La evolución es un hecho, no un relato. Y las moléculas nos hablan de ello.
escrito por C. Menor-Salvan | 17 diciembre, 2024
C. Menor-Salván 12/2024
Creacionismo y evolucionismo no son creencias o hipótesis alternativas; ni siquiera son conceptos establecidos con las mismas reglas. Son los magisterios no solapados tal como explicaba Stephen Jay-Gould. Así, un supuesto debate o confrontación entre supuestos «científicos» creacionistas y «creyentes en la teoría de la evolución» no tiene sentido.
En primer lugar, ¿de qué hablamos cuando hablamos de «teoría»? La evolución es un fenómeno natural. Por lo tanto, es observable. Sobre un fenómeno natural, los científicos construimos un marco teórico que permite explicar las observaciones y realizar predicciones; éste se actualiza al ir afinando nuestra capacidad para realizar observaciones y experimentos. Pero el hecho ocurre estemos nosotros o no para observarlo y construir hipótesis, modelos o teorías para explicarnoslo.
En todas las teorías hay puntos de especial dificultad. En el marco teórico sobre la evolución tenemos, por ejemplo, la cuestión del origen de la vida, donde observamos que las moléculas de la vida, y sus antecesoras, también siguen reglas de selección, adaptación y supervivencia que aún estamos entendiendo.
No creemos en la evolución; la evolución es un hecho observable
Usando un ejemplo quizá mas fácil de entender: la teoría que explica el funcionamiento de las enzimas también tiene puntos oscuros y aún hay discusión científica en torno a ello. Pero nadie pone en duda que las enzimas existen y su acción es un hecho, sea cual sea la teoría que construyamos para explicarlo. Este marco teórico ha ido cambiando, desde el obsoleto modelo de llave-cerradura hasta modelos como Circe o el de estabilización del estado de transición.
Los científicos no «creemos» en la evolución, del mismo modo que no «creemos» en la enzimas. Son fenómenos reales que tratamos de comprender y explicar. Decir «creo en la evolución» es, simplemente, absurdo.
Como tal hecho observable, las ideas en torno a la evolución no son algo nuevo. Según el historiador romano Diógenes Laercio, Anaxágoras de Clazomene enseñaba que
«los seres vivos se formaron de la humedad, el calor y sustancias terrosas; despues, se propagaron por generación unos de otros».
Esta idea evolutiva rudimentaria, lanzada por Anaximandro de Mileto, el maestro-abuelo de Anaxágoras, hace 2500 años, creaba una disonancia cognitiva con el creacionismo, lo que llevó a San Hipólito de Roma a recogerla en sus «Refutaciones de todas la Herejías«. Gracias a ello, tenemos testimonio de las ideas de la escuela de Anaximandro.
Aún hoy, la evolución es considerada pecaminosa por muchos grupos religiosos. Yo mismo he sido testigo de alguna manifestación en contra de la evolución en EEUU, recibida con bastante humor por parte de los científicos y estudiantes que estábamos en el campus, hay que decir.
Observando la evolución sin salir de tu barrio
La evolución no sólo es observable, sino que la llevamos utilizando siglos en nuestro beneficio. Basta comparar las variedades de lechuga cultivada que podemos encontrar en el supermercado, con su pariente silvestre más cercano y, posiblemente, su ancestro: la amarga e indigesta, aunque comestible lechuga silvestre (Lactuca serriola).
La lechuga silvestre. Antepasado de la lechuga cultivada. Foto: Olga Pokotilo/PlantNet
Todo el proceso de domesticación de plantas y animales llevado a cabo por los humanos durante milenios debería probar, en sí mismo, que la evolución es un hecho. Y no es exclusivo de los humanos. En la evolución de la vida terrestre, unas especies han ejercido presión selectiva sobre otras, condicionando su evolución en una compleja red.
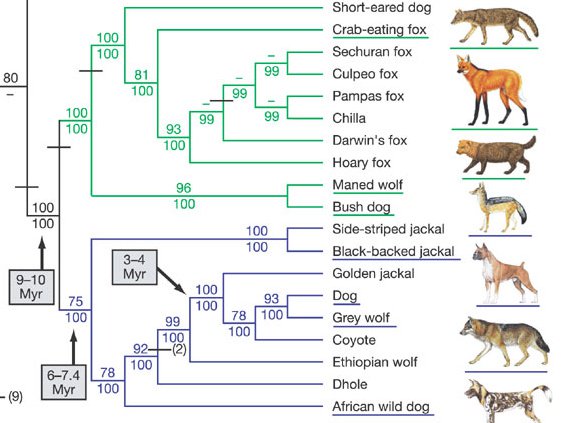
El perro original, que surgió de un ancestro común con el lobo gris actual, se ha diversificado un complejísimo arbol filogenético con todas las razas de perro doméstico. Un ejemplo de evolución divergente impulsada por la presión de selección ejercida por humanos. ¿cual es la frontera entre raza y nueva especie?
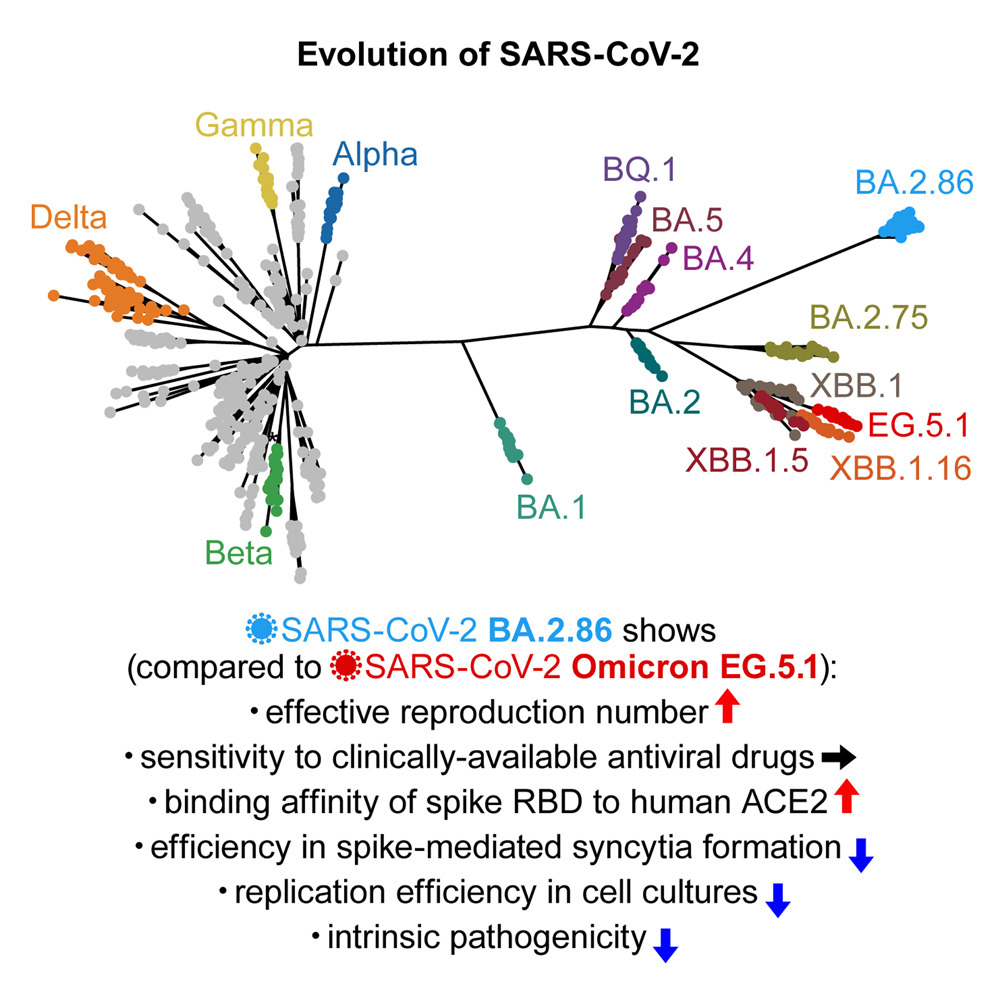
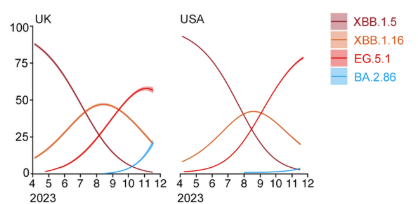
Tenemos un hecho evolutivo aún mas reciente: la pandemia de SARS-CoV-2. Nunca antes se había seguido la evolución de una especie viral tan de cerca y en tiempo real. Desde el inicio de la pandemia, gracias a la velocidad de replicación de los virus, fueron surgiendo variantes, creándose un complejo árbol filogenético. Durante el proceso, muchas variantes se extinguieron y otras han sobrevivido. Actualmente, tanto el virus como la enfermedad COVID son diferentes a los que existían en marzo de 2020. Este proceso de diversificación se basa en pequeños cambios en las estructuras moleculares que ocurren, inevitablemente, durante la replicación. Estos cambios, a veces, son silenciosos, si se mantiene la funcionalidad estructural; pero, a veces, llevan a las variantes a su desaparición, si la funcionalidad de las estructuras disminuye en las condiciones de ese momento, o a su prevalencia, si le aportan una funcionalidad ventajosa en las condiciones del momento. Con el tiempo, puede llegar a surgir una nueva especie.
