Los tratamientos conocidos hasta el momento contra la enfermedad del Alzheimer
Por Andrea Rufat Verdú y Ana Verdugo Abril. Grado en Biología Sanitaria, Universidad de Alcalá
- Alzheimer y su histopatología
La enfermedad de Alzheimer (EA) es un trastorno neurológico que provoca la muerte de las células nerviosas del cerebro. A medida que avanza la enfermedad, se van deteriorando las capacidades cognitivas, entre ellas la capacidad para tomar decisiones y llevar a cabo las tareas cotidianas. Además, pueden surgir modificaciones de la personalidad, así como conductas problemáticas. En sus etapas avanzadas, la enfermedad de Alzheimer conduce a la demencia y finalmente a la muerte. [1] Siendo esta enfermedad una de las principales causas de muerte a nivel mundial. Además, se espera que esto aumente en los próximos años por lo que sería interesante conseguir un diagnóstico temprano para la enfermedad, así como, un tratamiento eficaz.
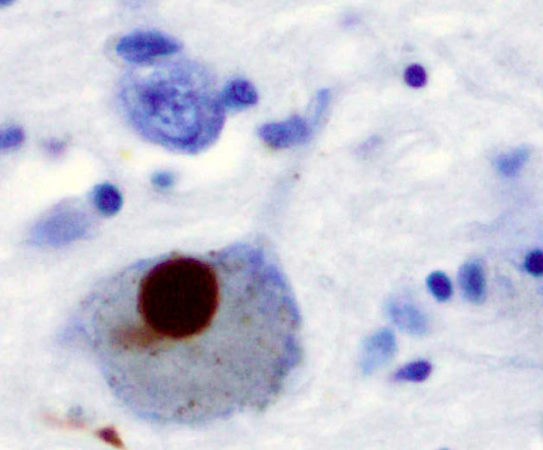
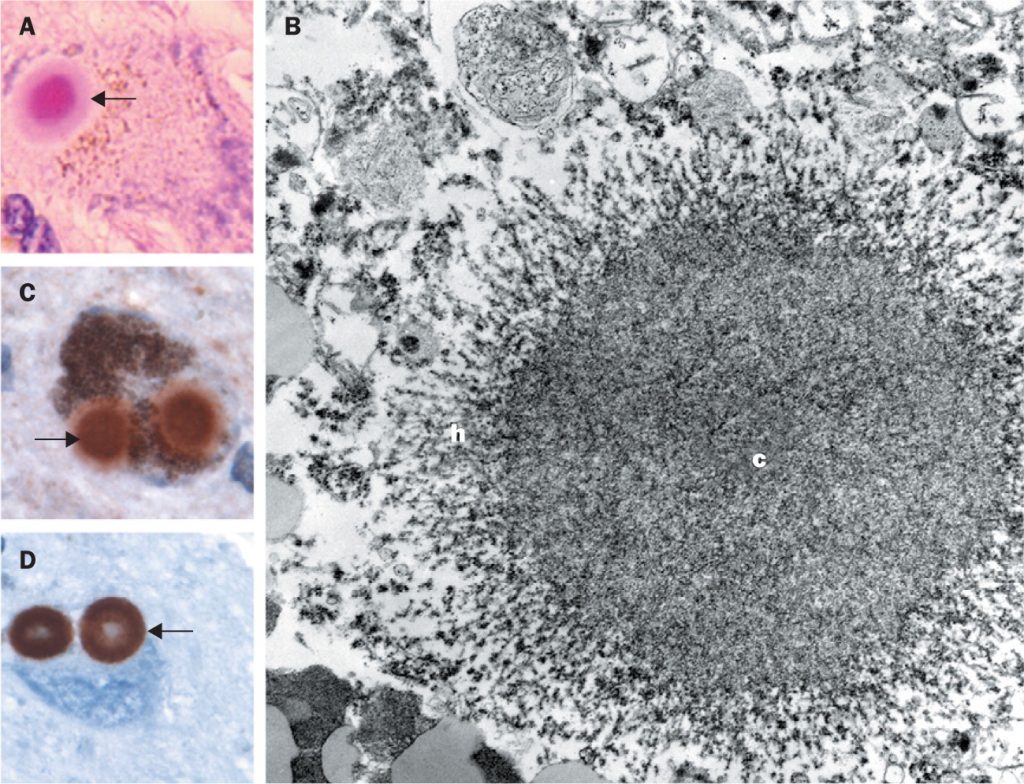
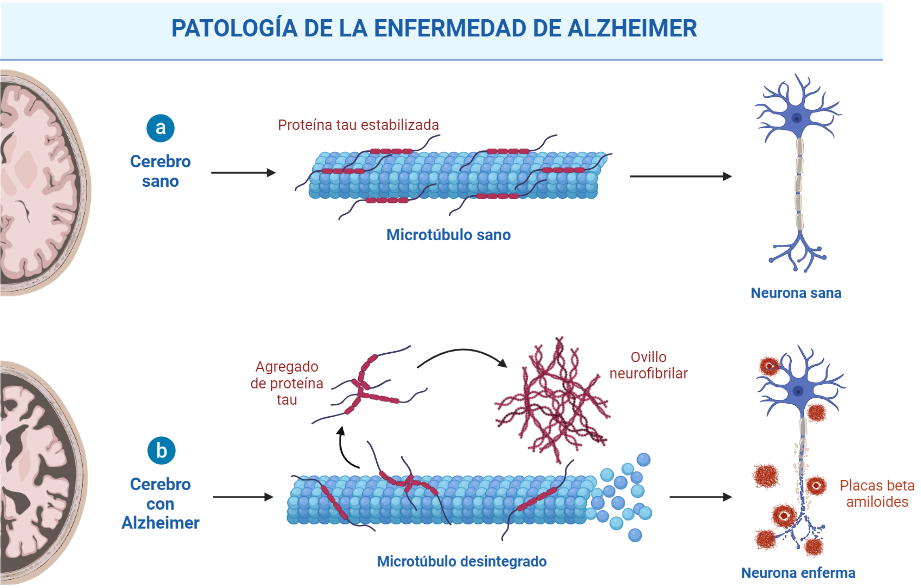
A nivel neuropatológico, la EA se caracteriza principalmente por la acumulación del péptido β-amiloide agregado en forma de placa (placas amiloides o placas de Aβ),y por la presencia de ovillos neurofibrilares (NFTs) que contienen a la proteína tau hiperfosforilada y agregada [2].
Las placas β-amiloides son depósitos extracelulares de proteínas, principalmente el péptido β-amiloide en su forma insoluble, mientras que los ovillos neurofibrilares son depósitos intraneuronales de la proteína asociada a microtúbulos Tau. Pese a que ambas son manifestaciones de la enfermedad del Alzheimer, la primera es característica también de otras patologías, como la arteriosclerosis, y en general del proceso de envejecimiento. [3]
