capacidad de la proteína VHL para unirse al factor de transcripción HIF1A puede verse afectada.
El HIF1A es una proteína que se une a ADN y regula la expresión de ciertos genes. Cuando hay una falta de oxígeno en las células, el HIF1A se activa y ayuda a las células a adaptarse al cambio. La proteína VHL normalmente ayuda a regular la actividad del HIF1A, impidiendo que se una a ADN cuando hay suficiente oxígeno. Sin embargo, si hay una mutación en la proteína VHL que impide que se una al HIF1A, el HIF1A puede seguir activo incluso cuando hay suficiente oxígeno, lo que puede llevar a cambios en la expresión génica y potencialmente a problemas de salud. En resumen, la mutación que cambia la tirosina por asparragina en la posición 98 de la proteína VHL puede afectar la capacidad de la proteína VHL para regular la actividad del HIF1A, lo que puede tener consecuencias para la salud. [11]
La importancia de las mutaciones.
Una mutación que cambia una arginina por una asparragina en una proteína puede tener diversos efectos en la estructura y función de la proteína.
La arginina es un aminoácido con un grupo amino positivamente cargado y un grupo carboxilo negativamente cargado, lo que le da una cierta reactividad química y le permite participar en interacciones con otros aminoácidos y moléculas.
La asparragina, por otro lado, es un aminoácido con un grupo amino negativamente cargado y un grupo carboxilo positivamente cargado, lo que le da una carga eléctrica y una reactividad química diferentes.
En general, una mutación que cambia una arginina por una asparragina puede afectar la estructura tridimensional de la proteína y, por lo tanto, su función. Por ejemplo, la arginina puede participar en interacciones de puentes salinos y en la formación de estructuras secundarias como hélices alfa, mientras que la asparragina puede tener dificultades para hacerlo. También puede haber cambios en la afinidad de la proteína por otras moléculas o en su estabilidad estructural debido a la mutación.
En resumen, la mutación de arginina a asparragina puede afectar la estructura y función de la proteína de diversas maneras y puede tener efectos en el organismo que dependen de la proteína afectada y de su papel en el cuerpo.[12]
iASPP, el oncogen que se une a VHL y evita la degradación de HIF-1α.
iASPP es un oncogen inhibidor de la proteína simuladora de apoptosis de p53. iASPP y HIF-1α se unen a la misma región de VHL.
La unión de VHL con iASPP evita que se una con HIF-1α, lo que aumenta la estabilidad y señalización de la proteína HIF-1α en las células cancerosas. Se ven promovidas la angiogénesis, la reprogramación metabólica y la glucólisis).
Se ha comprobado que iASPP contribuye a la activación constitutiva de HIF-1α en cáncer, y, por tanto, la modulación de la expresión de iASPP o su interacción con VHL es una estrategia terapéutica potencial con la que modular las actividades de la vía HIF. [13],[14]
La enfermedad de Von Hippel-Lindau (VHL), un trastorno genético.
El gen VHL, ubicado en el cromosoma 3, codifica para la proteína de Von Hippel-Lindau. Es un gen supresor de tumores y su mutación conduce al desarrollo de cáncer.
La enfermedad de Von Hippel-Lindau fue descrita por primera vez y por separado por Von Hippel en 1911 y por Lindau en 1926. Su incidencia se estima aproximadamente en 1/36000 nacido vivos. [8],[15]
Es un trastorno genético hereditario autosómico dominante caracterizada por la aparición a una edad temprana de tumores muy vascularizados. Se pueden clasificar en lesiones del SNC (hemangioblastomas del SNC y retinianos) y en lesiones viscerales (carcinomas renales, feocromocitomas y tumores neuroendocrinos pancreáticos).
La mayoría de los síndromes de VHL se acaban sometiendo a operación quirúrgica. En el caso de los feocromocitomas es conveniente usar previamente a la cirugía un bloqueador adrenérgico preoperatorio (el más común es la fenoxibenazamida). [9][10],[16]
Bibliografía.
Autores. Título. Revista. Vol. página inicial-página final (año).
Smith, B. E., Wang, S. L., Jaime-Figueroa, S., Harbin, A., Wang, J., Hamman, B. D., & Crews, C. M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nature Communications, 10(1), (2019). https://doi.org/10.1038/s41467-018-08027-7
An S, Fu L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine (553-563), 36, (2018).
Liu, J., Ma, J., Liu, Y., Xia, J., Li, Y., Wang, Z. P., & Wei, W. PROTACs: A novel strategy for cancer therapy. Seminars in Cancer Biology, 67, 171–179, (2020). https://doi.org/10.1016/J.SEMCANCER.2020.02.006
Kraemer, A., Doelle, A., Knapp, S., Structural Genomics Consortium. Structure of human WDR5 and pVHL:ElonginC:ElonginB bound to PROTAC with PEG linker (conformation #2), (2022) doi: 10.2210/pdb8BB2/pdb
Iwai, K., Yamanaka, K., Kamura, T., Minato, N., Conaway, R. C., Conaway, J. W., Klausner, R. D., & Pause, A. Identification of the von Hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc. Natl. Acad. Sci. U.S.A., 96(22), 12436–12441, (1999). https://doi.org/10.1073/pnas.96.22.12436
Yalla, K., Elliott, C., Day, J. P., Findlay, J., Barratt, S., Hughes, Z. A., Wilson, L., Whiteley, E., Popiolek, M., Li, Y., Dunlop, J., Killick, R., Adams, D. R., Brandon, N. J., Houslay, M. D., Hao, B., & Baillie, G. S. FBXW7 regulates DISC1 stability via the ubiquitin-proteosome system. Molecular Psychiatry, 23(5), 1278–1286, (2018). https://doi.org/10.1038/MP.2017.138
Foxler, D. E., Bridge, K. S., James, V., Webb, T. M., Mee, M., Wong, S. C. K., Feng, Y., Constantin-Teodosiu, D., Petursdottir, T. E., Bjornsson, J., Ingvarsson, S., Ratcliffe, P. J., Longmore, G. D., & Sharp, T. v. The LIMD1 protein bridges an association between the prolyl hydroxylases and VHL to repress HIF-1 activity. Nature Cell Biology, 14(2), 201–208, (2012). https://doi.org/10.1038/ncb2424
Chittiboina P, Lonser R. Von Hippel-Lindau disease. Handbook of Clinical Neurology, 139-139, 132, (2015).
Ben-Skowronek I, Kozaczyk S. Von hippel-lindau syndrome. Hormone Research in Paediatrics, 145-152, 84 (3), (2015).
Julio Pérez Márquez. Patología Celular. Factores nutricionales. Hipoxia y anoxia. Sensores celulares de oxígeno, (2022).
Knauth, K., Bex, C., Jemth, P., & Buchberger, A. Renal cell carcinoma risk in type 2 von Hippel-Lindau disease correlates with defects in pVHL stability and HIF-1alpha interactions. Oncogene, 25(3), 370–377, (2006). https://doi.org/10.1038/sj.onc.1209062
Nelson, D. L., Cuchillo Foix, C. M., Lehninger, A. L., & Cox, M. M. Lehninger: Principios de Bioquímica (4a. ed.). Barcelona: Omega, (2005).
Dong Zhao , Shanliang Zheng , Xingwen Wang, Hao Liu, Kunming Zhao, Li Li and Ying Hu. iASPP is essential for HIF-1α stabilization to promote angiogenesis and glycolysis via attenuating VHL-mediated protein degradation. Springer Nature Limited, (2022).
Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000;19:4298–309.
de Heer EC, Jalving M, Harris AL. HIFs, angiogenesis, and metabolism: elusive enemies in breast cancer. J Clin Investig. 130:5074–87, (2020).
Elias, R., Zhang, Q., & Brugarolas, J. The von Hippel-Lindau Tumor Suppressor Gene: Implications and Therapeutic Opportunities. Cancer Journal (Sudbury, Mass.), 26(5), 390–398, (2020). https://doi.org/10.1097/PPO.0000000000000480
DESCUBREN QUE EL FÁRMACO PARA DEJAR DE FUMAR ES CANCERÍGENO. Pero, ¿es más cancerígeno que el propio tabaco?
escrito por aitimarta_3C | 28 enero, 2023
Tabaquismo, Vareniclina y el fármaco definitivo para dejar de fumar.
Por Aitana López López y Marta de la Hoz Marchado. Grado en Biología Sanitaria, Universidad de Alcalá.
La vareniclina, comercialmente conocida como Champix®, es un potente fármaco para dejar de fumar (1). Sin embargo, en julio de 2021, la FDA solicitó su retirada voluntaria del mercado tras detectar la contaminación del fármaco con sustancias cancerígenas llamadas nitrosaminas (2) (Ver tabla 1). En concreto se detectaron impurezas de N-nitroso-vareniclina, sustancia con potencial mutagénico y cancerígeno (3). La farmacéutica Pfizer fue retirando progresivamente el fármaco. No obstante, el tratamiento solo dura 6 meses mientras que los efectos negativos se asocian al uso del fármaco a largo plazo (2,4). La FDA insistía en que los beneficios de dejar de fumar superaban el riesgo de cáncer por tomar vareniclina mientras la ingesta no superase los 37 ng de nitrosamina/día (2). Con lo cual comenzaba el siguiente debate: ¿Causa más cáncer fumar o el fármaco para dejar de fumar? En este blog enfrentaremos los riesgos del tabaco frente a los de la vareniclina y presentaremos un fármaco que podría ser más prometedor que el Champix para tratar a aquellos que quieren dejar de fumar, la citisina.
Tabla 1. Nitrosaminas cancerígenas. Tabla obtenida y adaptada de: Riesgos estimados de cáncer asociados con la contaminación por nitrosamina en medicamentos de uso común (2021). (3)
Sustancia
Fórmula
Carcinogenicidad
Reconocidas por
Casos
NDMA
N[1]nitrosodimethylamine
Sustancia carcinógena
IARC, US EPA, NT y California´s Proposition
40-126 casos adicionales de cancer por cada 100.000 individuos expuestos
NDEA
N-nitrosodiethylamine
Sustancia carcinógena
IARC, US EPA, NT y California´s Proposition
12-48 casos adicionales de cancer por cada 100.000 individuos expuestos
NMBA
N[1]nitroso-N-methyl-4-aminobutyric acid
Sustancia que induce cáncer de vejiga y de riñón
N-nitroso-vareniclina
N-nitroso-vareniclina
Faltan datos concluyentes de que sea una sustancia carcinógena aunque tiene potencial mutagénico y cancerígeno
1. TABACO
Fumar tabaco es un hábito extendido en todo el mundo. Según el INE el 16,4% de las mujeres y el 23,3% de los hombres fuman a diario (5). Las cifras siguen en aumento pese a que cada vez se sabe más sobre sus efectos negativos. Para estudiar por qué causa tanta adicción y a la vez tanto daño iremos desgranando sus componentes.
Figura 1. Consumo de tabaco. Imagen obtenida de: Encuesta Nacional de Salud (2020). MSCBS-INE. (5)
1.1 NICOTINA
La nicotina es el alcaloide primario en los productos del tabaco y capaz de atravesar fácilmente las membranas biológicas y la barrera hematoencefálica. Es un agonista exógeno del receptor colinérgico nicotínico (6,7). Son canales iónicos dependientes de ligando. Aquellos que se encuentran en el cerebro y que están formados por 2 subunidades α4 y 3 subunidades β2 presentan mayor afinidad por la nicotina (6,8). La nicotina ejerce un efecto estimulante en el locus ceruleus y un efecto de recompensa en el sistema límbico (7). La respuesta ocurre en milisegundos provocando la liberación de acetilcolina, norepinefrina, serotonina y dopamina.
Figura 2. Molécula de nicotina. Imagen obtenida de PubChem (16).
Figura 3. Receptor colinérgico nicotínico. A la izquierda observamos la molécula al completo. Hacia la derecha vemos un aumento de la zona de unión del receptor a la nicotina. En la imagen de la izquierda del todo vemos las molécula de nicotina (amarilla) interaccionado con los aminoácidos de las cadenas proteicas del receptor por la unión de 16 puentes de hidrógeno (líneas discontinuas azules). Fuente: Imagen obtenida con software UCSF Chimera gracias a César Menor Salván y adaptada con Biorender.
1.2 ADICCIÓN Y TOLERANCIA
En el cerebro la nicotina actúa sobre las neuronas dopaminérgicas en las vías cortico-límbicas estimulando la liberación de dopamina. Esta activa el sistema dopaminérgico, en concreto las neuronas dopaminérgicas del núcleo accumbes, centro de la recompensa. Genera una sensación de euforia y placer. Por ello el fumador es reforzado a repetir el comportamiento que ha motivado la activación dopaminérgica, es decir, fumar (8). Debido al efecto ansiolítico de la nicotina (9) se ha demostrado que si el consumo de nicotina cesa provoca un cambio negativo en el comportamiento (10).