Arbol filogenético del SARS-CoV-2. Evolución en vivo. Fuente: DOI 10.1016/j.chom.2024.01.001Dinámica de poblaciones de variantes virales. Estas curvas guardan una gran semejanza con las curvas de poblaciones de moléculas en procesos químicos como reacciones metabólicas o evolución química.
Ante todas las evidencias en torno a la evolución de la vida terrestre, la idea del creacionismo y el diseño inteligente se adaptan y, lejos de ser la herejía que añadía San Hipólito, la evolución se vuelve compatible con la idea religiosa de la creación. Así, en el diálogo público, el creacionismo se muestra con frecuencia como un _Deus Ex Machina_ que rellena convenientemente puntos oscuros en nuestra comprensión sobre la vida y el universo, dejando resueltas cuestiones espinosas, como el origen de la vida o del universo. Ello se combina a veces con una visión incorrecta sobre la evolución, cayendo en la trampa teleológica de que conduce hacia la inteligencia como máxima expresión de un proceso perfeccionador, puesto en marcha a partir de unas constantes físicas ajustadas cuidadosamente por un «diseñador» divino con este fin (el principio antrópico fuerte).
Sin embargo, la evolución biológica no es un proceso de mejora dirigido hacia un destino final. Su base es la preservación de estructuras supramoleculares funcionales, que sostienen la continuidad de la vida y que surgieron también mediante un proceso de evolución prebiótica.
La biología molecular es un tratado sobre la evolución
La biología molecular nos ofrece muchos ejemplos para ilustrar la evolución. Dos de los más interesantes son el ribosoma, cuya estructura relata la evolución de la vida desde su origen, y las polimerasas de DNA y RNA, que conectan todos los organismos y cuentan la historia evolutiva de los virus. El problema es que explicar brevemente para el público los complejos detalles moleculares de la evolución no es trivial. Voy a intentarlo, sin embargo, con un ejemplo sencillo (y simplificado), relacionado con un suceso que, desgraciadamente, se repite todos los inviernos: la toxicidad del monóxido de carbono.
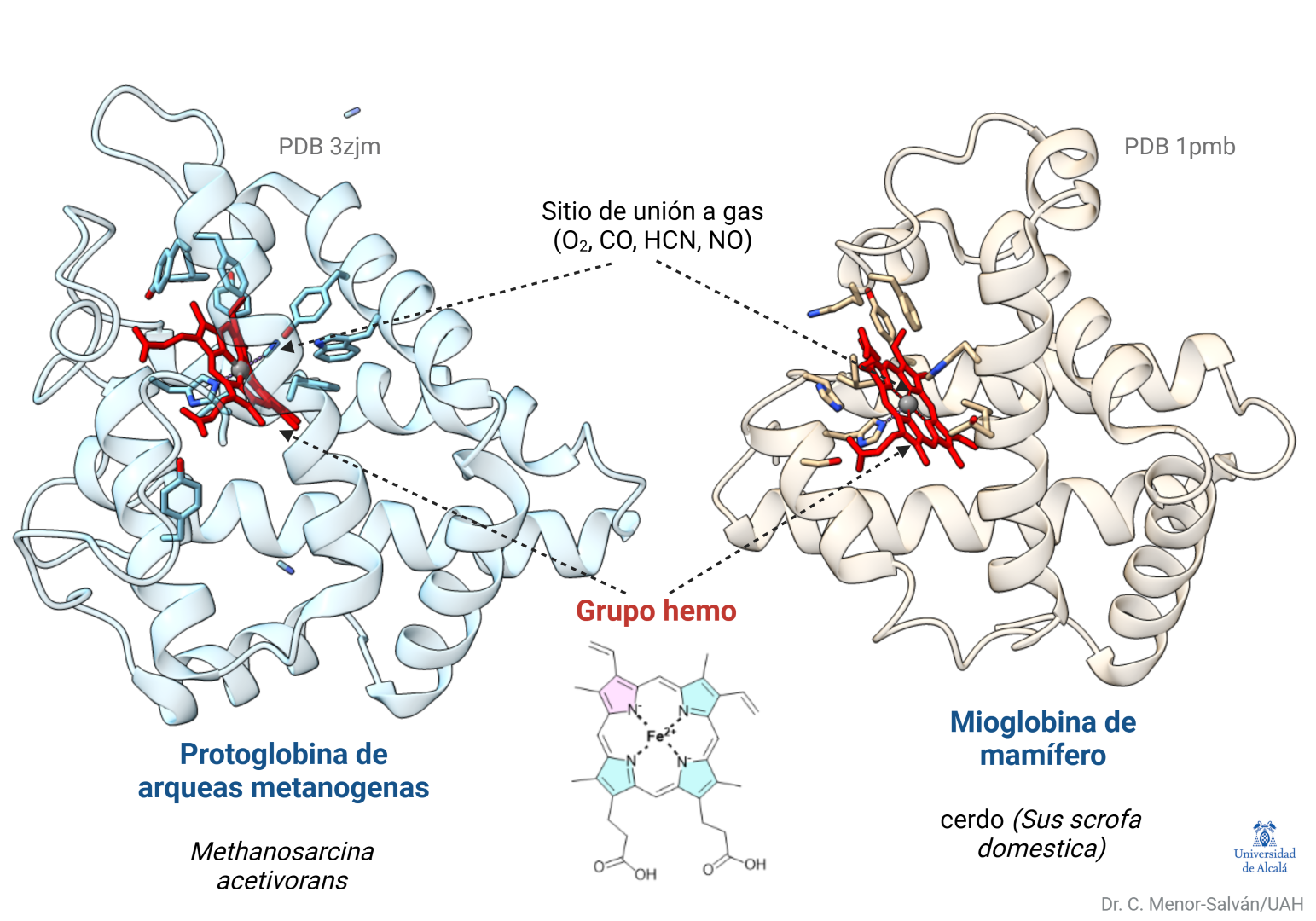
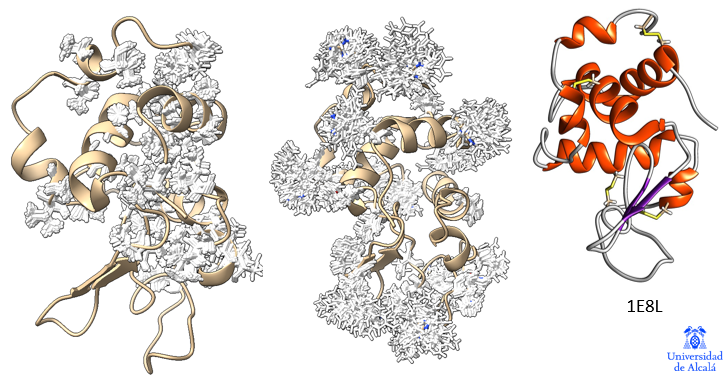
Estructuras de la protoglobina de arqueobacterias y la mioglobina de cerdo, mostrando el grupo hemo en su posición. Ambas proteínas, conectadas evolutivamente, tienen alta similitud estructural y gran afinidad por el monóxido de carbono.