Existen dos tipos de EA, que se diferencian en la edad de inicio, en la causa principal que provoca su aparición e incidencia. Sin embargo, comparten los mismos síntomas y lesiones histopatológicas.
Así encontramos la EA familiar o de inicio temprano, que afecta a individuos menores de 65 años, se asocia con la herencia mendeliana y representa alrededor del 5% de los casos de EA; y la EA esporádica o de inicio tardío, sin un modo de transmisión consistente que afecta a las personas mayores de 65 años y representa el mayor número de casos entre las personas mayores (90-95% de los casos de la EA). [2]
Created with BioRender.com
2. Intervenciones terapéuticas
Los tratamientos disponibles hasta el momento para la EA pueden lograr una mejoría sintomática y en la calidad de vida de los pacientes, pero ninguno consigue revertir, frenar o curar la fatal progresión de la enfermedad [4].
2.1 Tratamientos farmacológicos
En su tratamiento se encuentran los inhibidores de la enzima acetilcolinesterasa (IACE): donepezilo, rivastigmina y galantamina y los inhibidores de glutamato como el namenda (memantina), además de plantas como Melissa officinalis y Gingko biloba como técnicas de la Medicina Natural China. [5] También se ha estudiado el posible uso de la Huperzina A, un alcaloide natural que atraviesa la barrera hematoencefálica con un posible papel como neuroprotector [6].