En los fumadores la exposición a nicotina es prologada. Además, la nicotina permanece más tiempo en la hendidura sináptica que la acetilcolina (agonista endógeno). Por ello, se produce una desensibilización de los receptores que conlleva a la tolerancia a la nicotina. Se requiere consumir cada vez más cantidad de nicotina para obtener los mismos efectos. (8)
Figura 4. Liberación de dopamina en fumadores. (18)
1.3 TABACO Y CÁNCER
Se ha demostrado que la nicotina es capaz de inhibir la apoptosis celular, inducir estrés oxidativo en las células y estimular la proliferación celular. Por ello, podría estimular procesos de neoplasia aumentado el riesgo de padecer cáncer (6,11). Sin embargo, son el resto de las sustancias contenidas en el tabaco las que le hacen un potente cancerígeno (12).
Un cigarrillo contiene 250 sustancias nocivas, de las cuales, 50 son cancerígenas. Estos agente químicos carcinógenos están incluidos en el “Grupo I de carcinógenos humanos” por la International Agency for Research on Cancer (IARC) y son : Benceno, cadmio, arsénico, níquel, cromo, 2-naftil-amino, clorovinil, 4 aminobifenil y Berilio. Pero las sustancias cancerígenas del tabaco más potentes son los hidrocarburos aromáticos policíclicos y las nitrosaminas. (12,13,15)
El tabaquismo causa un tercio de las muertes por cáncer (tracto aerodigestivo, esófago, estomago, hígado, riñón, cervix, colorrectal) en los países occidentales. Se estima que el tabaco provoca el 25% de los cánceres en hombres y el 4% en mujeres. Además, es causa del 16% de los canceres en países desarrollados y del 10% en países subdesarrollados. Sin embargo, fumar causa más muertes por patologías cardiovasculares y respiratorias que por cáncer. Por ello, el tabaquismo se considera una epidemia. (14)
Figura 5. Mortalidad atribuible al consumo de tabaco en España en 2016. (19)
2. VARENICLINA
Como hemos mencionado arriba, la vareniclina, comercializada como Champix®, es una droga que contiene tartrato de vareniclina. Es un agonista parcial de los receptores nicotínicos (20).
Los receptores nicotínicos pasan por tres fases:
Reposo: canal iónico cerrado por ausencia de ligando.
Activo: la unión del agonista produce la apertura del canal y la correspondiente despolarización.
Desensibilizado: periodo refractario en el que el canal permanece cerrado aunque haya ligando.
Figura 6. Estados del receptor colinérgico de nicotina. A la izquierda el receptor en reposo presenta el canal cerrado por la cremallera de leucina con los sitios de unión de antagonista vacíos. En el medio vemos como entra la acetilcolina (ligando endógeno) a sus sitios de unión provocando un cambio conformacional que abre el canal permitiendo el paso de cationes que despolarizarán la célula y con ello se estimula la secreción de vesículas con neurotransmisores. A la derecha el canal se encuentra en estado de reposo, con el canal cerrado y la acetilcolina aún unida. Tras la salida de la acetilcolina el receptor vuelve a estado de reposo preparado para recibir nuevas moléculas agonistas. Fuente: Elaboración propia, Created with BioRender.com.
No obstante, cuando hay bajas concentraciones de nicotina, se puede producir el paso directo de estado de reposo a desensibilizado, mientras que, altas dosis de la droga inducen el paso de desensibilizado a activado.
La vareniclina inhibe competitivamente la unión de nicotina a los receptores. Esta, al unirse, activa los receptores nicotínicos y produce liberación de dopamina, por lo que se produce una sensación placentera similar a la generada por la nicotina, pero mantiene los receptores en estado desensibilizado por más tiempo cuando hay una sobreexposición a la droga. De este modo, se logra reducir la urgencia por fumar, los síntomas de la abstinencia y disminuye la recompensa ante la nicotina porque la vareniclina está bloqueando a los receptores. Además, no altera la farmacodinámica de otros fármacos al no ser metabolizado por el citocromo p450.
Figura 7. En en panel A se muestra el modo de acción de la nicotina sobre el receptor colinérgico nicotínico de la neurona causando la liberación de dopamina. En el panel B observamos el modo de acción de la vareniclina sobre el receptor colinérgico nicotínico, en este caso se observa una liberación de dopamina menor que en el caso A. En el panel C se muestra cómo sería la activación de dicho receptor en presencia de ambas sustancias siendo la nicotina retirada por la vareniclina ya que esta última tiene más afinidad por el receptor. Fuente: Elaboración propia con Biorender, Created with BioRender.com
Mediante ensayos in vitro en ratas se descubrió que la vareniclina tiene alta afinidad por los receptores α4β2, pero no por otros receptores nicotínicos. Además mediante ensayos clínicos se verificó su gran efecto contra la abstinencia puntual, comparado con otros fármacos como el bupropión, pero no se observaron resultados homogéneos en cuanto a disminuir la urgencia puntual. Por este motivo, Pfizer comercializó la vareniclina como el principal fármaco para dejar de fumar porque, a pesar de poseer efectos secundarios, como náuseas, no eran suficientes para condicionar su retirada.
Figura 8. Abstinencia puntual de 7 días confirmado por COesp. Jorenby y Gonzalez en ambos estudios la diferencia de abstinencia puntual fue significativa (p<0,001) contra placebo y en las semanas 12 y 24 contra bupropion (p<0,001 y <0,01). Solo en el segundo reporte la diferencia fu sisgnificativa en la semana 52 contra bupropion (p=0,05). (26)
2.2 LA CARA OSCURA DE LA VARENICLINA.
Recientemente se ha descubierto que este fármaco posee nitrosaminas (21), en concreto, N-nitroso-vareniclina. Las nitrosaminas son compuestos orgánicos cancerígenos que se forman mediante reacciones químicas entre nitritos con aminas. Son los metabolitos de las nitrosaminas los que interaccionan con el ADN y producen los efectos tóxicos. Las nitrosaminas no solo se encuentran en fármacos sino que también las podemos encontrar en productos de látex, caucho, maquillaje o en los conservantes usados para conservar alimentos cárnicos. Como no podía ser de otra forma, también se encuentran en la nicotina, esta, en concreto origina tanto el carcinógeno más potente, NNK, como el más débil, NNN. Estos compuestos se han visto implicados en el desarrollo de distintos cánceres, como el de estómago o faringe, por lo que se ha establecido una cantidad diaria máxima para evitar estos efectos nocivos. Champix® contiene una dosis más alta de lo permitido diariamente. Por ello se decidió retirarlo del mercado.
La cuestión es,¿qué resulta más cancerígeno, el tabaco per se o el fármaco para dejar de fumar? ¿Merece la pena asumir los riesgos de tomar vareniclina con tal de poner fin a la adicción por la nicotina? Bien pues, en primer lugar debemos comparar la toxicidad de las nitrosaminas producidas por la nicotina con las N-nitroso-vareniclinas del Champix®.Las nitrosaminas de la vareniclina se incluyen según la IARC dentro del grupo II (“Probablemente cancerígeno para los seres humanos”). Mientras que todos los componentes cancerígenos del tabaco se encuentran dentro del grupo 1 (“Cancerígeno para los seres humanos”) (Ver tabla 2). Podemos concluir que el tabaco es más cancerígeno, que la vareniclina en sí (15). Así mismo, la gran efectividad de la vareniclina en lograr el cese del tabaquismo en tiempos desde tres meses a un año (22) marca la diferencia, ya que ningún otro fármaco anterior había logrado tasas del 80% de éxito con efectos secundarios tolerables. Además, se han conseguido resultados similares disminuyendo la dosis del fármaco. En conclusión, el Champix® resulta tan eficaz en la erradicación del tabaquismo, incluso previniendo la recaída (20) (primer fármaco que logra esto), que realmente merece la pena asumir los riesgos de ingerir las nitrosaminas que contiene la vareniclina con tal de dejar de introducir todos los carcinógenos presentes en el tabaco.
Tabla 2. Comparación agentes carcinógenos del tabaco y del fármacos según la IARC. Tabla adaptada de Lista de Clasificaciones de sutancias cancerígenas de la IARC (13, 14)
Dónde se encuentra
Agente carcinógeno
Grupo IARC
Tabaco
4-aminobifenil
I
Tabaco
Arsénico
I
Tabaco
Benceno
I
Tabaco
Berilio
I
Tabaco
Cadmio
I
Tabaco
Cromo y derivados
I
Tabaco
2-naftilamina
I
Tabaco
Níquel
I
Tabaco
Cloruro de vinilo
I
Fármaco
N-nitrosodimetilamina
2A
Fármaco
N-nitrosodietilamina
2A
Fármaco
Ácido N-nitroso-N-metil-4-aminobutírico
No evaluado
3. CITISINA
Actualmente hay en vigor un nuevo fármaco para dejar de fumar, la citisina (23), comercializada como Tabex® o Todacitan®. Es un alcaloide natural derivado de la familia de plantas Fabaceae, en la que se incluyen los géneros Laburnum y Cytisus. Es un tratamiento contra el tabaquismo no solo más efectivo que otros sino también más económico. La citisina, también conocida como sophorina o baptitoxina, actúa, al igual que la vareniclina, es un agonista parcial de los receptores nicotínicos, especialmente el α4β2. Este fármaco, de igual manera que el Champix®, previene la completa activación del sistema nicotínico-dependiente de la liberación de dopamina en el sistema mesolímbico (disminuye el placer generado al fumar) y reduce los principales síntomas de la abstinencia tanto centrales, al aumentar ligeramente la liberación de dopamina en el cerebro, como periféricos, estimulando la liberación de catecolaminas por la glándula suprarrenal, evitando recaídas.
En definitiva, centrándonos en su modo de acción, la citisina parece ser una sustituta ejemplar de la vareniclina, pero, ¿resulta igual de eficaz? Bien pues, no solo existen estudios que corroboran que esta droga proporciona resultados competentes respecto a la vareniclina sino que, además, resulta extremadamente más productiva al presentar un coste de producción significativamente menor (24). Por lo tanto, podemos concluir que, en efecto, la citisina se lleva el podio de los fármacos contra el tabaquismo.
Figura 9. Tabex® (citisina). Fármaco para dejar de fumar (27)
4. CONCLUSIÓN
Resulta evidente que debemos poner fin al problema del tabaquismo en nuestra sociedad, en vista de la gran cantidad de eventos nocivos que tiene para nuestro organismo al contener potentes carcinógenos. Por ello, en un principio, merecía la pena asumir el riesgo que presentan las N-nitroso-vareniclinas cancerígenas que presenta el Champix®, el fármaco más eficaz que existía hasta la fecha para dejar de fumar. No obstante, con la aparición de la citisina, una droga con un modo de acción idéntico a la vareniclina, que no solo presenta la misma eficacia, sino que también es más barata, el Champix® queda totalmente excluido como tratamiento de elección a la hora de abandonar este hábito nocivo tan socialmente aceptado como es el tabaquismo.
Figura 10. Pódium de los fármacos para dejar de fumar. Elaboración propia.
BIBLIOGRAFÍA
Fouz Rosón, N. (2016). Impacto del tratamiento con vareniclina a bajas dosis vs estándar, ambas en pautas cortas, en la tasa de abstinencia tabáquica, adherencia al tratamiento y efectos adversos. (Tesis doctoral inédita). Universidad de Sevilla, Sevilla.
US FDA. FDA Alerts Health Care Professionals and Patients to a Voluntary Recall of Varenicline (Chantix) to the Warehouse Level. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-alerts-health-care-professionals-and-patients[1]voluntary-recall-varenicline-chantix-warehouse?utm_medium=email&utm_source=govdelivery (accessed on 2 July 2021).
Li, K., Ricker, K., Tsai, F. C., Hsieh, C. J., Osborne, G., Sun, M., … & Sandy, M. S. (2021). Riesgos estimados de cáncer asociados con la contaminación por nitrosamina en medicamentos de uso común. Revista internacional de investigación ambiental y salud pública, 18(18), 9465.
Instituto Nacional de Estadística. (2020). Determinantes de Salud (Consumo de Tabaco). INE.es. https://www.ine.es/ss/Satellite?L=es_ES&c=INESeccion_C&cid=1259926698156&p=1254735110672&pagename=ProductosYServicios%2FPYSLayout
Yildiz, D. (2004). La nicotina, su metabolismo y una visión general de sus efectos biológicos. Toxicon, 43(6), 619-632.2.
Treviño, L. J., Fernández, M. T. B., GONZÁLEZ, M. P. G., Martínez, P. A. S., & BOUSOÑO, M. (2004). La nicotina como droga. Monografía Tabaco, 16(suplemento 2), 143.
Salas, R., Pieri, F., Fung, B., Dani, J. A., & De Biasi, M. (2003). Respuestas alteradas relacionadas con la ansiedad en ratones mutantes que carecen de la subunidad β4 del receptor nicotínico. Revista de Neurociencia, 23(15), 6255-6263.
al’Absi, M., Amunrud, T., & Wittmers, L. E. (2002). Efectos psicofisiológicos de la abstinencia de nicotina y los desafíos conductuales en fumadores habituales. Farmacología Bioquímica y Comportamiento, 72(3), 707-716.