Cuando la vida estaba en sus inicios y la atmósfera no contenía oxígeno, unas arqueobacterias productoras de metano ya estaban dotadas de la proteína protoglobina. Hoy día podemos encontrarlas en lugares tales como sistemas hidrotermales, aguas residuales, algunas minas, donde forman ecosistemas con otros procariotas que usan minerales (sulfuros metálicos) para obtener energía, o en fondos de lagos y mares, donde contribuyen a generar metano atmosférico.
Estas arqueas usan un sistema ancestral para metabolizar monóxido de carbono y usarlo como fuente de energía y carbono. En los inicios de la vida, posiblemente la protoglobina era un sensor de monóxido de carbono y, tal vez, de cianuro, otro componente quizá presente en aquel ambiente. Ambos gases se unen fuertemente al grupo hemo de la protoglobina.
Nosotros tenemos mioglobina y hemoglobina, que almacenan y transportan oxígeno. Estas proteínas evolucionaron a partir de aquellas globinas ancestrales de las arqueas, de quienes heredamos muchas estructuras moleculares, pues somos el resultado de una unión entre arqueas y bacterias que ocurrió hace unos 2000 millones de años.
Hace unos 600 millones de años, comenzó la evolución de la hemoglobina, que posibilitó el transporte de oxígeno desde el ambiente a los órganos, favoreciendo la evolución de los animales. Una consecuencia de este proceso de evolución es que el monóxido de carbono es muy tóxico para nosotros.
Esta toxicidad se debe a que nuestras mioglobina y hemoglobina tienen mucha afinidad por el monóxido de carbono, como la protoglobina arqueal; quizá, es un recuerdo de aquel lejano ancestro, que vivía en un ambiente sin oxígeno, alimentándose del monóxido de carbono. La mioglobina y hemoglobina no son el resultado de un «diseño inteligente», sino que resultan de la evolución de algo que surgió en un ambiente sin oxígeno y cuya función original se convirtió en un problema millones de años después.
Y no es el único problema. La vida nació en un ambiente sin oxígeno; cuando este empezó a aumentar en la atmósfera, tuvo lugar una de las primeras grandes extinciones. Los supervivientes desarrollaron adaptaciones moleculares que «parcheaban» los problemas que surgieron. En un ambiente sin oxígeno, el hierro reducido de la protoglobina era estable; en el ambiente con oxígeno, el hierro de la mioglobina y hemoglobina se oxida, por lo que tuvo que evolucionar un sistema antioxidante.
Otro de estos parches es una modificación en nuestras globinas que reduce ligeramente su afinidad por el monóxido de carbono. Ello reduce lo suficiente la toxicidad del gas como para que los fumadores puedan dar las gracias por ello. Sin esa modificación, los animales no soportaríamos un poco de humo.
La histidina 64, presente en la hemoglobina pero no en la protoglobina, estabiliza la unión del oxígeno al grupo hemo de la hemoglobina, que se produce formando un ángulo, mientras que obstaculiza ligeramente la unión del monóxido de carbono. Aunque el CO sigue teniendo mucha más afinidad por la hemoglobina que el O2, este pequeño cambio aportó una ligera ventaja a los animales.
Este ejemplo ilustra una característica básica de la evolución: el mantenimiento y adaptación de estructuras funcionales, no la mejora hacia un diseño óptimo. La mioglobina de mamífero y la protoglobina de arqueas sólo tienen en común un 13% de la secuencia de sus genes. Ese pequeño porcentaje permite que sus estructuras y función básica (la unión de estos gases) se hayan preservado y guardan una gran similitud estructural.
Las moléculas biológicas nos conectan a todos los organismos en un gran árbol cuya raíz está en el origen de la vida, hace más de 4000 millones de años. Y esto es un hecho, no un relato.
HEXOKINASA: ESTRUCTURA, EVOLUCIÓN Y PAPEL EN EL CÁNCER
escrito por cmfc5_1A | 17 diciembre, 2024
Redactado por María Arrondo Sánchez y Carolina Amil Zamorano
INTRODUCCIÓN
La hexoquinasa es una enzima transferasa (D-hexose-6-phosphotransferase), del grupo de las quinasas, encargada de fosforilar hexosas. Sin embargo, presenta mayor afinidad por la glucosa, puesto que la Km de esta es menor que la de otras hexosas como la fructosa. Esta proteína presenta cuatro isoformas, que han ido surgiendo de forma gradual. En dicho proceso de evolución ocurren determinados cambios que son claves en la estructura y que han permitido que la hexokinasa en dos de sus isoformas se oligomerice. La estructura de la proteína va a contar con dos dominios de unión a los sustratos (glucosa y ATP) y su actividad va a estar regulada alostéricamente mediante un mecanismo de ajuste inducido provocado por la propia glucosa.
Así mismo, esta enzima va a presentar un papel clave en el cáncer. En este artículo se abordará el normal funcionamiento de la hexokinasa así como su papel tumoral por diferentes vías, profundizando, además, en posibles estudios futuros y nuevos campos que se abren en la investigación contra el cáncer que emiten un rayo de esperanza en el estudio biomédico.
PAPEL BIOLÓGICO
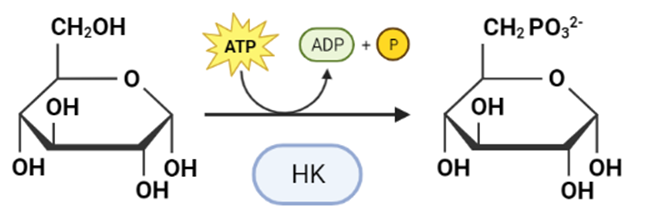
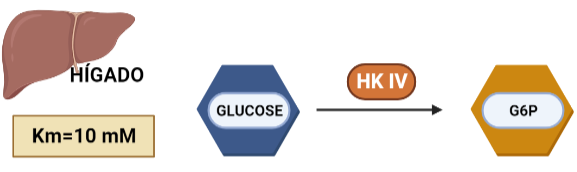
Figura I: reacción esquemática de la Hexokinasa, que fosforila la glucosa, produciendo Glucosa-6-Fosfato.