Los IACE inhiben la acción de una enzima que destruye la acetilcolina, un químico cerebral implicado en la memoria y otros procesos cognitivos y afectivos, por lo que su consecuencia será el aumento de la acetilcolina. Se ha observado que la enfermedad de Alzheimer afecta desde muy temprano a las neuronas que producen acetilcolina, de ahí que una de las primeras estrategias terapéuticas ha sido crear fármacos que impidan la degradación de la acetilcolina. [5] Estos medicamentos protegen contra el estrés oxidativo y la toxicidad amiloide,
pero son costosos y pueden dañar las membranas neuronales [6].
Por otro lado, los inhibidores de glutamato actúan sobre este neurotransmisor que se produce en grandes cantidades por las células dañadas por Alzheimer de forma que se adhiere a receptores NMDA que aceleran el daño celular. [5]
También se pueden usar como tratamiento combinaciones de medicamentos como Namzaric que contiene tanto donepezilo como memantina.
Asimismo, se ha investigado acerca del uso de los receptores muscarínicos M1 como blancos terapéuticos en el tratamiento de la enfermedad. Por ejemplo, la xanomelina es un agonista tanto de receptores M1 como M4 que atraviesa la barrera hematoencefálica. Se ha demostrado que mejora la función cognitiva, sin embargo como cualquier fármaco tiene efectos secundarios indeseables, en concreto, sobre el sistema gastrointestinal y cardiovascular. [6]
2.2 Otros remedios: antioxidantes, antiinflamatorios…
En la literatura científica se ha asociado la neurotoxicidad del péptido ß-amiloide a la generación de radicales libres desencadenando así un estado de estrés oxidativo con daños celulares como la peroxidación lipídica. [7] El cerebro es un órgano particularmente vulnerable a este tipo de daños por estrés oxidativo, debido a su alto consumo de oxígeno, o bajos niveles de antioxidantes, entre otros. [8] Por ello, no se descarta el uso de antioxidantes como tratamiento o incluso, el hábito saludable de una buena dieta mediterránea, puesto que pueden prevenir la degeneración neuronal eliminando especies reactivas de oxígeno (ROS) o previniendo su formación. Algunos son la vitamina E, la selegilina o la melatonina. [6]
Cabe mencionar el uso de antiinflamatorios para la inflamación generada por las placas seniles, o el de quelantes de hierro como la desferroxamina. Aunque ambos palian los efectos de la EA, pueden causar reacciones adversas como toxicidad en la retina o en el hígado. [6]
La curcumina, sustancia presente en la cúrcuma, también tiene un papel importante en la prevención y el tratamiento de esta enfermedad. Cuenta con propiedades antioxidantes, lipofílicas e incluso antiinflamatorias que permiten la mejora de las funciones cognitivas en los pacientes con Alzheimer. [9]