Sasco, A. J., Secretan, M. B., & Straif, K. (2004). Tabaquismo y cáncer: una breve revisión de la evidencia epidemiológica reciente. Cáncer de pulmón, 45, S3-S9.
Ruiz, A. M., Gómez, I. R., Rubio, C., Revert, C., & Hardisson, A. (2004). Efectos tóxicos del tabaco. Revista de toxicología, 21(2-3), 64-71.
Smith CJ, Livingston SD, Doolittle DJ. Una encuesta de literatura internacional de «carcinógenos del Grupo I de la IARC» reportados en el humo del cigarrillo convencional. Alimentos Químicos Toxicol. 1997 Oct-Nov;35(10-11):1107-30. doi: 10.1016/s0278-6915(97)00063-x. PMID: 9463546.
Sasco, A. J., Secretan, M. B., & Straif, K. (2004). Tabaquismo y cáncer: una breve revisión de la evidencia epidemiológica reciente. Cáncer de pulmón, 45, S3-S9.
PubChem. (s/f). Nicotine. Nih.gov. Recuperado el 29 de diciembre de 2022, de https://pubchem.ncbi.nlm.nih.gov/compound/89594
.PDB. (s/f). 6PV7. Receptor nicotínico de acetilcolina alfa3beta4 humano en complejo con nicotina. Banco de Datos de Proteínas. Recuperado el 29 de diciembre de 2022, de https://www.rcsb.org/structure/6PV7.
LOS EFECTOS DE LA NICOTINA EN EL CEREBRO. (s/f). Diadmbe.es. Recuperado el 29 de diciembre de 2022, de https://www.diadmbe.es/los-efectos-de-la-nicotina-en-el-cerebro/
Pérez-Ríos, M., Schiaffino, A., Montes, A., Fernández, E., López, M. J., Martínez-Sánchez, J. M., Sureda, X., Martínez, C., Fu, M., García Continente, X., Carretero Ares, J. L. y Galán, I. (2020). Mortalidad atribuible al consumo de tabaco en España 2016. Archivos de Bronconeumologia, 56(9), 559–563. https://doi.org/10.1016/j.arbres.2019.11.021
GONZÁLES D, RENNARD SI, NIDES M, ONCKEN C, AZOULAY S, BILLING CB, et al. Vareniclina, un agonista parcial del receptor de acetilcolina de nicotina α4β2 versus bupropión de liberación sostenida y placebo para dejar de fumar. JAMA 2006; 296: 47-55.
Schuller HM. Nitrosamines as nicotinic receptor ligands. Life Sci. 2007 May 30;80(24-25):2274-80. doi: 10.1016/j.lfs.2007.03.006. Epub 2007 Mar 19. PMID: 17459420; PMCID: PMC1987356.
Fouz Rosón, N. (2016). Impacto del tratamiento con vareniclina a bajas dosis vs estándar, ambas en pautas cortas, en la tasa de abstinencia tabáquica, adherencia al tratamiento y efectos adversos.
Cytisine. Uses, Interactions, Mechanism of Action | DrugBank Online. (n.d.). Retrieved December 31, 2022, from https://go.drugbank.com/drugs/DB09028
Leaviss, J., Sullivan, W., Ren, S., Everson-Hock, E., Stevenson, M., Stevens, JW, … y Cantrell, A. (2014). ¿Cuál es la eficacia clínica y la rentabilidad de la citisina en comparación con la vareniclina para dejar de fumar? Una revisión sistemática y evaluación económica. Evaluación de tecnologías sanitarias (Winchester, Inglaterra) , 18 (33), 1-120.
¿Se puede obtener fármacos contra el cáncer a partir de sustancias presentes en animales marinos?
escrito por HoLa_3D | 28 enero, 2023
Laura Saelices y Hortensia Rivera. Grado en Biología Sanitaria, Universidad de Alcalá.
Introducción:
A lo largo de la evolución, muchos organismos marinos han desarrollado numerosos productos químicos con diversos fines, como por ejemplo defensivos, protectores o de comunicación entre ellos. Es por esto que las formas de vida marinas van a ser una fuente de compuestos que podrán ser usados de forma terapéutica, por ejemplo, en el tratamiento de diversos cánceres. Para la investigación de estos fármacos va a ser fundamental la ampliación del conocimiento y conservación de la gran diversidad marina albergada en nuestro planeta.
¿Qué es el cáncer?
El cáncer es una patología biológica y genética de las células que componen los tejidos de nuestros órganos. Este conjunto de enfermedades se producen cuando los estrictos mecanismos de control que posee la célula para realizar diversos procesos como la replicación fallan, dando lugar a un acúmulo de mutaciones en genes encargados de controlar el crecimiento, la proliferación, la división o la muerte celular. (1)
Imagen 1: Las seis características distintivas del cáncer y las capacidades complementarias que permiten el crecimiento del tumor. Hanahan, D., & Weinberg, R. A. (2000). The hallmarks of cancer. cell, 100(1), 57-70.
Los genes que van a ser alterados van a ser de tres tipos:
Oncogenes: su mutación produce un aumento descontrolado del crecimiento celular
Genes supresores de tumores: cuya mutación da lugar a división incontrolada
Genes de reparación de DNA: su mutación provocará acumulación de aberraciones en la estructura del DNA
Los tumores, tanto benignos como malignos, se clasifican dependiendo del tipo de célula que procedan, siendo los mayoritarios los carcinomas de las células epiteliales, los sarcomas de células del músculo, hueso, cartílago y tejido fibroso, y por último las leucemias y linfomas de células hematopoyéticas y del sistema inmune.
Debido a la difusión de los tumores malignos que los hace más resistentes al tratamiento local, las formas de tratarlos van a ser la cirugía, la radioterapia y la quimioterapia, siendo estos tres no excluyentes y frecuentemente usados de forma simultánea o secuencial.
En nuestro caso nos vamos a centrar en la quimioterapia, que es el uso de fármacos para destruir las células cancerosas, concretamente en el uso de sustancias producidas por organismos marinos con este fin.
Mecanismos de acción de los antitumorales de origen marino:
La gran variedad de compuestos químicos encontrados en la fauna marina poseen una amplia heterogeneidad de acción, actuando a muy diferentes niveles. Los principales mecanismos de acción antineoplásicos de estos bioactivos descubiertos son los siguientes:
Inhibición del proceso de angiogénesis
Inducción del proceso de muerte celular programada o apoptosis
Inhibición de proteínas específicamente destinadas a regular el ciclo celular.
Inactivación de topoisomerasas
Inhibición de la formación de microtúbulos
(1)
A continuación, veremos más detenidamente alguno de estos mecanismos de actuación centrándonos en tres de estos compuestos antitumorales.
Citarabina:
La citarabina es un análogo de nucleósido de pirimidina sintético, denominado arabinósido de citosina o Ara-C, desarrollado a partir de la espongotimidina, que fue aislada en la demosponja Tectitethya crypta. Este antimetabolito se usa en el tratamiento de leucemia; en concreto se usa en su forma convencional para el tratamiento de leucemia linfocítica aguda, leucemia mieloide aguda y las fases de crisis blástica de la leucemia mieloide crónica y la leucemia meníngea, y en su forma liposomal en el tratamiento de meningitis linfomatosa. (2)
Imagen 2: Foto de la demosponja Tectitethya crypta.Tectitethya crypta. (s. f.). guide.poriferatreeoflife.org. https://guide.poriferatreeoflife.org/sp_35.htm Imágenes 3 y 4: Estructura de la citarabina. Cytarabine. (s. f.). Pubchem. https://pubchem.ncbi.nlm.nih.gov/compound/6253#section=2D-Structure
Este agente citotóxico afecta especialmente a las células que se encuentran en fase S del ciclo celular. La citarabina es, por tanto, un antimetabolito análogo de la citidina (nucleósido de citosina con anillo de ribosa), el cual tiene beta-D-arabinosa en lugar de ribosa. Este es convertido en la célula en citarabina trifosfato, que será el metabolito activo, y entonces competirá con la citidina en su incorporación al ADN. La replicación quedará parada debido a que la arabinosa induce una rotación en el ADN, lo que hará que pare el ciclo celular. Además, este compuesto también inhibe la ADN polimerasa alfa y beta, haciendo que descienda la tasa de replicación y de replicación del ADN. (3)
Imagen 5: Representación del mecanismo de acción del antitumoral citarabina. Realizada con BioRender https://biorender.com/
Además, este compuesto ha demostrado tener cierta capacidad antiviral e inmunosupresora, pero esto ha sido sólo demostrado en experimentos in vitro.
Yondelis
Este es el nombre comercial que ha recibido el antitumoral cuyo principio activo es ecteinascidina-743, un alcaloide de tetrahidroisoquinolina marina aislado de Ecteinascidia turbinata.
Imagen 6y 7: Ecteinascidia turbinata. (s. f.). Canal mar menor. https://canalmarmenor.carm.es/inventario-ecologico/fauna/ascidia-de-manglar-ecteinascidia-turbinata/. Imagen 8: Estructura de la ecteinascidina-743. (s. f.). Pubchem. https://pubchem.ncbi.nlm.nih.gov/compound/108150#section=2D-Structure
El mecanismo de acción de este compuesto es bastante novedoso, puesto que se une al surco menor del DNA formando aductos covalentes en la posición N2 de la guanina; esta unión al surco menor permite que el fármaco interactúe con proteínas específicas de las células cancerosas, impidiendo la copia celular y provocando roturas de doble cadena en el ADN (1,4)
Se cree que el fármaco afecta a varios factores de transcripción implicados en la proliferación celular, en particular a través del sistema de reparación por escisión de nucleótidos acoplados a la transcripción. De este modo interfiere con la división celular, los procesos de transcripción genética y la maquinaria de reparación del ADN.
En concentraciones relativamente altas también puede provocar la desorganización del ensamblaje de microtúbulos y la red de microfilamentos intermedios; provocando primero una pérdida de la organización de los microtúbulos en la porción distal, que continúa con la aparición de microtúbulos colapsados alrededor del núcleo celular.
Así mismo, se ha observado que inhibe la sobreexpresión del gen de resistencia a múltiples fármacos (MDR-1) que es un factor importante responsables de que las células desarrollen resistencia a los fármacos contra el cáncer (4).
En condiciones de laboratorio y concentraciones nanomolares, ET-743 presenta actividad frente a una variedad de líneas celulares de tumores sólidos, incluido el melanoma, tumores de pulmón, ovario y colón; siendo las células en la fase G1 del ciclo celular y las células del sarcoma de tejidos blandos, especialmente sensibles a la muerte inducida por este compuesto. (1)
Actualmente, Yondelis está autorizado para comercializarse como tratamiento para el sarcoma de tejidos blandos avanzado o metastásico y para el tratamiento del cáncer de ovario recurrente platino-sensible en combinación con otro fármaco (DOXIL/Caelyx). También se están llevando a cabo ensayos en fase II para cáncer de mama, cáncer de próstata y para tumores pediátricos. (1)
Imagen 9:Mecanismo de acción de Yondelis. (2016, 31 agosto). Curetoday. https://www.curetoday.com/view/medical-illustration-marinederived-cancer-treatments
Zalypsis:
Zalypsis (PM00104) es un alcaloide sintético relacionado con el compuesto natural marino Jorumycina, del molusco Jorunna funebris, y con la familia de las Renieramycinas, de las esponjas Neopetrosia. El mecanismo de acción de este citotóxico es multifactorial e implica tanto el bloqueo del ciclo celular como la estimulación de diferentes cascadas apoptóticas. (5)
Imagen 10: Foto del molusco Jorunna funebris. Jorunna funebris. (s. f.). nudibranchdomain.org. https://nudibranchdomain.org/product/jorunna-funebris/Imagen 11: Estructura de Zalypsis. PM00104. (s. f.). Pubchem. https://pubchem.ncbi.nlm.nih.gov/compound/Zalypsis#section=2D-Structure
Este compuesto utiliza su grupo carbinolamina para unirse al surco menor del ADN mediante unión covalente a restos de guanina en tripletes. Esta interacción da lugar a un aducto que inhibe las fases más tempranas de la transcripción y puede dar lugar a double-stranded breaks. Debido a esto va a detener las células en la fase S. (5,6)
Además, como ya hemos comentado, Zalypsis también va a estar relacionado con algunas rutas apoptóticas, lo cual está probablemente relacionado con los daños en el ADN que provoca. Así, está relacionado con activación de cascadas dependientes de caspasas como algunas que incluyen a la enzima PARP y otras que conducen a la activación de p53. (6)
Imagen 12: Representación del mecanismo de acción del antitumoral Zalypsis. Realizada con BioRender https://biorender.com/
En numerosos estudios preclínicos ha demostrado una fuerte actividad contra el cáncer de mama, de próstata, gástrico y renal. Además, actualmente está en fase II en mieloma múltiple, vejiga y sarcoma de Ewing. (1)
Producción sostenible de recursos marinos
Todos estos compuestos poseen un enorme potencial económico; sin embargo aparecen en los organismos en cantidades muy escasas, lo que requiere el desarrollo de métodos de producción que aseguren el adecuado suministro y comercialización del producto sin alterar las poblaciones y ecosistemas.