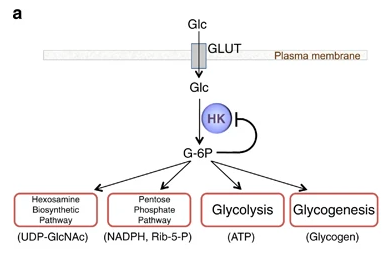
La Hexokinasa participa en la primera reacción irreversible de la glucólisis, que es la primera etapa del metabolismo de la glucosa. Tras hidrolizar el ATP, transfiere el grupo fosfato a la glucosa, para dar glucosa-6-fosfato (G6P), y así manteniendo el gradiente de glucosa que permite que haya un flujo de la misma mediado por los transportadores GLUT. La G6P es un inhibidor competitivo del ATP, por tanto se trata de un fenómeno de feedback negativo, en el que el mismo producto regula alostérica y negativamente su reacción de síntesis. Además, el grupo fosfato (Pi) liberado de la hidrólisis del ATP puede antagonizar la inhibición de G6P o sumarse al efecto inhibidor, según la isoenzima que haya llevado a cabo la reacción.
El producto (G6P) puede seguir varias rutas o vías celulares y funcionales:
Metabolismo catabólico: se introduce la glucosa en la glucólisis, para llevar a cabo un metabolismo oxidativo y obtener energía.
Metabolismo anabólico: la G6P es destinada a la vía de las pentosas fosfato, para sintetizar NAPDH y Ribulosa-5-Fosfato; o puede ser convertido a sus formas poliméricas (glucógeno), mediante la gluconeogénesis.
Figura II: vías del producto Glucosa-6-Fosfato, que se introduce en vías como la glucólisis, la ruta de las pentosas fosfato, o la glucogénesis.
ESTRUCTURA
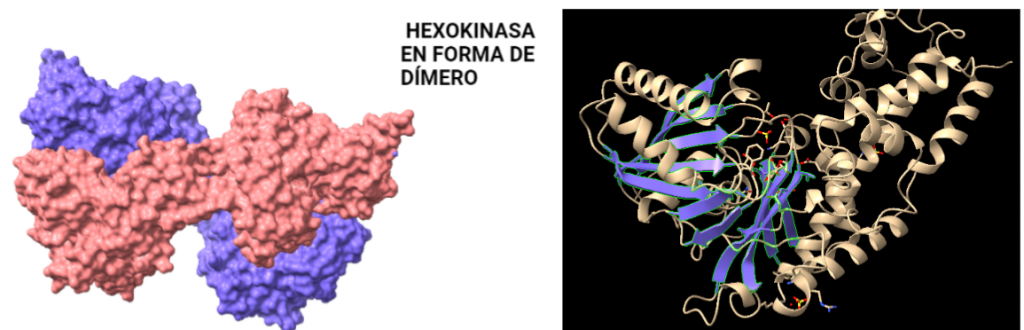
La estructura de las Hexokinasas más comunes (las isoformas I, II y III) cuenta con dos lóbulos muy similares de unos 50KDa cada uno. Algunas de ellas, como la HK I, son monoméricas, pero cuando se une a la membrana externa de la mitocondria se oligomerizan. De esta manera, la Hexokinasa cuenta con dos dominios principales, uno regulador y otro catalítico. La estructura dimérica y por tanto cuaternaria está presente en todas las isoformas salvo en la IV, que es la más ancestral.
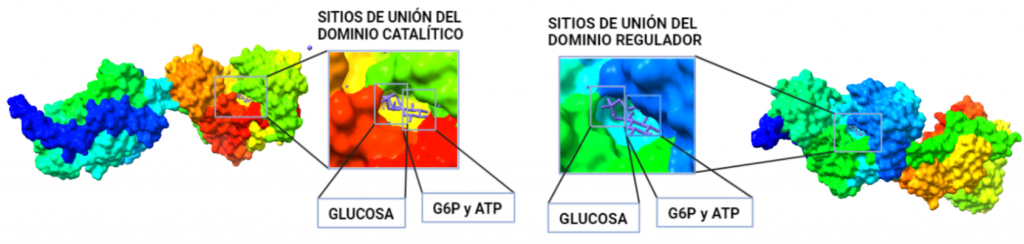
Figura III: HexokinasaI dimérica, con cada monómero de un color. Hecho con BioRender y Chimera, a partir de PDB 1BG3. Figura IV: Dominio de unión del ATP, se observan cuatro láminas paralelas y una antiparalela. Hecho con Chimera
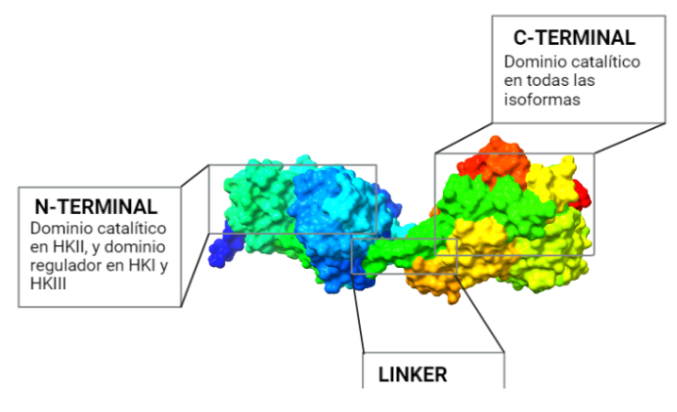
El dominio N-terminal se considera el dominio regulador en las isoenzimas I y II, y contiene el motivo de unión a la mitocondria. Además, está unido al dominio C-terminal (que es el dominio catalítico) a través de una hélice alfa. Ambos dominios presentan sitios de unión con la glucosa, G6P y ATP, y la inhibición de G6P en el dominio regulador se contagia al dominio catalítico por medio del contacto por la hélice alfa entre los dominios. La estructura terciaria de la hexoquinasa se basa en un plegamiento alfa/beta abierto. El dominio de unión al ATP está compuesto por cinco láminas beta y tres hélices alfa en el cual cuatro de las láminas beta son paralelas y una es antiparalela. Por otro lado, la hexoquinasa requiere de iones de magnesio para poder llevar a cabo la actividad catalítica. El magnesio (Mg2+) va a ser el cofactor de la enzima y se encuentra formando un complejo con el ATP (MgATP2-), que estabiliza la catálisis y reduce la energía de activación de la reacción.
Figura V: esquema general de la Hexokinasa en forma de monómero, que presenta un dominio catalítico y otro regulador, unidos por una hélice alfa. Hecho con BioRender, a partir de PDB 1BG3.Figura VI: Sitios de unión de la Hexokinasa con la glucosa y el inhibidor G6P, en ambos dominios. Hecho con BioRender, a partir de PDB 1BG3.