En la patogénesis de la EA se observa que el péptido Αβ puede interrumpir los canales de calcio en la membrana, provocando una pérdida del flujo (o influjo) que conlleva al desequilibrio del ion y así a altas concentraciones de calcio intracelular. Las concentraciones de calcio demasiado altas o bajas tienen efectos tóxicos, como lo es la alteración sobre la producción y transmisión de neurotransmisores en las células nerviosas. Finalmente esto conlleva a un proceso de muerte celular. Es por eso que los antagonistas del calcio también han sido utilizados como intervención terapéutica. Un ejemplo es el del nimodipino, que inhibe el influjo de calcio y mejora la circulación sanguínea cerebral. [6]
2.3 Inmunoterapias
Como se ha mencionado con anterioridad, el péptido Αβ amiloide tiene un papel clave en esta enfermedad ya que es neurotóxico, altera la función sináptica y produce neurodegeneración. [4] Por lo tanto, las estrategias dirigidas a Aβ podrían frenar eficazmente la progresión de la EA. En la actualidad, los mecanismos de acción de los fármacos anti-Aβ incluyen principalmente la reducción de la producción de Aβ, la prevención de la agregación de Aβ y la promoción de la eliminación de Aβ. [10]
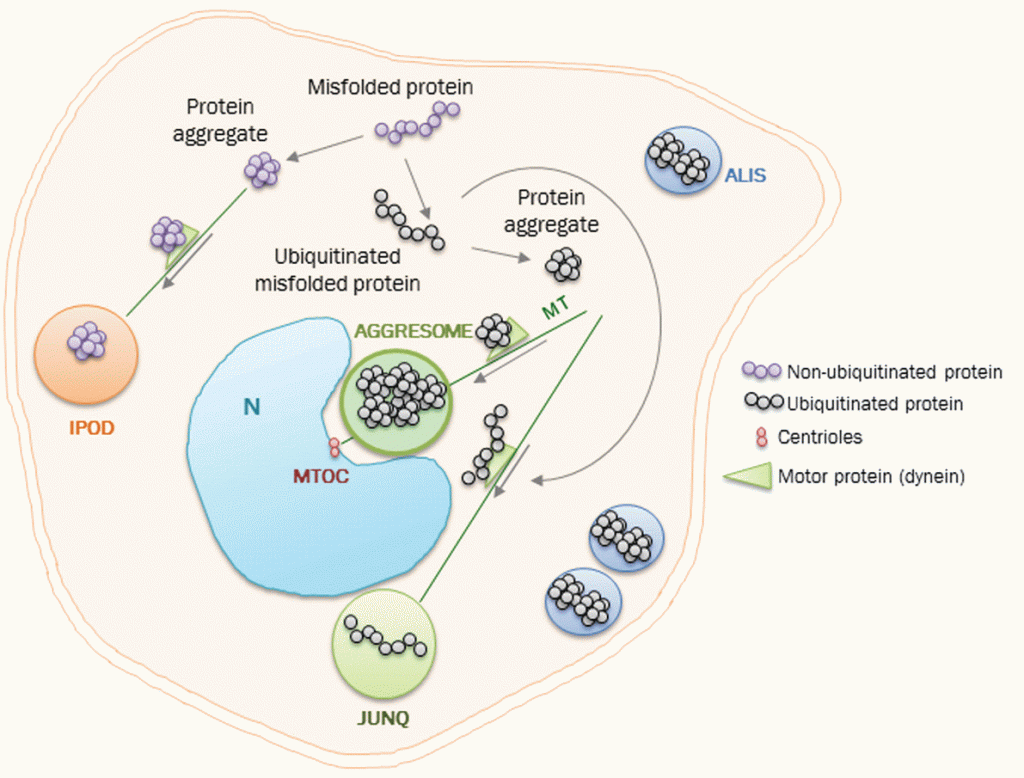
Las inmunoterapias anti-Aβ más elaboradas son las vacunas y los anticuerpos exógenos, conocidas como inmunoterapia activa y pasiva, respectivamente. La inmunización activa estimula el sistema inmunitario mediante la administración de Aβ o sus fragmentos, lo que desencadena una respuesta inmunitaria para producir anticuerpos endógenos contra Aβ. Sin embargo, estas vacunas presentan una baja reactividad y la aparición de reacciones adversas dependientes de células T, por lo que en la actualidad se desarrolla la inmunoterapia pasiva utilizando anticuerpos monoclonales humanizados o inmunoglobulinas policlonales para promover la eliminación de Aβ [10].
En cuanto a inmunización activa se han desarrollado varias vacunas como AN1792, amilomotida y UB-311.
Aducanumab, donanemab, lecanemab, solanezumab, crenezumab y gantenerumab son anticuerpos monoclonales humanizados que se están estudiando como tratamientos de la enfermedad de Alzheimer.
Recientemente ha sido aprobado el anticuerpo monoclonal Lecanemab en Estados Unidos. Es el primer fármaco que realmente modifica el curso de la enfermedad, pues reduce hasta un 27% el empeoramiento de los síntomas del Alzheimer después de administrarse durante 18 meses [11].