Estas técnicas de producción deben ser adecuadas para cada caso, basándonos en una serie de factores como la fuente del compuesto, complejidad de la molécula, abundancia del organismo en la naturaleza, condiciones de crecimiento, etc.
Variación intraespecífica de la actividad:
Los compuestos de los que hemos estado hablando tienen una gran variabilidad tanto entre especies como dentro de un solo organismo. La biosíntesis de estos productos en los organismos en los que son producidos está muy influenciada por factores tanto externos, condiciones ambientales o presencia de depredadores, como internos, estado de desarrollo o la masa corporal. Lo que produce está variabilidad no está del todo claro pero el mayor candidato es la heterogeneidad ambiental, es decir, el contenido químico de las especies depende estrechamente de dónde y cuándo es recolectado el organismo. Por esto, es posible que se encuentren metabolitos nuevos en especies ya investigadas si las condiciones de cultivo son diferentes. (1)
Conclusiones:
Como hemos visto en los animales marinos podemos encontrar una gran cantidad de metabolitos que pueden ser de gran utilidad para la cura de enfermedades tan importantes como el cáncer. Es por esto que es importante conocer en profundidad la biodiversidad marina, además de otras razones como la conservación e investigación del potencial genético.
Como conclusión, es muy posible que, bajo el mar, muchos animales marinos tengan ya solucionados problemas que nosotros seguimos intentando resolver.
Bibliografía:
(1) Navia, A. J. L., & San Sebastián, M. M. (2015). Drogas marinas: los animales marinos como fuentes de compuestos antitumorales. AmbioCiencias: revista de divulgación, (13), 35-51.
(2) U.S. National Library of Medicine. (n.d.). Cytarabine. National Center for Biotechnology Information. PubChem Compound Database. Retrieved January 4, 2023, from https://pubchem.ncbi.nlm.nih.gov/compound/6253
(3) Betcher, D. L., & Burnham, N. (1990). Cytarabine. Journal of Pediatric Oncology Nursing, 7(4), 154-157.
(4) Trabectedin: Uses, Interactions, Mechanism of Action | DrugBank Online. (s. f.). DrugBank. https://go.drugbank.com/drugs/DB05109
(5) Petek, B. J., & Jones, R. L. (2014). PM00104 (Zalypsis®): A marine derived alkylating agent. Molecules, 19(8), 12328-12335.
(6) Ocio, E. M., Maiso, P., Chen, X., Garayoa, M., Álvarez-Fernández, S., San-Segundo, L., … & Pandiella, A. (2009). Zalypsis: a novel marine-derived compound with potent antimyeloma activity that reveals high sensitivity of malignant plasma cells to DNA double-strand breaks. Blood, The Journal of the American Society of Hematology, 113(16), 3781-3791.
BRCA1 Y BRCA2: MUTACIONES Y CÁNCER
escrito por GRUPO11 | 28 enero, 2023
Noelia Ares Bóveda, Belén Asenjo Lajusticia e Irene Cañadilla González. Biología Sanitaria. Universidad de Alcalá.
Poco sabía Mary-Claire King en 1994 que su descubrimiento revolucionaría la biología molecular del cáncer: el gen BRCA1 que, junto con el gen BRCA2 descubierto un año después, abrió la puerta a la investigación de diferentes tipos de cáncer y su enfoque en clínica.
Estos dos genes juegan un papel fundamental en el mantenimiento de la estabilidad del genoma. Ante su pérdida de funcionalidad, falta o mutación, pueden conducir a fenómenos de tumorigénesis, además de otras patologías como la anemia de Fanconi.
Sin embargo, no toda mutación implica riesgo de aparición de tumores, y aquí subyace la complejidad de su estudio.
¿Cómo son estos genes y cómo funcionan? ¿En qué tipos de cáncer están implicados? ¿Qué aproximaciones terapéuticas existen a día de hoy?
FUNCIÓN Y ESTRUCTURA
BRCA1/2 (Breast Cancer Genes) son genes supresores de tumores que juegan un papel fundamental en el mantenimiento de la estabilidad genómica. Cuando se producen roturas en la doble hebra de DNA (DSB), ocasionando así una interrupción en la lectura del mismo, entran en juego estos genes: las proteínas que codifican llevan a cabo el proceso de recombinación homóloga (HR), uno de los mecanismos de reparación fundamentales que tienen lugar en la fase S del ciclo celular.
La inactividad de estos genes desencadenaría una recombinación homóloga defectiva que en último término podría desembocar en una predisposición al tumor, especialmente en la mama.
Figura 1: se ilustra el papel supresor de tumores de BRCA1/2 cuando se encuentra activo, mientras que ante su pérdida de funcionalidad se genera una predisposición a la formación de tumores. Created with BioRender.com.
A pesar de tener una función similar, BRCA1 y BRCA2 presentan diferencias en cuanto a su biología molecular, sus interacciones con proteínas y su relación con el cáncer.
BRCA1
El gen BRCA1 se sitúa en el brazo largo del cromosoma 17 (17q21), y fue descubierto e identificado en 1994. Este gen codifica la proteína BRCA1 supresora de tumores (1863 aminoácidos).
En la proteína BRCA1 encontramos tres clusters asociados al cáncer de mama (BCCR: “Breast Cancer Cluster” Region) y un único cluster asociado al cáncer de ovario (OCCR: “Ovarian Cancer Cluster” Region). Mutaciones en estas regiones aumentan la posibilidad del desarrollo de un cáncer en dichos tejidos específicos.
Figura 2: estructura de la proteína BRCA1 (dominios, clusters) y las proteínas con las que interacciona señaladas con un recuadro. Extraída de la referencia [1].
Esta proteína presenta en su extremo N-terminal un dominio RING rico en cisteína con el que interacciona con la proteína BARD1. Así forma un complejo BRCA1-BARD1 que tiene actividad ubiquitina ligasa. En caso de que haya una mutación en el dominio RING de la proteína BRCA1, esta no podrá unirse a BARD1 y por tanto, se perderá la función ubiquitina ligasa, pudiendo suponer una predisposición al desarrollo de cáncer.
En su extremo C-terminal presenta el dominio BRCT, que interacciona con la RNA polimerasa II relacionada con el proceso de transcripción. Además, este dominio interactúa con tres proteínas: Abraxas, BACH1, CtIP; estas uniones permitirán la formación de tres complejos distintos implicados en la reparación de DSBs:
La proteína BRCA1 se asocia a las Abraxas a través de RAP80, y cuando hay un daño en el DNA este complejo interacciona con las histonas dañadas.
El complejo BRCA1-BACH1 participa en la recombinación homóloga. La proteína BACH1 también es conocida como BRIP1 y tiene función helicasa.
La proteína CtIP unida a BRCA1 forma un complejo encargado de la resección de DSBs en pasos tempranos de la reparación.
Figura 3: esquema en el que se ilustran las interacciones de diferentes proteínas con BRCA1 y las funciones de los complejos mencionados. Created with BioRender.com.
BRCA2
El gen BRCA2 se encuentra en el cromosoma 13 (13q12), y fue descubierto e identificado en 1995. Este gen codifica la proteína BRCA2 supresora de tumores, menos conocida pero de mayor tamaño que la codificada por BRCA1 (3418 aminoácidos).
La proteína ácida pequeña DSS1 se une a una región de la proteína BRCA2, formando un complejo proteico que se asociará a las zonas dañadas de DNA.
La proteína BRCA2 presenta unas repeticiones BRC que permitirán la unión directa a RAD51, una enzima de recombinación necesaria en el proceso. El complejo formado por las dos proteínas juega un papel importante en el reconocimiento de DSBs y el inicio de la recombinación.
Los clusters ya mencionados localizados en la proteína BRCA1 también se encuentran en la proteína BRCA2.
Figura 4: esquema de la proteína BRCA2, se pueden observar los cluster (BCCR y OCCR). Extraído de referencia [2].
Además, se ha demostrado que el gen BRCA2 tiene un papel fundamental en la citocinesis, ya que mutaciones en este gen conllevan anomalías cromosómicas.
Figura 5: ilustra la unión de las proteínas RAD51 y DSS1 a la proteína BRCA2, así como las interacciones de sus estructuras moleculares (PDB: 1N0W y PDB: 1IYJ). Created with BioRender.com and Chimera.
CONEXIÓN ENTRE BRCA1 Y BRCA2: PALB2
Pero, ¿de qué manera se conectan las proteínas codificadas por BRCA1 y BRCA2? Esto es posible gracias a la proteína PALB2 (Partner And Localizer of BRCA2). La interacción entre las tres proteínas ocurre de la siguiente forma, tal y como se ilustra en el esquema:
El extremo N-terminal coiled-coil de PALB2 interacciona con el dominio coiled-coil de BRCA1.
El extremo N-terminal de BRCA2 se conecta con el extremo C-terminal de PALB2.
Esto daría lugar a la formación del complejo BRCA1-PALB2-BRCA2 el cual es esencial para la recombinación homóloga, así como la activación y mantenimiento del punto de control G2/M del ciclo celular.
PALB2 es susceptible a sufrir mutaciones que podrían conducir a la pérdida de estabilidad genómica y, en consecuencia, un posible desarrollo de cáncer.
Figura 6: se ilustran las regiones de la proteína PALB2 (Partner And Localizer of BRCA2) con las que interaccionan las proteínas BRCA1 y BRCA2, formando el complejo BRCA1-PALB2-BRCA2. Además, se puede observar la interacción ya mencionada de BRCA1 con la proteína BRIP1 (también conocida como BACH1). Esquema extraído de referencia [5].
RELACIÓN CON p53
Se cree que la proteína producto del gen BRCA1 lleva a cabo su función mediante la formación de grandes complejos. En estos complejos participan diversas proteínas, entre las cuales se encuentra la p53, que juega un papel crucial en el mantenimiento de la integridad genómica por su función supresora de tumores.
En muchos de los tipos de cáncer debidos a mutaciones de los genes BRCA, se ha visto que ha habido interrupciones en la relación entre algunos componentes de estos complejos, provocando que la función de los genes BRCA se vea alterada y por tanto se inhiba la supresión del tumor, dando como resultado la aparición de cáncer.
Figura 7: alteraciones en el complejo proteico en el que intervienen BRCA1/2 y p53 pueden conducir a la formación de un tumor, al inhibirse su supresión. Created with BioRender.com.
Por ejemplo, el complejo BRCA1-BARD1 está implicado en la activación de ciertos puntos de control del ciclo celular. Estas tareas complementarias de BRCA1 en los puntos de control y su rol estabilizador de p53 podrían ayudar en su función de mantenimiento de la estabilidad genómica.
Muchos estudios sugieren que en la formación del tumor existe una estrecha relación entre la pérdida de función de BRCA1/2 y la pérdida de función de la p53.
Se cree que la rotura de la molécula de DNA (debida a esa falta de funcionalidad de BRCA) hace que se activen puntos de control dependientes de p53 y/o apoptosis para evitar la tumorigénesis.
MUTACIONES Y CÁNCER
Cuando consultamos la página web del BRCA Exchange, una iniciativa internacional de la Global Alliance for Genomics and Health, encontramos a mes de febrero de 2021 un total de 40389 variantes de estos genes, clasificadas según su significado en clínica en cuanto a su patogenicidad (una escala de patogénico a benigno o sin significado clínico).
Esta cifra nos ilustra uno de los desafíos y la complejidad que supone el estudio de BRCA1 y BRCA2: no todas las mutaciones implican cáncer.
Las mutaciones en BRCA1/2 implicadas en tumorigénesis generalmente afectan a la mama y al ovario. Pero, ¿a qué se debe el tropismo por estos tejidos?
Ambos tienen gran susceptibilidad a sufrir estimulación hormonal por señales fuertes de crecimiento. La enzima aromatasa está implicada en la síntesis de estrógenos y está regulada negativamente por el gen BRCA1. Además, se ha observado un incremento de los niveles de estrógenos en sangre en personas portadoras de mutaciones en BRCA1, así como de progesterona. Por ello, actualmente se encuentra en estudio la correlación entre niveles hormonales y la predisposición a tumores cuando BRCA1 se encuentra mutado.
Además, en menor medida estas mutaciones están relacionadas con el cáncer de páncreas, entre otros.
CÁNCER DE MAMA
Tal y como nos sugiere su nombre, estos genes están asociados con el desarrollo de cáncer de mama, de manera que el riesgo de padecer este tipo de cáncer a lo largo de la vida cuando se produce una pérdida de funcionalidad de BRCA2 es del 45%, mientras que cuando se trata del gen BRCA1 alcanza el 57%.
Un aspecto a remarcar en el desarrollo del cáncer de mama es la importancia de la predisposición genética: cuando se estudian casos de esta enfermedad debidos a la herencia de una mutación, entre todos los genes de susceptibilidad al cáncer de mama descritos, la mayoría de estas mutaciones se han encontrado en BRCA1 y BRCA2.