EVOLUCIÓN
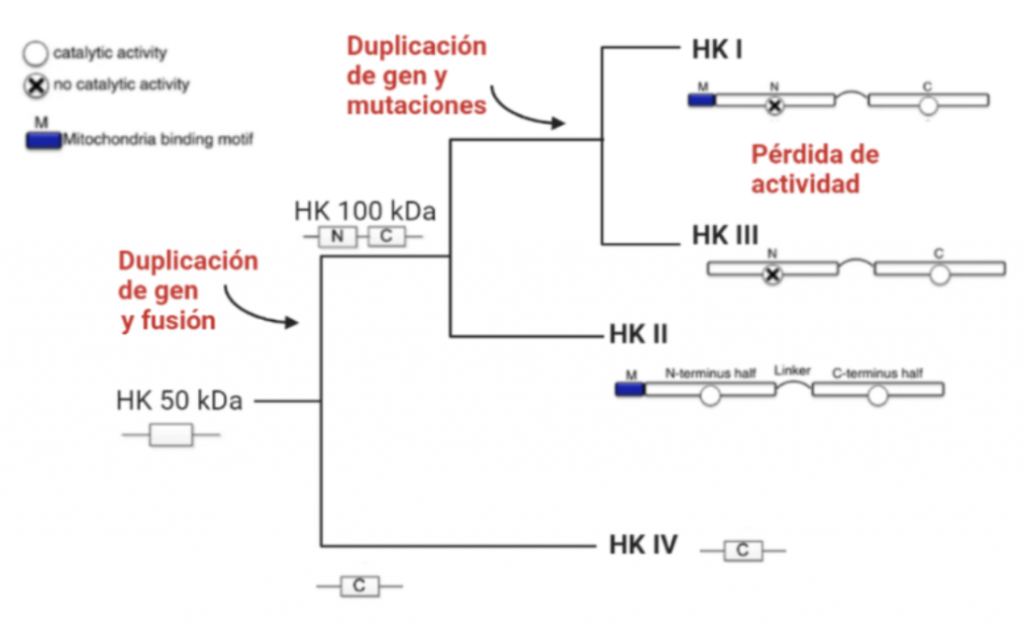
Todas las isoenzimas de la Hexokinasa provienen de una Hexokinasa de 50 kDa, susceptible a la inhibición por el producto G6P, por tanto, todas las isoenzimas presentan esta característica. A partir de la duplicación y fusión del gen que codificaba esta forma ancestral, surgieron las isoenzimas I, II y III, que ya son moléculas de 100kDa.
Figura VII: Esquema de la evolución de las isoformas de la Hexokinasa, hecho con BioRender e imágenes de D J Roberts y S. Miyamoto
La isoforma más próxima evolutivamente a la Hexokinasa original es la Tipo IV, que no sufrió la duplicación y fusión génica. Una vez que esto ocurrió, la segunda isoenzima que apareció fue la Hexokinasa II, que mantiene la actividad catalítica en ambos extremos terminales de la proteína, al igual que la Hexokinasa ancestral.
Una consecuente duplicación tuvo como resultado la aparición de la isoforma III. Posteriormente, las mutaciones de genes que codificaban la Hexokinasa 100 kDa, produjeron que el extremo N-terminal se diferenciara funcionalmente, perdiendo la actividad catalítica, y adquiriendo una función reguladora (con un sitio de unión para el inhibidor G6P). Esta diferenciación dio lugar a las en las Hexokinasas I y III.
Además, en las HK I y II, el extremo N-terminal presenta un dominio hidrofóbico que permite a estas integrarse en la membrana de la mitocondria. Concretamente, se unen a las porinas (VDAC) de la membrana mitocondrial externa, las cuales interaccionan con los ANT (Translocadores de Nucleótidos de Adenina). Esto es esencial para el mecanismo enzimático de la HK, puesto que es el sitio de salida del ATP producto de la fosforilación oxidativa (que usará la HK), y el sitio de entrada del ADP resultante de la reacción enzimática de la hexokinasa. Por tanto, existe una coordinación entre la introducción de la glucosa al metabolismo glucolítico y las últimas etapas de este en la mitocondria (la fosforilación oxidativa), para que se den a un ritmo adecuado a las necesidades celulares.
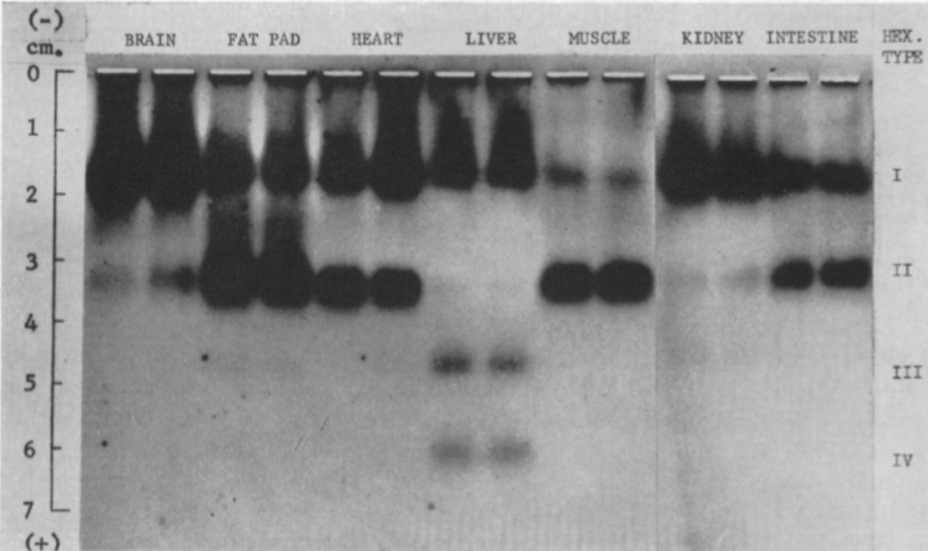
Hay cuatro isoenzimas de la Hexokinasa (HK) en los tejidos de mamíferos, con una estructura similar, pero expresión en diferentes tejidos:
Figura VIII: Electroforesis de las isoformas de la Hexokinasa en diferentes tejidos, que muestra que la HK I se encuentra presente de manera general, mientras que la HK II aparece en el músculo y tejido adiposo, y la HK III y IV, en el hígado. Imagen de H M Katzen and R T Schimk.
HEXOKINASA I (HKI)
Fundamentalmente en el cerebro, donde la tasa metabólica es muy exigente, pero expresada de manera general.
HEXOKINASA II (HKII)
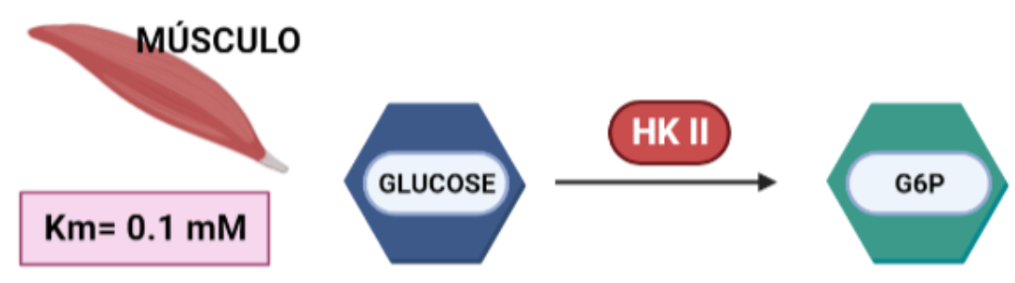
Más limitada en su expresión, aparece en tejidos sensibles a insulina, como es el caso del tejido adiposo y el músculo esquelético. En el músculo, es necesaria una alta tasa de glucólisis, y por tanto, lo que ocurre es que esta isoforma presenta una gran afinidad por la glucosa (menor Km), para que con concentraciones muy bajas de glucosa, se alcancen altas velocidades de la enzima.