Los péptidos Aβ existen en varios estados conformacionales, incluidos monómeros solubles, agregados solubles de tamaño creciente y fibrillas y placas insolubles. Los agregados de Aβ solubles, como las protofibrillas de Aβ, son más tóxicos que los monómeros o las fibrillas insolubles. El modo de acción del Lecanemab consiste en unirse a las protofibrillas beta amiloides, por tanto, es capaz de reducir los niveles de beta amiloide patógeno (Aβ) y prevenir el depósito de Aβ. [12]
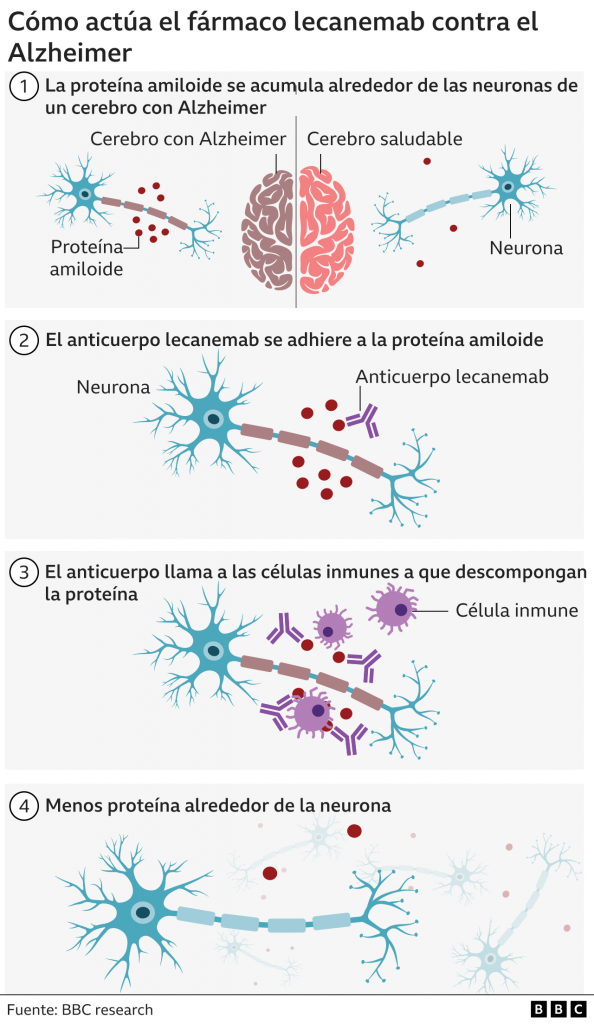
Tomada de: BBC News Mundo (2022) «Alzheimer: el medicamento aclamado como un avance trascendental en la lucha contra la enfermedad», BBC News Mundo, 30 noviembre. Disponible en: https://www.bbc.com/mundo/noticias-63806450.
Como ya se ha explicado, los ovillos neurofibrilares están formados por la proteína tau anormalmente fosforilada. Esta es una proteína citoplasmática que puede estabilizar los microtúbulos a través de la unión a la tubulina durante su polimerización en condiciones normales. Sin embargo, en la EA está hiperfosforilada, lo que implica que tenga una capacidad reducida para unirse a los microtúbulos y que, en ocasiones, cause la formación de ovillos neurofibrilares y la generación de agregados. Al igual que sucede con los péptidos Αβ amiloides, también existen estrategias anti-tau que previenen la fosforilación anormal de tau, inhiben la agregación de tau y promueven la eliminación de agregados de tau. Actualmente, la mayoría de los agentes anti-tau en ensayos clínicos son inmunoterapias. [10]
Aβ y la proteína tau fosforilada son reconocidos por receptores en la superficie de la microglía, lo que promueve la liberación de factores inflamatorios en los cerebros con EA. Los factores inflamatorios, a su vez, aumentan la formación de depósitos de Aβ y ovillos neurofibrilares, creando así un círculo vicioso que exacerba el proceso de la enfermedad. Existen genes como TREM2 que se sobreexpresan en la microglía y que se pueden utilizar como blancos en tratamientos de inmunoterapia como es el caso del anticuerpo monoclonal AL002. [10]
3. Conclusiones
- Es imprescindible el estudio de la neuropatología del Alzheimer con el fin de valorar posibles dianas farmacológicas que nos permitan desarrollar futuros fármacos eficaces contra la enfermedad.
- A día de hoy no se conoce ningún fármaco aprobado capaz de mejorar la EA al 100%. Sin embargo, muchos pueden retrasar el progreso de la enfermedad, como lo son aquellos aprobados por la FDA (U.S Food and Drug Administration). Incluyendo en este grupo al Lecanemab como el último fármaco aprobado por dicha organización, capaz de reducir hasta un 27% el empeoramiento de la enfermedad del Alzheimer.