A modo de curiosidad, en un estudio se ha observado que la expresión tanto de BRCA1 como de BRCA2 es menor en trabajadores a turnos (incluyen jornadas laborales nocturnas) que en trabajadores en turno de día y, además, cuanto mayor es el número de noches trabajadas por mes, menor es la expresión de los genes. Estos resultados llevaron a algunos autores a proponer que esta podría ser la causa de que la alteración del ritmo circadiano pueda conducir a un incremento del riesgo de desarrollo de cáncer de mama (tal y como han comprobado diferentes estudios), y a plantear que los genes BRCA podrían incluirse dentro de los conocidos como “clock-controlled genes”.
CÁNCER DE OVARIO
El ovario, junto a la mama, es uno de los órganos más afectados por las mutaciones en los genes BRCA. Se piensa que esto puede deberse al estrés oxidativo derivado de los ciclos menstruales, así como al papel de la regulación hormonal (especialmente por estrógenos).
Por ello, la alta predisposición en estos tejidos a sufrir daños en su DNA implica la importancia de los mecanismos de reparación en los que intervienen los genes BRCA. Las mutaciones en el gen BRCA1 suponen un aumento del 11% en el riesgo de sufrir cáncer de ovario y un 40% en el caso de pérdida de funcionalidad del gen BRCA2.
Figura 8: created with BioRender.com.
CÁNCER DE PÁNCREAS
Se trata del tercer tipo de cáncer más común asociado a la mutación de BRCA.
Se caracteriza por tener una alta letalidad, debido potencialmente a que presenta una gran resistencia hacia los tratamientos.
Sólo un pequeño porcentaje de pacientes con cáncer de páncreas presenta mutaciones germinales en BRCA1/2 (aproximadamente el 7%). Por tanto, el cáncer de páncreas causado por mutaciones en BRCA1/2 es poco común; y cabe destacar que presenta una “ventaja” ya que al poseer características biológicas únicas (diferentes a las del cáncer de páncreas común) se pueden crear tratamientos específicos.
Las mutaciones en BRCA1 o en BRCA2 aumentan de manera diferente el riesgo de padecer cáncer de páncreas: si es BRCA2 el afectado, el riesgo de padecer la enfermedad aumenta mucho más que si se trata de mutaciones de BRCA1.
TERAPIAS INNOVADORAS
INHIBIDORES DE PARP (PARPi)
Los inhibidores de PARP (PARPi) se han usado en tratamientos de cáncer en pacientes sensibles a quimioterapia con platino.
PARP (Poli-ADP-ribosa polimerasa) es una enzima con papel fundamental en el reclutamiento del complejo proteico encargado de restaurar los daños del DNA. Las proteínas BRCA, entre otras, forman parte de estos complejos.
Como ya se ha tratado, una mutación en los genes BRCA resultaría en la transcripción de las proteínas BRCA1 y BRCA2 con una pérdida de funcionalidad y por consecuente, una errónea reparación.
Figura 9: participación de PARP en la formación del complejo proteico. Created with BioRender.com.
Este tratamiento basado en el uso de inhibidores de PARP consiste en la “captura” (PARP-trapping) de esta enzima. Esto ocasiona la inhibición de los mecanismos de reparación, ya que en caso de que se produjese una mutación en BRCA1/2, la reparación errónea podría conducir a la formación de tumores. De esta forma, al no actuar los mecanismos de reparación, se activa la apoptosis celular y esas células morirían.
Figura 10: esquema ilustrativo de la función de PARPi. Created with BioRender.com.
TERAPIAS BASADAS EN G-CUADRUPLEXOS
Un enfoque terapéutico de gran interés actualmente para tumores en los que el gen BRCA2 se ve implicado se centra en los G-cuadruplexos, unas estructuras formadas por tres láminas de tétradas de guanina unidas por apareamientos de Hogsteen y estabilizadas por un catión metálico. Podemos encontrar estas estructuras al final de las secuencias teloméricas, interfiriendo con su replicación, y se ha observado que BRCA2, entre otras proteínas, interviene en la replicación de los telómeros (de forma que estos no se acortan).
En trabajos experimentales se ha demostrado que tratamientos de células con pérdida de funcionalidad de BRCA2 con compuestos estabilizadores de G-cuadruplexos disminuye la viabilidad de las mismas, conduciendo de alguna manera a un aumento de la letalidad específica al incrementar la fragilidad de los telómeros. Concretamente, se utilizó la piridostatina (PDS). Otro aspecto que genera interés de este trabajo es que también afecta a aquellas células con pérdida de funcionalidad de BRCA2 que muestran resistencia a los tratamientos con inhibidores de PARP ya mencionados.
Figura 11: en este gráfico se representa un descenso de la viabilidad de las células deficientes en BRCA2 (línea roja) cuando son tratadas con piridostatina (PDS), un estabilizador de G-cuadruplexos. Extraído de los resultados de la referencia [13].
Además, otro compuesto estabilizador de G-cuadruplexos conocido como CX-5461 entró en 2016 en fase I de ensayo clínico para el tratamiento de tumores relacionados con el gen BRCA.
Con BRCA1 y BRCA2 surgió una nueva manera de concebir el estudio del genoma y el cáncer: pocas décadas atrás existía cierto rechazo en el campo de la ciencia a vincular la aparición de tumores a cambios en el material genético; pero esto cambió con este descubrimiento que tuvo gran impacto en la investigación contra el cáncer.
REFERENCIAS
[1] Takaoka, M. and Miki, Y. (2018) ‘BRCA1 gene: function and deficiency’, International Journal of Clinical Oncology. Springer Japan, 23(1), pp. 36–44. doi: 10.1007/s10147-017-1182-2.
[2] Venkitaraman, A. R. (2019) ‘How do mutations affecting the breast cancer genes BRCA1 and BRCA2 cause cancer susceptibility?’, DNA Repair. Elsevier, 81(July), p. 102668. doi: 10.1016/j.dnarep.2019.102668.
[3] Semmler, L., Reiter-Brennan, C. and Klein, A. (2019) ‘BRCA1 and breast cancer: A review of the underlying mechanisms resulting in the tissue-specific tumorigenesis in mutation carriers’, Journal of Breast Cancer, 22(1), pp. 1–14. doi: 10.4048/jbc.2019.22.e6.
[4] Bracci, M., Ciarapica, V., Zabaleta, M. E., Tartaglione, M. F., Pirozzi, S., Giuliani, L., Piva, F., Valentino, M., Ledda, C., Rapisarda, V., Stevens, R. G. and Santarelli, L. (2019) ‘BRCA1 and BRCA2 gene expression: diurnal variability and influence of shift work’, Cancers, 11(8), pp. 1–15. doi: 10.3390/cancers11081146.
[5] Murphy, C. G. and Moynahan, M. E. (2010) ‘BRCA Gene Structure and Function in Tumor Suppression’, The Cancer Journal, 16(1), pp. 39–47. doi: 10.1097/ppo.0b013e3181cf0204.
[6] López-Urrutia, E., Salazar-Rojas, V., Brito-Elías, L., Coca-González, M., Silva-García, J., Sánchez-Marín, D., Campos-Parra, A. D. and Pérez-Plasencia, C. (2019) ‘BRCA mutations: is everything said?’, Breast Cancer Research and Treatment. Springer US, 173(1), pp. 49–54. doi: 10.1007/s10549-018-4986-5.
[7] Roy, R., Chun, J. and Powell, S. N. (2012) ‘BRCA1 and BRCA2: Different roles in a common pathway of genome protection’, Nature Reviews Cancer. Nature Publishing Group, 12(1), pp. 68–78. doi: 10.1038/nrc3181.
[8] Luo, G., Lu, Y., Jin, K., Cheng, H., Guo, M., Liu, Z., Long, J., Liu, C., Ni, Q. and Yu, X. (2015) ‘Pancreatic cancer: BRCA mutation and personalized treatment’, Expert Review of Anticancer Therapy, 15(10), pp. 1223–1231. doi: 10.1586/14737140.2015.1086271.
[9] E. Filippini, S. and Vega, A. (2013) ‘Breast cancer genes: beyond BRCA1 and BRCA2’, Frontiers in Bioscience, 18, pp. 1358–1372.
[10] Venkitaraman, A. R. (2014) ‘Cancer suppression by the chromosome custodians, BRCA1 and BRCA2’, Science, 343(6178), pp. 1470–1475. doi: 10.1126/science.1252230.
[11] Simhadri, S., Vincelli, G., Huo, Y., Misenko, S., Foo, T. K., Ahlskog, J., Sorensen, C., Oakley, G., Ganesan, S., Bunting, S., Xia, B. (2018) ‘PALB2 connects BRCA1 and BRCA2 in the G2/M checkpoint response’. Oncogene. doi:10.1038/s41388-018-0535-2.
[12] Xu, H., Di Antonio, M., McKinney, S., Mathew, V., Ho, B., O’Neil, N. J., Santos, N. Dos, Silvester, J., Wei, V., Garcia, J., Kabeer, F., Lai, D., Soriano, P., Banáth, J., Chiu, D. S., Yap, D., Le, D. D., Ye, F. B., Zhang, A., Thu, K., Soong, J., Lin, S. C., Tsai, A. H. C., Osako, T., Algara, T., Saunders, D. N., Wong, J., Xian, J., Bally, M. B., Brenton, J. D., Brown, G. W., Shah, S. P., Cescon, D., Mak, T. W., Caldas, C., Stirling, P. C., Hieter, P., Balasubramanian, S. and Aparicio, S. (2017) ‘CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours’, Nature Communications, 8(205). doi: 10.1038/ncomms14432.
[13] Zimmer, J., Tacconi, E. M. C., Folio, C., Badie, S., Porru, M., Klare, K., Tumiati, M., Markkanen, E., Halder, S., Ryan, A., Jackson, S. P., Ramadan, K., Kuznetsov, S. G., Biroccio, A., Sale, J. E. and Tarsounas, M. (2016) ‘Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds’, Molecular Cell. The Authors, 61(3), pp. 449–460. doi: 10.1016/j.molcel.2015.12.004.
[14] Rudkin, T. M. and Foulkes, W. D. (2005) ‘BRCA2: Breaks, mistakes and failed separations’, Trends in Molecular Medicine, 11(4), pp. 145–148. doi: 10.1016/j.molmed.2005.02.003.
[15] Mittica, G., Ghisoni, E., Giannone, G., Genta, S., Aglietta, M. and Valabrega,G. (2018) ‘PARP Inhibitors in Ovarian Cancer’, Recent Patents on Anti-Cancer Drug Discovery, 13(4), pp. 392–410. doi: 10.2174/1574892813666180305165256.
[16] Neff, R. T., Senter, L., Salani, R. (2018) ‘BRCA mutation in ovarian cancer: testing, implications and treatment considerations’, Therapeutic Advances in Vaccines, 9(6), pp. 259–261. doi: 10.1177/https.
[17] Venkitaraman, A. R. (2002) ‘Cancer susceptibility and the functions of BRCA1 and BRCA2’, Cell, 108(2), pp. 171–182. doi: 10.1016/S0092-8674(02)00615-3.