Figura IX: Afinidad por la glucosa de la HKII en músculo. Creado con BioRender
La insulina aumenta la actividad de esta isoforma, induciendo la transcripción del gen que la codifica, y de esta forma, favoreciendo la metabolización y eliminación de glucosa. Por esta razón, en los individuos con diabetes de tipo II, la expresión de la HKII se ve reducida, acentuando la hiperglucemia.
Al igual que la isoenzima Tipo I, incluye un dominio hidrofóbico en el extremo N-terminal que permite que se inserte en la membrana externa mitocondrial, y también usa ATP intramitocondrial.
Cabe destacar su predominancia en células tumorales, puesto que estas se caracterizan por presentar un aumento anormal del metabolismo, en el que la reacción que lleva a cabo la Hexokinasa es esencial para la obtención de energía.
HEXOKINASA III (HKIII)
A diferencia de las dos isoenzimas anteriores, la Hexokinasa III no está unida a la mitocondria, puesto que carece del dominio hidrofóbico en el extremo N-terminal. Se piensa que se expresa en el citoplasma, o que incluso tiene una localización perinuclear, en células del hígado.
HEXOKINASA IV (HK IV)
En hepatocitos y células beta pancreáticas, y es conocida como glucoquinasa. La G6P que produce está destinada a la síntesis deglucosa. Esto es un proceso que se lleva a cabo cuando la cantidad de sustrato (glucosa) es alta, por tanto, tiene sentido que esta isoenzima presente una mayor Km, porque necesitará altas concentraciones de glucosa para realizar la reacción a una velocidad alta.
Figura X: Afinidad por la glucosa de la HK IV en hígado. Creado con BioRender.
MECANISMO
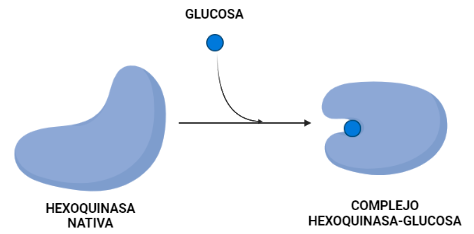
La hexoquinasa sufre un cambio conformacional que es regulado por la propia glucosa que va a ser esencial para la catálisis. En este proceso se observa que la superficie en contacto con el solvente del complejo hexoquinasa-glucosa es más pequeña que la hexoquinasa nativa.
Utilizando dicho cambio en el área de superficie que se encuentra expuesta se ha podido estimar la contribución hidrofóbica a los cambios de energía libre tras la unión de la glucosa. De esta manera se descubre que el efecto hidrofóbico por sí solo favorece la conformación activa de la hexoquinasa en presencia y ausencia de azúcar. La estabilidad observada de la conformación inactiva de la enzima en ausencia de sustratos puede resultar de una deficiencia de interacciones complementarias dentro de la cavidad que se forma cuando los dos lóbulos se unen.
El cambio conformacional que sufre la hexoquinasa mantiene la estructura terciaria prácticamente igual excepto por un gran cambio en la orientación de los dos lóbulos. Para demostrar este cambio lo que se hizo fue superponer los carbonos alfa de cada lóbulo usando un procedimiento de mínimos cuadrados y tratando a los carbonos como cuerpos rígidos. Esta superposición mostró que cada lóbulo se comporta como un cuerpo rígido durante el cambio conformacional entre la forma nativa de la proteína y el complejo con la glucosa.
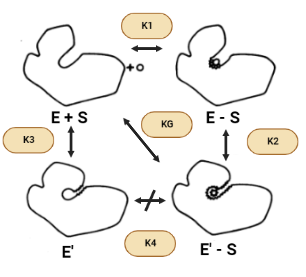
Figura XI: Cambio conformacional de la HK inducido por la glucosa y cambio en la superficie de contacto con el solvente.
Tal y como venimos viendo, la superficie accesible de la hexoquinasa se reduce cuando se une la glucosa para formar el complejo enzima-sustrato (ES) y se reduce aún más por el cambio a E’(inactiva)-S. Por lo tanto, se puede esperar que las fuerzas hidrofóbicas favorezcan la conformación activa en presencia de azúcar, asumiendo que todos los donantes y aceptores de enlaces de hidrógeno están satisfechos en E’-S. Sin embargo, el área superficial también se reduce cuando la conformación activa se forma en ausencia de azúcar (E’ – E). Sin embargo, debido a que hay menos de un factor de dos diferencias entre los cambios en la superficie accesible para las transiciones E-S → E’-S y E’-E, el efecto hidrofóbico no puede explicar la gran diferencia en las constantes de equilibrio conformacional K2 y K3 en presencia y ausencia de azúcar.
Figura XII: Cambios conformacionales de la Hexoquinasa en presencia y ausencia de azúcar. Esquema creado con Biorender.
Otra cuestión que surge es por qué la enzima no permanece en el estado activo, E, en ausencia de ligandos sabiendo que el efecto hidrófobo, de manera individual, predice que la estructura E debería ser más estable. La respuesta a esta pregunta reside en que cuando la enzima carece de la presencia de la glucosa contiene una cavidad en la que entran las moléculas de agua y donde quedan encerradas. Además, tanto los puentes de hidrógeno como las fuerzas de Wan der Waals contribuyen muy poco a la estabilidad de la proteína y del complejo proteína-ligando. El hecho de no obtener estas interacciones complementarias dentro de la cavidad daría como resultado entalpías desfavorables causadas por la pérdida de los puentes de hidrógeno o fuerzas de Van der Waals en relación con los que se producen en la estructura abierta. También puede haber alguna pérdida de entropía traslacional al atrapar una pequeña cantidad de moléculas de agua en la cavidad.
Presuntamente, el agua misma desestabiliza la forma activa mediante la creación de interacciones favorables con la estructura abierta inactiva. Solamente el ligando correcto puede proporcionar las fuerzas de Van der Waals y los puentes de hidrógeno necesarios para que se active la estructura.
Con ello, concluimos que hay al menos dos posibles funciones para el cambio conformacional inducido por la glucosa: permitir un «mecanismo de acogida» o proporcionar especificidad.
PAPEL BIOMÉDICO: HEXOKINASA II EN CÁNCER
El metabolismo de las células tumorales se caracteriza por una alta actividad glucolítica: metabolizan anaeróbicamente grandes cantidades de glucosa en ácido láctico, incluso en presencia de oxígeno, aumentando la velocidad de la glucólisis y de la síntesis de ATP. Esto es lo que se conoce como el efecto Warburg. Por tanto, la actividad de cualquier enzima glucolítica como la HK será esencial en un tumor.
La Hexokinasa II aparece sobreexpresada en células tumorales, satisfaciendo estas altas velocidades de la glucólisis. La introducción de la glucosa en el metabolismo glucolítico es crucial para la producción de energía, y la síntesis de precursores de nucleótidos (derivados de glucosa) por la vía de las pentosas fosfato, destinados a la síntesis de ADN para la proliferación del tumor.