- La EA no se trata únicamente con la administración de medicinas, sino que también es importante que las personas que la padezcan adopten en su vida cotidiana hábitos saludables, como una dieta mediterránea o la incorporación de especias como la cúrcuma gracias a los efectos de la curcumina. Porque pese a que no tengan un gran efecto sobre el progreso de la enfermedad, son capaces de colaborar en su mejoría.
- La histopatología de la enfermedad de Alzheimer se debe principalmente a la formación de placas amiloides y ovillos neurofibrilares, los cuales son las principales dianas de los tratamientos de esta enfermedad. Sin embargo, la patogénesis de la EA también puede ser tratada consiguiendo un aumento de acetilcolina, disminuyendo el exceso de glutamato y fomentando la actividad antiinflamatoria de la microglía y suprimiendo la proinflamatoria.
- Cabe destacar que la inmunoterapia actualmente está en pleno desarrollo como una de las formas más efectivas para conseguir un tratamiento para la cura del Alzheimer. Por otro lado, es necesario seguir con la investigación en este campo ya que muchos de los anticuerpos monoclonales y vacunas que se han conseguido han fracasado en sus últimas fases de ensayo.
- No solo es necesario encontrar un buen tratamiento para la EA, sino que también hay que seguir investigando para contar con un diagnóstico temprano, ya que estos fármacos resultan más efectivos con una temprana administración.
4. Bibliografía
[1]: Del Huerto Paredes, N. M., Nissen, M. D., Parquet, C. A. & Romano, M. F., 2007. ENFERMEDAD DE ALZHEIMER. Revista de Posgrado de la VIa Cátedra de Medicina, Issue 175, pp. 9-12.
[2]: Lara Ureña, N. (2020) Papel de HIF1 y PHD3 en la microglía de la enfermedad de Alzheimer. Universidad de Sevilla.
[3]: Heras Garvín, A. (2015) Microglía e hipoxia: Implicaciones en la enfermedad de Alzheimer. Universidad de Sevilla.
[4]: Rojas Delgado, K., Salazar Nassar, J. y Torrealba Acosta, G. (2019) “Alzheimer e Inmunoterapia: revisión de tres anticuerpos monoclonales humanizados dirigidos contra el Aβ amiloide (bapineuzumab, solaneuzumab y aducanumab)”, Revista Médica de Costa Rica, 84(627), pp. 2–7.
[5]: Gómez Tejeda, J. J., Hernández Pérez, C. y Iparraguirre Tamayo, A. E. (2020) “Tratamientos paliativos en la Enfermedad de Alzheimer”, 16 de Abril, 59(275) p. 727.
[6]: Cabrera, M.J.A. et al. (2014) «Patogenia y tratamientos actuales de la enfermedad de Alzheimer», Revista Cubana de Farmacia, 48(3), pp. 508-518.
[7]: Manzano-León, N. y Mas-Oliva, J. (2006) «Estrés oxidativo, péptido β-amiloide y enfermedad de Alzheimer», Gaceta Medica De Mexico, 142(3), pp. 229-238.
[8]: Bello-Medina, P. C. et al. (2022) “Estrés oxidativo, respuesta inmune, plasticidad sináptica y cognición en modelos transgénicos de la enfermedad de Alzheimer”, Neurología (English Edition), 37(8), pp. 682–690
[9]: Mishra, S. y Palanivelu, K. (2008) «The effect of curcumin (turmeric) on Alzheimer′s disease: An overview», Ann Indian Acad Neurol, 11(1), pp. 13-19.
[10]: Song, C., Shi, J., Zhang, P. et al. (2022) «Immunotherapy for Alzheimer’s disease: targeting β-amyloid and beyond», Translational Neurodegeneration, 11(18).
[11]: Periódico, E. (2023) «EEUU da luz verde al lecanemab, el fármaco que ralentiza el avance del alzhéimer», elperiodico, 6 enero. Disponible en: https://www.elperiodico.com/es/sociedad/20230106/eeuu-aprueba-lecanemab-farmaco-alzheimer-eisai-biogen-80796419.
[12]: Lecanemab: Uses, Interactions, Mechanism of Action | DrugBank Online [en línea], (sin fecha). DrugBank Online | Database for Drug and Drug Target Info. Disponible en: https://go.drugbank.com/drugs/DB14580