Telómeros, cáncer y envejecimiento
escrito por patricia.tato | 28 enero, 2023
Yuliia Fatych, Patricia Tato Moreno. 3º Biología Sanitaria. Universidad de Alcalá
Introducción histórica
En 1938, Hermann J. Müller observó que en los extremos de cromosomas expuestos a rayos X de Drosophila melanogaster no se producían mutaciones como deleciones o inversiones, mientras que esto sí ocurría en el resto del genoma. Esto se debía a la presencia de un “casquete protector” al que denominó “gen terminal” y, más tarde, “telómero”.1
Dos años después, Barbara McClintock, que realizaba estudios genéticos con maíz, describió la formación de cromosomas dicéntricos mediante la ruptura de los cromosomas y su posterior adhesión y fusión de sus extremos. Gracias a estas investigaciones, McClintock demostró que los extremos de los cromosomas se podían restaurar gracias a la obtención de un nuevo telómero.1
Sin embargo, la investigación sobre los telómeros no se volvió a retomar hasta treinta años más tarde, cuando James Watson identificó el problema de la replicación del DNA en los extremos de los cromosomas. Debido a que las DNA-polimerasas sólo pueden sintetizar DNA en sentido 5’ → 3’, la cadena 5’ → 3’ se forma mediante la síntesis de fragmentos de Okazaki, cada uno de los cuales necesita un cebador. Esto hace que el extremo 3’ del cromosoma no se pueda replicar por completo y, por tanto, en cada replicación, éste se acorta. Esto limita la capacidad de replicación de la célula. Olovnikov descubrió que la senescencia celular se producía como consecuencia del sobrepaso de ese límite, lo que provoca la alteración de la célula.1
Elizabeth Blackburn, Carol Greider y Jack Szostak (de izquierda a derecha). The Nobel Prize in Physiology or Medicine 2009. (s.f.). The Nobel Prize in Physiology or Medicine 2009. [Imágenes]. Recuperado el 24 de noviembre de 2019 de https://www.nobelprize.org/prizes/medicine/2009/summary/
Más tarde, tras muchos años de trabajo e investigación sobre los telómeros del protozoo Tetrahymena thermophila y de la levadura Saccharomyces cerevisiae, Elizabeth Blackburn, Jack Szostak y Carol Greider, descubrieron la existencia de una actividad enzimática que denominaron “transferasa telómero terminal”. Esta actividad estaba presente en la enzima telomerasa, a la que le atribuyeron el papel de la replicación del DNA telomérico, impidiendo el acortamiento progresivo de los telómeros en cada división celular.1 Finalmente, en el año 2009, Blackburn, Szostak y Greider recibieron el Premio Nobel de Medicina y Fisiología por sus estudios sobre los telómeros y el descubrimiento de la telomerasa.2
Estructura de los telómeros
Los telómeros son unas estructuras que se ubican en los extremos de los cromosomas lineales eucarióticos y que están compuestos por proteínas y secuencias de DNA no codificante repetidas en tándem. La secuencia repetida es la secuencia (TTAGGG)n, lo que hace que en los telómeros exista una hebra rica en nucleótidos de guanina, conocida como hebra G, y otra hebra rica en nucleótidos de citosina. La hebra G es la que se encuentra orientada en dirección 5’ → 3’ y, en su extremo, sobresale de la cadena de DNA, por lo que no se aparea con la hebra antiparalela, debido a que ésta es más corta. Este fragmento de DNA simple de la hebra G se conoce como overhang-3’ y tiene una longitud que varía según la especie. Además, la longitud de los telómeros también es variable y en cada cromosoma la cantidad de DNA telomérico puede ser diferente.3,4
Estructura del telómero. Pierce, B. A. (2016). Genética: Un enfoque conceptual. [Figura]
Es importante señalar la existencia de estructuras complejas en el extremo 3’ rico en G, denominadas G-cuadruplexos. Dichas estructuras se pueden encontrar en diferentes conformaciones, unidas por planos cuadrados que contienen 4 guaninas (que a su vez interaccionan por puentes de hidrógeno de Hoogsteen). Esta estructura ordenada impide la acción de las nucleasas, las cuales trabajan sobre las hebras sueltas de DNA.5
Por otro lado, en los telómeros también podemos encontrar unas secuencias repetidas, conocidas como secuencias asociadas a los telómeros. Estas estructuras varían según la especie en cuanto a su longitud, secuencia y complejidad. Además, parece ser que no tienen un papel importante en la estabilidad del cromosoma y todavía no se conoce cuál es exactamente su función.4
Esta estructura especial de los telómeros, hace que éstos tengan la función de evitar que se fusionen con los extremos de otros cromosomas, lo que se conoce como fusión telomérica. Además, también van a tener otras funciones como preservar la región codificante del DNA de la acción enzimática, permitir la interacción entre los cromosomas y la matriz nuclear, e intervenir en la transcripción de genes subteloméricos que regulan el ciclo celular. 6
Los telómeros de mamíferos se encuentran asociados a un complejo multiproteico formado por seis proteínas que recibe el nombre de shelterina, cuya función es favorecer la formación de un lazo (“loop T”) que permite que el telómero se doble, secuestrando el extremo terminal de los cromosomas. Esto evita que el DNA telomérico sea dañado por nucleasas. Además, dicho complejo multiproteico impide que se lleve a cabo un mecanismo de reparación de DNA (MRA) en los telómeros, y regula la actividad de la telomerasa.5
Las proteínas que forman el complejo de la shelterina son TRF1, TIN2, TRF2, RAP1, POT1 y TPP1. La shelterina es reclutada por los telómeros a través de TRF1 Y TRF2. La proteína TRF1 (factor 1 de unión a las repeticiones teloméricas) se une a las secuencias repetidas TTAGGG de doble cadena e interacciona con TIN2 (factor 2 nuclear de interacción con TRF2). TRF2 también se une a las repeticiones teloméricas de doble cadena y, a su vez, interacciona con RAP1 (proteína 1 represora/activadora). Además, TIN2 también se encuentra asociada a TRF2. Sin embargo, a diferencia de TRF1 y TRF2, POT1 (protección del telómero 1) se puede unir a las secuencias TTAGGG formadas por una hebra simple y se conecta a TRF1 y TRF2 a través de la proteína TPP1 (proteína homóloga de la displasia adrenocortical). Al mismo tiempo, TPP1 también se asocia a TIN2.7,8
Además del “loop T”, en el cual el DNA monocatenario se enrosca alrededor de un círculo estabilizado por shelterina, existe al final de éste otro bucle, el “loop D”. Este último loop consiste en una estructura de triple hebra denominado como bucle de desplazamiento, en el cual el DNA telomérico monocatenario se entrelaza con una región de DNA bicatenario.5
Aspectos moleculares de la telomerasa: estructura, función, regulación.
Arquitectura funcional de la telomerasa
La telomerasa es una enzima de tipo ribonucleoproteína que participa en la síntesis de las secuencias repetitivas de DNA de los telómeros, estabilizando su longitud.6
La enzima telomerasa humana posee una subunidad catalítica (TERT), una subunidad de RNA (hTR, también llamado TERC o TER) que proporciona el molde para la adición de la secuencia telomérica (TTAGGG)n, y proteínas accesorias. Estas proteínas participan en la regulación de la biogénesis de la telomerasa, así como en su localización dentro de la célula y su funcionamiento in vivo. 5
Las proteínas accesorias que intervienen en la arquitectura de la telomerasa son5:
La disquerina, el NHP2 y la NOP10 son 3 de las proteínas accesorias implicadas en la estabilidad y la acumulación de TER (RNA de la telomerasa humana). Por otro lado, la disquerina y GAR1 asociados a TER, posibilitan que la telomerasa sea funcional.5
A su vez, nos encontramos con dos ATPasas (pontina y reptina), que se asocian a TERT (región catalítica) en la fase S del ciclo celular. Su ausencia, dificulta la acumulación de la telomerasa, por lo cual son dos proteínas muy importantes para el montaje de la enzima. Además, se ha visto que también ayudan a la estabilidad de TER durante el ensamblaje de la telomerasa.5
Se cree que una vez terminado el acoplamiento de todos los componentes de la telomerasa, la pontina y la reptina se disocian, dejando libre la enzima activa en su actividad catalítica.5
Se cree que TCAB1 sería la proteína accesoria encargada de la ubicar a la telomerasa dentro de la célula.5
Estructuras en alta resolución de los subdominios TER Y TERT
TER
La longitud de TER puede variar dependiendo del organismo en el que nos fijemos: 150 nucleótidos en ciliados, 450 nucleótidos en vertebrados, y hasta 1300 nucleótidos en algunas levaduras. A pesar de esto, se ha descubierto que todas las subunidades TER contienen 2 estructuras secundarias conservadas: un dominio central de pseudonudo (“pseudoknot-template core domain”), catalíticamente esencial, y un tallo-bucle (“stem-loop element”), denominado CR4-CR5 en vertebrados. Dichas estructuras interaccionan directamente con TERT.9
Gracias a la comparación de secuencias genéticas codificantes para TERT de distintos organismos, se ha descubierto la existencia de un dominio muy conservado en cuanto a su organización y tamaño (de unos 1100 aminoácidos).9
Dicha región de la subunidad catalítica TERT está formada desde el sentido N-terminal hasta el C-terminal por9:
-Un dominio esencial de extensión N-terminal (“essential N-terminal extension domain” o TEN), el cual tiene afinidad por el DNA telomérico monocatenario. A su vez, contacta directamente con TPP1/TIN2.
-Una región de enlace flexible (“linker”). Es el sitio de unión de TEN y TRBD.
-Un dominio de unión a RNA (“RNA binding domain” o TRBD). Interacciona con la región tallo-bucle o CR4-CR5 de TER.
-Un dominio central de la transcriptasa inversa (RT), el cual posee homología estructural y funcional con la transcriptasa inversa retroviral.
Para esclarecer la estructura tridimensional de TERT se han utilizado escarabajos (Triboliumcastaneum). Teniendo en cuenta que no poseen el dominio TEN, la estructura resultante genera una forma de anillo gracias a que TRBD y CTE se acercan en el espacio y forman un túnel catalítico. El DNA se uniría a CTE y el molde de RNA a RT, posicionando el extremo 3’ del G-overhang en el túnel catalítico para la adición de nucleótidos.9
Estructuras tridimensionales de la telomerasa humana y de Tetrahymena
La estructura de las telomerasas se ha determinado mediante microscopía electrónica de partículas individuales (EM) en tinción negativa, con resolución de 25 Å.9
Se pueden observar diferencias a nivel de subunidades, a la vez que similitudes en la organización de TERT.9
La telomerasa humana consiste en un dímero, conteniendo cada monómero una subunidad TER y una subunidad TERT, además de proteínas accesorias. Dichos monómeros están unidos por una región de bisagra flexible. Los autores sugieren que la telomerasa humana debe ser dimérica para poder extender dos extremos teloméricos en paralelo, facilitando que las cromátidas hermanas presenten la misma longitud en sus telómeros.9
Por otro lado, la telomerasa de Tetrahymena es monomérica, y es funcional en esa forma. Contiene una subunidad TERT, una subunidad TER y proteínas accesorias. La subunidad TERT es próxima a la TER, y además, posee semejanzas estructurales en relación a la telomerasa humana.9
La replicación del DNA romosómico se realiza gracias a DNA polimerasas que pueden extender las cadenas a partir de un RNA cebador y en extremo 5’ → 3’. Dichos cebadores se reemplazarán por DNA y se unirán a las nuevas cadenas de DNA por las DNA ligasas.10
Cuando llegamos a los extremos del cromosoma se plantea un problema. La hebra cuyo molde es la cadena conductora 3’ → 5’, seguirá sintetizándose desde el último origen de replicación hacia el extremo final en dirección 5’ → 3’.10
Por otra parte, la hebra copiada a partir de la cadena retardada 5’ → 3’, contiene fragmentos de Okazaki. Se plantea la dificultad de que en el último fragmento de Okazaki, el RNA cebador no puede ser reemplazado por DNA, ya que al no haber una secuencia adyacente, no hay posibilidad de que actúen las DNA polimerasas. Debido a esto, la hebra sintetizada de la cadena retrasada, perderá entre 50 y 100 nucleótidos en su extremo 5’ con respecto a su molde.10
Para evitar la pérdida de información genética, ya se ha comentado que en los extremos 3’ de las hebras cromosómicas se encuentra el DNA telomérico rico en G, adyacente a la última secuencia de DNA que se puede replicar por la DNA polimerasa. Aunque el DNA telomérico contiene secuencias repetidas en tándem, podría desaparecer después de varios ciclos de replicación. Debido a esto, encontramos la enzima telomerasa, que va a prolongar la secuencia telomérica.10
En el primer paso de la síntesis de DNA telomérico, la telomerasa se recluta a través de la interacción de TPP1 de la shelterina con el dominio N-terminal de su subunidad catalítica TERT. Además, el extremo 3’ G-overhang del telómero se coloca en el sitio activo de TERT alineándose con el RNA (TER) a través de la formación de pares de bases.9 Este RNA es complementario a la cadena de DNA rica en G y aparea parcialmente con ella, proporcionando un molde para la síntesis de copias de la unidad repetida. Los desoxinucleótidos se añaden de novo al extremo 3’ de la cadena rica en G. Después de que se hayan añadido varios nucleótidos se produce una translocación de la telomerasa hacia el extremo del telómero y se reinicia el proceso.10
Se producen unos 10.000 pares de nucleótidos en el extremo 3’, siendo dicha hebra más larga que la complementaria. A su vez, esta elongación de la cadena retardada 5’ → 3’, sirve para producir un nuevo espacio para la creación de un fragmento de Okazaki: se sintetiza un RNA cebador, y la DNA polimerasa elonga la cadena en dirección 5’ → 3’.10
Explicación de la función de la telomerasa. Paniagua, R., Nistal, M., Sesma, P., Álvarez-Uría, M., Fraile, B., Anadón, R. & Sáez, F. J. (2002). Citología e histología vegetal y animal. [Figura].
En humanos, la telomerasa está activa en las células embrionarias pluripotenciales y las células madre germinales, sanguíneas o de tejidos adultos en continua renovación. Por otra parte, está reprimida en el tejido somático, limitándose así su capacidad de división. Sin embargo, en los procesos tumorales esta enzima se reactiva, permitiendo su proliferación y desarrollo.6,10
Los procariotas constan de un cromosoma circular, por lo que se podrá hacer una copia de todos los nucleótidos, ya que como la cadena no se interrumpe en ningún momento, el RNA cebador que se generó al comienzo de la copia, podrá ser sustituido por DNA una vez que la copia alcance el final de la cadena.10
Se ha visto que la actividad telomerasa puede encontrarse en las fases G1, S y G2 a lo largo de un ciclo celular. A su vez, se da una represión cuando las células entran en G0 debido a6:
Una falta de factores de crecimiento.
Inhibición por contacto de la división celular.
Inducción de la senescencia por reversión en una línea celular inmortalizada.