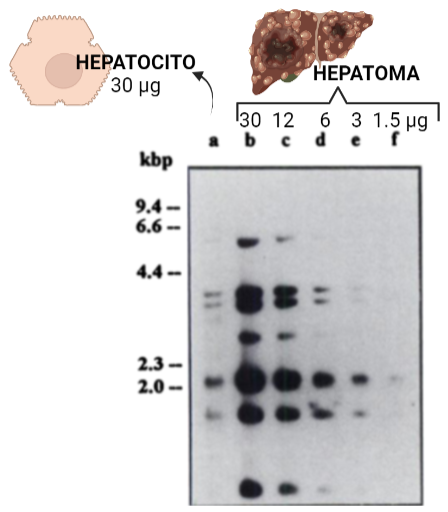
Fig. XIII: Amplificación del gen de la HKII en hepatoma y hepatocitos. Hay unas 5-10 copias más de lo normal en el hepatoma, ya que la intensidad de una tira de ADN de hepatocito de 30 μg se asemeja a la de ADN de hepatoma de 6-3 μg Imagen: Annette Rempel
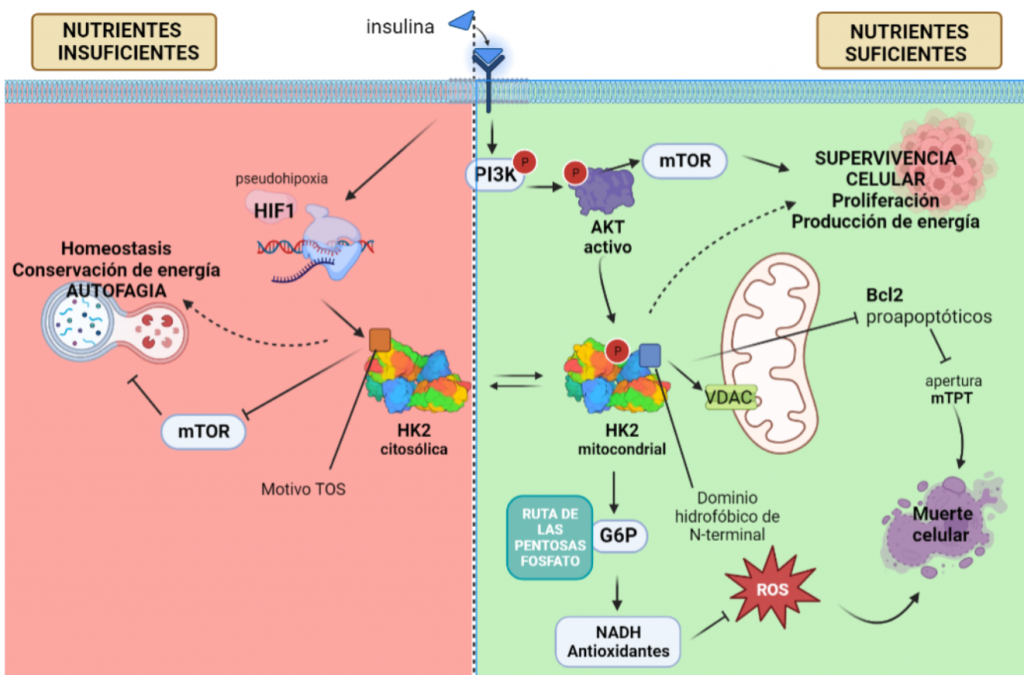
La regulación por AKT de la HKII es el factor determinante para que esta isoforma sea la esencial en el metabolismo tumoral, puesto que controla la unión de esta a la mitocondria, y con ello, fija la función de la HK, que varía en función de si aparece unida a la mitocondria o no.
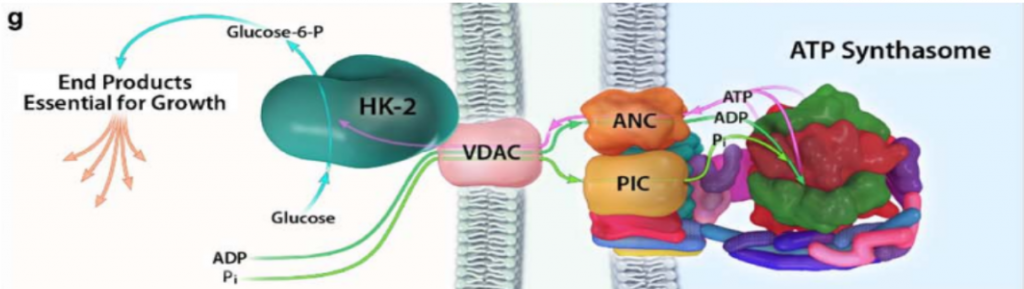
Con el objetivo de crecimiento del tumor, y cuando hay disponibilidad de nutrientes, AKT une la HKII a la mitocondria (fosforilando su residuo Thr-473), conectándola con VDAC. Esto le permite a la enzima tener un acceso privilegiado al ATP que sintetiza la mitocondria, y la célula sigue un metabolismo de proliferación y de producción de energía, mediante la glucólisis.
Figura XIV: Unión de la HKII con el VDAC de la membrana externa de la mitocondria, y cómo esta le posibilita acceder al ATP sintetizado por la ATP sintasa. Esta imagen señala la relación entre el primer paso de la glucólisis y el último paso del metabolismo oxidativo. Imagen: Pedersen, P.L.
La HK II mitocondrial, además, lleva a cabo una función protectora en tumores con acceso a nutrientes, promoviendo la supervivencia celular: inhibe a los miembros pro-apoptóticos de la familia Bcl2, e introduce la G6P en la ruta de las pentosas fosfato, cuyos productos son antioxidantes que reducen las ROS (especies reactivas de oxígeno).
Sin embargo, en una situación de isquemia, en la que no llegan suficientes nutrientes y oxígeno a la célula, disminuye la actividad de AKT y HKII mitocondrial, aumentando la HKII citoplásmica. Entonces, esta isoforma se une a mTORC (mediante el motivo TOS), inhibiéndola, e induciendo la vía autofágica, y la conservación de energía y homeostasis en ausencia de glucosa, pensando en el “bien mayor” del tumor.
Figura XV: Esquema explicativo del papel que juega la HK II en células tumorales, promoviendo la supervivencia celular si tiene acceso a nutrientes, pero induciendo indirectamente la autofagia en el caso contrario, con el fin de conservar la energía y homeostasis tisular.Creado con BioRender.
Esta regulación por AKT posibilita esta compleja acción de la HKII, que puede ser proapoptótica (HKII citoplásmica) o antiapoptótica (HKII mitocondrial), según la disponibilidad de recursos. Sin embargo, AKT no regula la HKI, ya que esta no presenta una secuencia de consenso para esta enzima, y por ello, esta isoenzima no se encuentra prevalentemente en tumores. Además, la HKI no puede satisfacer la alta demanda energética, al perder la actividad catalítica en el extremo N-terminal.
Según todas las vías beneficiosas para el tumor mencionadas anteriormente en las que participa la HKII, la eliminación de esta isoforma perjudicaría a la progresión del tumor, por lo tanto, es un frente esperanzador en terapias oncogénicas. Lo ideal sería encontrar un modo de inhibir únicamente esta isoforma, pero es difícil puesto que todas ellas son bastante similares.