Diferenciación.
Regulación
La telomerasa se regula por factores genéticos, epigenéticos y ambientales.11
La regulación de la telomerasa se puede dar a través del factor TRF1, que actuaría como un supresor de la elongación telomérica. Su mecanismo de acción consiste en la unión al DNA telomérico de doble cadena, impidiendo la acción de la telomerasa. Dicho factor se encarga de establecer un feedback negativo que estabiliza la longitud telomérica en la interfase y la mitosis.12 Para que este proceso se lleve a cabo se ha descubierto que se necesita que TRF1 interactúe con TIN2.6
El cese de la actividad de la telomerasa en los tejidos embrionarios se puede dar a través de la represión del gen hTERT o a través de empalmes alternativos del transcrito hTERT, que daría lugar a proteínas sin acción transcriptasa inversa.13
Existen factores epigenéticos tales como metilación de islas CpG, metilación de histonas y acetilación, que son importantes para la transcripción de TERT.11
Cáncer
El cáncer es un conjunto heterogéneo de trastornos que se caracterizan por la presencia de células que no responden a los controles normales de la división, lo que hace que estas células cancerosas se dividan rápidamente y de forma continua, generando tumores que privan de nutrientes a los tejidos sanos.2
Varios autores afirman que en las células somáticas humanas, el potencial de proliferación restringido permite unas 50-70 divisiones celulares, alcanzando después la senescencia.5 El concepto de senescencia celular engloba a todas las respuestas que genera una célula mitóticamente competente frente a estímulos capaces de inducir su malignización, es decir, generar un cambio neoplásico en ella. Por tanto, se produce la transformación morfológica y funcional de la célula a un fenotipo senescente y se permite una detención de su crecimiento. Esto hace pensar que la senescencia es un mecanismo de seguridad que surgió para evitar la tumorigénesis en células que posean riesgos neoplásicos (como acortamiento/ alteración telomérica, daños DNA, proteína RAS mutada).14
Respecto al acortamiento telomérico mencionado anteriormente, se ha visto que en hongos, los telómeros cortos posibilitan el daño al DNA, la liberación factores de transcripción y la remodelación de la heterocromatina.14
Otros estudios han visto que si la reducción telomérica supera un límite mínimo de longitud, se crean impedimentos para una correcta separación cromosómica en la mitosis, debido a la aparición de asociaciones teloméricas (tas). Esto provoca inestabilidad cromosómica, que podrá dar lugar a errores genéticos, importantes en procesos neoplásicos como amplificación génica y pérdida de heterocigosidad. Las células que vayan acumulando tas pueden acabar perdiendo regiones repetitivas ricas en guanina, favoreciendo así dichos mecanismos neoplásicos.6
Las tasconsisten en la unión de los extremos de dos o más cromosomas, pero sin pérdida aparente de material genético. Pueden producirse en una cromátida o en ambas (simple o doble cromátida).6
Algunos autores proponen que la formación de tas estaría asociada a replicaciones defectuosas de los telómeros, traduciéndose en pérdidas de las secuencias teloméricas tras cada ciclo, aumentando así la probabilidad de producir fusiones cromosomales. Los mecanismos por los cuales esto ocurre apuntan al fallo de la enzima telomerasa, tal y como se ha visto en células de ratones.6
Las tas se han descrito tanto en tumores sólidos, como en neoplasias hematológicas. A su vez también se han encontrado en células infectadas por virus, células con fenotipo senescente y patologías genéticas como síndrome de Turner. Sin embargo, no se ha visto este fenómeno en células normales, ya que están protegidas de la unión entre cromosomas mediante sus telómeros.6
La mayor parte de los tumores malignos presentan la enzima telomerasa activa, lo que hace que las células adquieran la capacidad de proliferar de manera ilimitada. Sin embargo, esto no ocurre en los tumores benignos, que se caracterizan por la ausencia de la telomerasa.15
El promotor del gen de hTERT es rico en guanina y citosina y presenta numerosos sitios de unión para múltiples proteínas, por lo que su expresión está muy controlada. Por ejemplo, este gen presenta sitios para la unión de los dedos de zinc del factor de transcripción SP1 y para la unión de otras proteínas como USF1/2, MYC, MAX, MXD1 o el factor de transcripción TFII-I. Algunos factores que producen una estimulación de la expresión de hTERT son c-MYC, SP1 y los factores de transcripción ETS.16
Las mutaciones del gen hTERT se pueden producir en dos sitios de la secuencia en los que tienen lugar transiciones C-T, y ambas dan lugar a un mismo ácido nucleico de 11 pares de bases, que presenta una secuencia para la unión de los factores ETS. Aunque el papel de estos factores todavía no está muy claro, se sabe que la unión del ETS-2 al promotor de hTERT está asociada con la activación de la telomerasa mediada por el factor de crecimiento epidérmico (EGF) en el cáncer de pulmón.16
Por otro lado, también se cree hTERT podría tener otro papel en el cáncer, además de intervenir en el alargamiento de los telómeros. Se ha observado que el aumento de la expresión de TERT daba lugar a linfomas de linfocitos T en células del timo de ratones sin cambios significativos en la longitud de los telómeros. Asimismo, se ha visto que TERT se asocia indirectamente con genes como el de la IL-6, la IL-8 o el TNFα (factor de necrosis tumoral), los cuales intervienen en los procesos de inflamación y de progresión del cáncer.17
Por tanto, una de las mutaciones que se producen en muchos cánceres, como en glioblastomas, liposarcomas o melanomas primarios, es la mutación del promotor proximal del gen de TERT de la telomerasa. Sin embargo, no se conoce por qué la frecuencia de esta mutación es menor en otros cánceres bastante comunes, como el cáncer de pulmón, el cáncer de colon, el cáncer de ovario o el cáncer de próstata. Por tanto, se piensa que no es necesario que las células presenten telomerasa activa para que se desarrolle el cáncer, sino que solo se requiere algún mecanismo que mantenga la longitud de los telómeros.15
De hecho, en algunos cánceres en los que no se detecta la telomerasa existe un mecanismo alternativo de alargamiento de los telómeros, el cual recibe el nombre de ALT (alternative lengthening of telomeres). Además, los tumores telomerasa-positivos pueden pasar a tener el mecanismo ALT y, por tanto, convertirse en resistentes a los fármacos dirigidos frente a la telomerasa. Este mecanismo ALT se basa en copiar la secuencia telomérica mediante recombinación homóloga. Las células que lo presentan suelen tener una longitud de los telómeros muy variada y gran cantidad de ADN telomérico extracromosómico.18
El alargamiento alternativo de los telómeros se ha encontrado en osteosarcomas, tumores pancreáticos neuroendocrinos, tumores astrocíticos, leidomiosarcomas y sarcomas pleomórficos indiferenciados. Se cree que este mecanismo se activa cuando se producen mutaciones en las proteínas ATRX o DAXX y/o en la histona H3.3 o por fallos en su expresión, lo cual se ha observado en tumores del sistema nervioso central y en tumores pancreáticos neuroendocrinos.18 La histona H3.3 se incorpora a regiones teloméricas y sitios de transcripción y se asocia con cromatina activa, para lo cual requiere la presencia de ATRX y DAXX, lo que impide la recombinación telomérica. Si existe una mutación en una de estas proteínas, se alterará la incorporación de la H3.3 a la cromatina, lo que modificará la heterocromatina telomérica. Esto conducirá a una desestabilización de los telómeros y a un aumento de la recombinación homóloga en ellos, facilitándose así el mecanismo ALT. 18, 19, 20 Además, la mutación de los genes que dan lugar a ATRX o a DAXX puede aumentar la expresión del RNA TERRA, que se trata de un RNA no codificante que inhibe la actividad de la telomerasa. Este RNA TERRA también estimula la formación de un “loop R” en los telómeros, lo que induce ALT.17
Envejecimiento.
Uno de los procesos que contribuye al envejecimiento es el acortamiento de los telómeros.2 Se propone que el DNA telomérico, que protege los extremos cromosómicos de la recombinación, actuaría como un “reloj mitótico”, el cual induciría la salida del ciclo celular mediante la senescencia una vez que los telómeros se acorten.6
Los telómeros se pueden considerar metafóricamente como un “reloj mitótico”. Imagen sin copyright. Recuperada de: https://pixabay.com/.
Para la comprensión del proceso del envejecimiento, se han llevado a cabo estudios en los que se han empleado ratones que no tenían un gen funcional de la telomerasa, por lo que en ellos no se expresaba esta enzima. Se observó que los telómeros de estos ratones se iban acortando progresivamente y, tras varias generaciones, los animales presentaban signos de envejecimiento prematuro (caída del pelo, aparición de canas o retraso en cicatrización de heridas).2
Además, en el año 2012, científicos de Reino Unido realizaron una investigación sobre la longitud de los telómeros en pinzones y observaron que los pájaros que tenían telómeros más cortos eran menos longevos que los pájaros que tenían telómeros más largos.2
Otros estudios han revelado que la disfunción telomérica (relacionada con las proteínas de respuesta al daño del ADN) aumentan con la edad en preparaciones in vivo en la piel de primates y el hígado, tracto digestivo y pulmones de ratones.21
Por tanto, según estos resultados, podemos afirmar que hay cierta relación entre el envejecimiento y la longitud de los telómeros.
La longitud inicial de los telómeros puede variar entre distintos individuos. En mamíferos, la tasa de acortamiento de los telómeros es igual en todos los tejidos del mismo organismo (entre 50 y 200 nucleótidos por duplicación).6, 22
Los telómeros se van acortando progresivamente en cada ciclo celular y esto hace que llegue un momento en el que no se pueda unir el complejo de la shelterina, por lo que se desestabiliza el “loop T”. Esto provoca la exposición del extremo del cromosoma, el cual es reconocido por la maquinaria de reparación del DNA como una rotura de la doble cadena de DNA. De hecho, algunos estudios han demostrado que la eliminación del TRF2 del complejo de la shelterina provocaba el reclutamiento y la activación de proteínas como 53BP1, ATM o la histona H2AX, que están implicadas en la respuesta al daño del DNA. Además, se ha visto que esta respuesta al daño, se produce específicamente en la región telomérica cuando POT1 se separa del telómero. Por otro lado, dicha maquinaria reparativa del DNA también puede dar lugar a la activación de la proteína p53, que estimula la reparación del DNA, la detención del ciclo celular, la senescencia y la apoptosis.21
Se han hallado evidencias de que la senescencia puede estar relacionada con el envejecimiento a través de la acumulación de células con fenotipo senescente en los tejidos, ya que éstas pueden resistir más los estímulos apoptóticos. A su vez, dichas células expresan en mayor frecuencia algunas moléculas de secreción (metaloproteinasas de la matriz junto a otras enzimas capaces de degradar, factores de crecimiento, citocinas inflamatorias), las cuales pueden alterar el entorno local del tejido al que se vierten. La acumulación de dichas células y su hipersecreción provocan la destrucción de la integridad y la funcionalidad tisular.14
Debido a la perturbación tisular, se ha sugerido que las células senescentes puedan facilitar la expresión de fenotipos neoplásicos de otras células mutadas.14
La extensión telomérica ha sido diana de numerosos estudios. Se ha visto que esta longitud telomérica es muy variable entre individuos de la misma edad. Además, dicha longitud pasará a ser más heterogénea entre los diferentes tejidos en la vejez.11
Otro dato de interés que se ha encontrado es que, aunque no haya diferencias de longitud telomérica entre los recién nacidos varones y mujeres afroamericanos y caucásicos, a medida que estas poblaciones pasan a edad adulta, los individuos afroamericanos conservan unos telómeros más extensos que los caucásicos. Además, generalmente los varones adultos tienen los telómeros más cortos que las mujeres adultas. Se piensa que la tasa de acortamiento telomérico es menor en la población femenina debido a la acción estimulante del estrógeno sobre la telomerasa, tal y como se ha comprobado in vitro.11
La longitud de los telómeros estará determinada por factores genéticos y ambientales. Al poseer muchos residuos de G, los telómeros son más susceptibles al estrés oxidativo. Además, se ha comprobado que en células como las del endotelio, el estrés oxidativo disminuye en gran medida la actividad telomerasa. Por lo tanto, si se añaden antioxidantes, se enlentece el acortamiento telomérico al promover a la enzima telomerasa (comprobado en cultivos celulares).11
De esto se deduce lo siguiente: si evitamos el estrés psicológico, se produce menos estrés oxidativo, y por lo tanto prolongamos más la actividad telomerasa, así como los telómeros. Estudios han demostrado que eventos adversos, maltrato infantil, enfermedades crónicas, también parecen ser la causa de unos telómeros más cortos de cara al futuro.11
A: Longitud media telomérica en individuos con estrés alto y con estrés bajo. B: actividad media de la telomerasa en individuos con estrés alto y con estrés bajo. Epel, E. S., Blackburn, E. H., Lin, J., Dhabhar, F. S., Adler, N. E., Morrow, J. D., & Cawthon, R. M. (2004). Accelerated telomere shortening in response to life stress. [Figura]. Recuperado de https://www.ncbi.nlm.nih.gov/pmc/articles/PMC534658/pdf/pnas-0407162101.pdf
Existen datos sobre los hábitos de vida, que también están relacionados con la longitud telomérica11:
Los fumadores tienen más estrés oxidativo, por tanto, menor longitud telomérica.
El deporte influye de manera positiva sobre dicha longitud.
Personas que, debido a su dieta, presentaban niveles más bajos de ácido docosahexaenoico y ácido eicosapentaenoico, sufrían de un acortamiento telomérico más veloz.
Hábitos de vida que influyen en la longitud telomérica. Mientras que los individuos fumadores sufrirán de un acortamiento teloméricos precoz, los individuos deportistas conservarán mejor la longitud de sus telómeros. Imágenes sin copyright. Recuperadas de: https://pixabay.com/.
Patologías relacionadas
Disqueratosis congénita/ Síndrome de Zinsser-Engman-Cole
La disqueratosis congénita fue la primera patología en la que se identificaron mutaciones en la telomerasa humana. Dicha enfermedad cursa con: pigmentación anormal de la piel, distrofia de las uñas, y leucoplasia oral (placa blanca localizada en la mucosa oral que puede ser un factor de riesgo para el cáncer oral23). Además hay otro síntomas como tales como retraso en el desarrollo, atrofia testicular, pérdida prematura del cabello e incapacidad funcional de algunos órganos (siendo la deficiencia de la médula ósea, la principal razón de mortalidad prematura).5
En esta patología se ha detectado la mutación del gen DKC1, situado en el cromosoma X, dando lugar a la disqueratosis congénita ligada al cromosoma X. La consecuencia de esto es una sustitución de aminoácidos en la posición 353 de Alanina por Valina, afectando a la disquerina. Dicha proteína es 1 de las 3 de las proteínas accesorias implicadas en la estabilidad y la acumulación de TER (RNA de la telomerasa humana). Debido a esto los niveles de TER están disminuidos, y por tanto, sus telómeros están más reducidos que sus respectivos controles normales.24,25
Se crea inestabilidad cromosómica que afecta más a tejidos de rápida proliferación, tales como: médula ósea, piel y mucosa gastrointestinal.24
Es un síndrome hereditario que cursa en la disqueratosis congénita autosómica dominante.26
Aparte de los síntomas descritos anteriormente de una disqueratosis congénita, la anemia aplásica produce un trastorno hematológico que viene dado por: la reducción de los eritrocitos, fallo de médula ósea, y patologías hepáticas y pulmonares.5
Este tipo de anemia es ocasionado por una mutación en el gen hTR que codifica para TER.25,26 También se han visto mutaciones en TERT en la disqueratosis congénita autosómica dominante.27
Es una patología crónica y progresiva que cursa con una fibrosis pulmonar irreversible.5 Desde el diagnóstico, los pacientes viven de promedio 3 años.27
Las bases patológicas radican en la mutaciones de genes codificantes para TER y TERT.27
Dicha patología también se asocia con la disqueratosis congénita.27
Síndrome de Werner
El síndrome de Werner es producido por una mutación del gen WRN, que codifica para la helicasa Recq, que es necesaria para la replicación de los telómeros. Cuando esta enzima es defectuosa, los telómeros se acortan de forma prematura, lo que hace que aparezcan signos de envejecimiento en la adolescencia y primeras etapas de la adultez, como piel arrugada, encanecimiento, cataratas o atrofia muscular.2
En conclusión, las enfermedades mencionadas anteriormente poseen telómeros más cortos que los controles (sin patologías) con los que son comparados.
Dianas farmacéuticas
Las posibles dianas farmacéuticas en situaciones de cáncer (supresión de telomerasa) y de envejecimiento (activación de la telomerasa), vendrían resumidas en la siguiente imagen28:
La telomerasa es una buena diana terapéutica ya que está muy expresada en las células cancerígenas en comparación con el resto de las células (por ejemplo, las células somáticas tienen expresión casi nula o nula de la telomerasa) . Además, los procedimientos terapéuticos van encaminados a la subunidad catalítica TERT.28
Algunas potenciales terapias son28:
Inhibidores de oligonucleótidos: son oligonucleótidos antisentido o ácidos nucleicos modificados químicamente. Actúan inhibiendo la telomerasa, concretamente sobre la subunidad TER o TERT, o sobre proteínas asociadas. El acortamiento telomérico produce apoptosis o senescencia. Un ejemplo bastante prometedor es el Imetestalt, cuya secuencia oligonucleotídica es complementaria a TER (hTR o TERC) de la telomerasa, inhibiendo dicha subunidad. Dicho compuesto ha sido testado con éxito en glioblastoma (tumor cerebral).28
Estructura y lugar de acción del Imetelstat en la telomerasa. Jafri, M. A., Ansari, S. A., Alqahtani, M. H., & Shay, J. W. (2016). Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. [Figura]. Recuperado de https://link.springer.com/content/pdf/10.1186%2Fs13073-016-0324-x.pdf
Inhibidores de molécula pequeña: a partir de inhibidor natural epigalactocatequina-3-galato (EGCG). Por ejemplo, la rapamicina, inhibidor de mTOR, una proteína serín/treonín quinasa encargada de la localización de la subunidad TERT.29
Terapia génica dirigida a la telomerasa: dicha terapia se dirige al promotor de genes de la telomerasa de células cancerígenas. Se usan Adenovirus, que al usar el promotor de hTERT, son capaces de replicarse y matar a la célula cancerígena infectada.28
Fitoquímicos: moléculas naturales de plantas que tienen efecto inhibitorio de la telomerasa en algunos cánceres (alicina, curcumina, sibilina, etc…). El mecanismo de acción no se conoce del todo, pero se sugiere que afecta a TERT, ya sea en su expresión, actividad o disociación de la Hsp90 co-chaperona.28
Otro fármaco de importante mención es la telomestatina, un potente inhibidor de la telomerasa extraído de Streptomyces anulatus.30
Dicha molécula posee similitud estructural con el G-cuadruplexo. Su habilidad inhibitoria permite crear con mayor facilidad los G-cuadruplexos o estabilizarlos intramolecularmente, en caso de que ya estuvieran formados.30
Estructura del G-cuadruplexo (A) comparada con estructura de la telomestatina (B). Kim, M. Y., Vankayalapati, H., Shin-Ya, K., Wierzba, K., & Hurley, L. H. (2002). Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular G-quadruplex. [Figura]. Recuperado de https://pubs.acs.org/doi/pdf/10.1021/ja017308q
De este modo, la telomerasa no puede alargar el telómero porque no es capaz de desorganizar los G-cuadruplexos, y se produce senescencia de tipo Hayflick, que es el envejecimiento celular provocado por desgaste de telómeros.
Modelo tridimensional del acoplamiento telomestatina/G-cuadruplexo. Kim, M. Y., Vankayalapati, H., Shin-Ya, K., Wierzba, K., & Hurley, L. H. (2002). Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular G-quadruplex. [Figura]. Recuperado de https://pubs.acs.org/doi/pdf/10.1021/ja017308q
Conclusión
Los telómeros y la telomerasa son unas estructuras moleculares muy importantes a la hora de comprender procesos naturales como el envejecimiento, y procesos patológicos como el cáncer.
La integridad de los cromosomas en sus extremos está mantenida por la telomerasa, y el acortamiento de dichas zonas produce senescencia, envejecimiento o cáncer.
En muchas ocasiones, el cáncer presentará aumentada la actividad telomerasa, por lo que dicha enzima es una diana terapéutica bastante prometedora a la hora de intentar erradicar patologías de origen tumoral.
Hay que tener en cuenta que la telomerasa es un elemento destacado en todos los procedimientos relacionados con los telómeros, pero no hay que olvidar la interacción que ejercen otros factores conocidos y los que faltan por descubrir.
De cara al futuro, la investigación en este campo permitirá una mejor comprensión global de los telómeros, con importantes repercusiones clínicas.
Bibliografía
Chuaire, L. (2006). Telómeros y telomerasa: breve recuento de una historia iniciada por Hermann Müller y Barbara McClintock. Colombia Médica, 37(4), 332-336.
Pierce, B. A. (2016). Genética: Un enfoque conceptual. Madrid, España: Ed. Médica Panamericana.
Hernández Fernández, R. A. (1999). Telómeros y telomerasas. Revista Cubana de Investigaciones Biomédicas, 18(2), 121-129.
Blackburn, E. H. (1991). Structure and function of telomeres. Nature, 350(6319), 569.
Gómez, D. E., Armando, R. G., & Farina, H. G. (2014). Telomerasa y telómero: su estructura y dinámica en salud y enfermedad. Medicina (Buenos Aires) 74, 69-76
Cottliar, A. S., & Slavutsky, I. R. (2001). Telómeros y actividad de telomerasa: su participación en el envejecimiento y el desarrollo neoplásico. Medicina (Buenos Aires), 61, 335-42.
Maciejowski, J., & de Lange, T. (2017). Telomeres in cancer: tumour suppression and genome instability. Nature reviews Molecular cell biology, 18(3), 175.
Shay, J. W. (2018). Telomeres and aging. Current opinion in cell biology, 52, 1-7.
Sandin, S., & Rhodes, D. (2014). Telomerase structure. Current opinion in structural biology, 25, 104-110.
Paniagua, R., Nistal, M., Sesma, P., Álvarez-Uría, M., Fraile, B., Anadón, R. & Sáez, F. J. (2002). Citología e histología vegetal y animal. Madrid, España: McGraw-Hill Interamericana.
Zhu, H., Belcher, M., & Van Der Harst, P. (2011). Healthy aging and disease: role for telomere biology? Clinical science, 120(10), 427-440.
Van Steensel, B., & De Lange, T. (1997). Control of telomere length by the human telomeric protein TRF1. Nature, 385(6618), 740.
Ulaner, G. A., Hu, J. F., Vu, T. H., Giudice, L. C., & Hoffman, A. R. (1998). Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer research, 58(18), 4168-4172.
Pardo Andreu, Gilberto, & Delgado Hernández, René. (2003). Senescencia celular y envejecimiento. Revista Cubana de Investigaciones Biomédicas, 22(3), 204-212.
Shay, J. W. (2016). Role of telomeres and telomerase in aging and cancer. Cancer discovery, 6(6), 584-593.
Jafri, M. A., Ansari, S. A., Alqahtani, M. H., & Shay, J. W. (2016). Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome medicine, 8(1), 69.
Akincilar, S. C., Unal, B., & Tergaonkar, V. (2016). Reactivation of telomerase in cancer. Cellular and Molecular Life Sciences, 73(8), 1659-1670.
Shay, J. W., Reddel, R. R., & Wright, W. E. (2012). Cancer and telomeres—an ALTernative to telomerase. Science, 336(6087), 1388-1390.
Heaphy, C. M., De Wilde, R. F., Jiao, Y., Klein, A. P., Edil, B. H., Shi, C., … & Offerhaus, G. J. (2011). Altered telomeres in tumors with ATRX and DAXX mutations. Science, 333(6041), 425-425.
Schwartzentruber, J., Korshunov, A., Liu, X. Y., Jones, D. T., Pfaff, E., Jacob, K., … & Hovestadt, V. (2012). Driver mutations in histone H3. 3 and chromatin remodelling genes in paediatric glioblastoma. Nature, 482(7384), 226.
Victorelli, S., & Passos, J. F. (2017). Telomeres and cell senescence-size matters not. EBioMedicine, 21, 14-20.
Bernadotte, A., Mikhelson, V. M., & Spivak, I. M. (2016). Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging (Albany NY), 8(1), 3.
Moles, M. G., & González-Ruiz, L. (2018). Leucoplasia oral, una revisión de los aspectos esenciales de su diagnóstico y tratamiento. Actualidad médica, 103(803), 44-46.
Smoje, G., Dal Borgo, A., Cuevas, M., Núñez, L., Bolte, C., & Martinez, W. (2004). Disqueratosis congénita ligada al cromosoma X. Revista chilena de pediatría, 75(6), 547-550.
Núñez Quintana, A., Nordet Carrera, I., Menéndez Veitía, A., & González Otero, A. (2004). Neutropenias congénitas. Revista Cubana de Hematología, Inmunología y Hemoterapia, 20(1)
Vulliamy, T., Marrone, A., Dokal, I., & Mason, P. J. (2002). Association between aplastic anaemia and mutations in telomerase RNA. The Lancet, 359(9324), 2168-2170.
Armanios, M. (2012). Telomerase and idiopathic pulmonary fibrosis. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 730(1-2), 52-58.
Jäger, K., & Walter, M. (2016). Therapeutic targeting of telomerase. Genes, 7(7), 39.
Miwa, S., & Saretzki, G. (2017). Telomerase and mTOR in the brain: the mitochondria connection. Neural regeneration research, 12(3), 358.
Kim, M. Y., Vankayalapati, H., Shin-Ya, K., Wierzba, K., & Hurley, L. H. (2002). Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular G-quadruplex. Journal of the American Chemical Society, 124(10), 2098-2099.