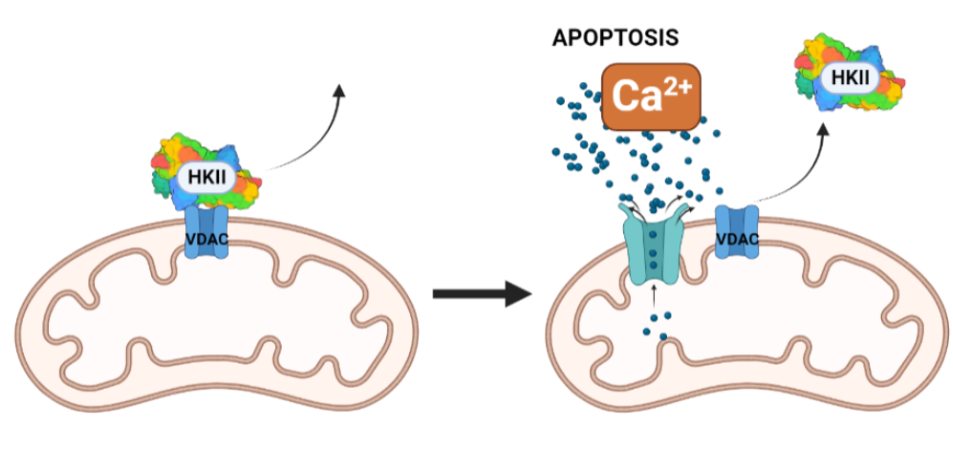
La inhibición de la HKII en células cancerosas puede darse por p53, o por un sustrato análogo a la glucosa, la 2-desoxiglucosa, que favorece la apoptosis. Una combinación de estos dos factores podría ser favorecedora. Otro posible campo a investigar sería la inhibición de la HKII por fosfato inorgánico, que sensibiliza la inhibición por G6P en esta, pero la antagoniza en HKI. Incluso, una alternativa más podría ser el uso de determinados péptidos que desplazaran la HKII de la mitocondria. Este desplazamiento parece producir un aumento de la concentración de Ca2+ citosólica, lo que abriría poros en la membrana mitocondrial e induciría a la célula a apoptosis.
Figura XVI: Posible terapia oncogénica sobre HK II, mediante péptidos que la separen de la mitocondria. Creado con BioRender.
Bennett, W. S., & Steitz, T. A. (1978). Glucose-induced conformational change in yeast hexokinase. Proceedings of the National Academy of Sciences, 75(10), 4848-4852. https://www.pnas.org/doi/abs/10.1073/pnas.75.10.4848
Gahr, M. (1980). Isoelectric Focusing of Hexokinase and Glucose-6-Phosphate Dehydrogenase Isoenzymes in Erythrocytes of Newborn Infants and Adults. British Journal of Haematology, 46(4), 529-535. https://doi.org/10.1111/j.1365-2141.1980.tb06009.x
Haruhiko Osawa, Calum Sutherland, R. Brooks Robey, Richard L. Printz, Daryl K. Granner (1996). Analysis of the Signaling Pathway Involved in the Regulation of Hexokinase II Gene Transcription by Insulin. https://www.sciencedirect.com/science/article/pii/S002192581831932X
Mulichak, A., Wilson, J., Padmanabhan, K. et a (1998)l. The structure of mammalian hexokinase-1. Nat Struct Mol Biol 5, 555–560. https://pubmed.ncbi.nlm.nih.gov/9665168/
R.L. Printz, S. Koch, L.R. Potter, R.M. O’Doherty, J.J. Tiesinga, S. Moritz, D.K. Granner (1993). Hexokinase II mRNA and gene structure, regulation by insulin, and evolution. Journal of Biological Chemistry. Volume 268, Issue 7 https://www.sciencedirect.com/science/article/pii/S0021925818535213
Reference for PDB-101: PDB-101: Educational resources supporting molecular explorations through biology and medicine. Christine Zardecki, Shuchismita Dutta, David S. Goodsell, Robert Lowe, Maria Voigt, Stephen K. Burley. (2022) Protein Science 31: 129-140 doi:10.1002/pro.4200
Reference for RCSB PDB: The Protein Data Bank H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne (2000) Nucleic Acids Research, 28: 235-242. doi:10.1093/nar/28.1.235
Rempel, A., Mathupala, S. P., Griffin, C. A., Hawkins, A. L., & Pedersen, P. L. (1996). Glucose Catabolism in Cancer Cells: Amplification of the Gene Encoding Type II Hexokinase1. American Association for Cancer Research. https://pubmed.ncbi.nlm.nih.gov/11557773/
Roberts, D., Miyamoto, S. (2015) Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ 22, 248–257. https://www.nature.com/articles/cdd2014173
UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE. Protein Sci. 2018 Jan;27(1):14-25.
UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. Protein Sci. 2021 Jan;30(1):70-82.
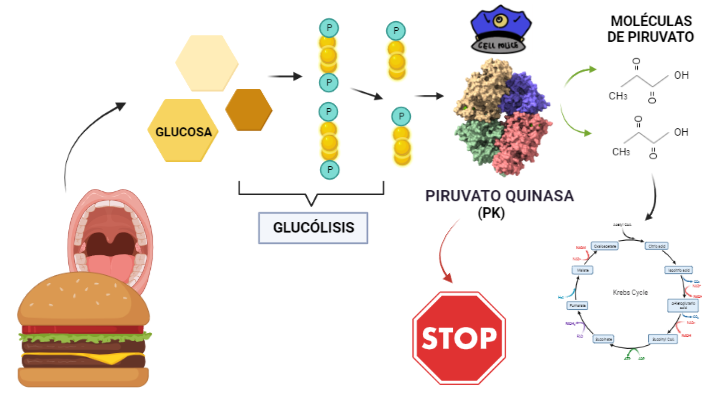
La Piruvato quinasa (PK) es una enzima que se encuentra al final de la ruta metabólica conocida como glucólisis y su función es permitir la salida del piruvato a la siguiente vía metabólica con el fin de obtener energía. Para entender la relevancia de esta molécula, quizás es conveniente explicar de forma concisa el recientemente popular término, conocido como “metabolismo”.
El metabolismo, al que hacen referencia desde dietistas y entrenadores hasta profesores de bioquímica, es quizás un proceso que para el ciudadano de a pie resulta abstracto en su relación con la ingesta de comida. Brevemente, se trata de un conjunto de reacciones que consisten en tomar una molécula grande (glucosa) con alto potencial energético y dividirla en paquetes más pequeños (piruvatos, en el caso de la glucólisis) con el fin de distribuir la energía por los distintos sistemas celulares y permitir que los organismos vivos se sigan considerando como tales. Esa glucosa, llega a las células por medio de la alimentación y se convierte en energía química útil mediante las distintas rutas metabólicas. De modo, que el correcto funcionamiento de la PK, es fundamental para que estos pequeños paquetes se introduzcan en la siguiente ruta metabólica (el ciclo de Krebs) que se encargará de distribuir la energía. De hecho, modificaciones de esta molécula podrían degenerar en condiciones tan serias como el Cáncer o el Alzheimer, dado que impediría que la energía alcanzase los destinos pertinentes.
PAPEL BIOLÓGICO
El rol que asume la PK a nivel biológico se traduce a la función que tiene por ejemplo el personal de seguridad a la entrada de un museo. Éste, en caso de que el turista no presente ninguna irregularidad (posesión de armas, comida, etc), procede a concederle la entrada al establecimiento; del mismo modo la PK permite el acceso al piruvato a la siguiente ruta, siempre que no suponga una amenaza contra el correcto funcionamiento de la célula. Al tratarse la PK de una quinasa, esto la convierte en una molécula con la capacidad de fosforilar (añadir grupos fosfato a otras moléculas). De modo que para concederle el acceso a la siguiente ruta, la PK retira los fosfatos de las triosas procedentes de la glucosa y los añade a moléculas de ADP, dando lugar a ATP (que es una molécula que transporta energía). Como resultado de este proceso, se sintetizan dos piruvatos, a los que finalmente les será concedido el acceso al ciclo de Krebs.
La PK en humanos, aparte de la función biológica general descrita anteriormente, también tiene una serie de isoformas cuya función e importancia biomédica es más específica. Las cuatro isoformas de la PK (PKM, PKK, PKR, PKL) fueron descubiertas en 1965 y reciben su nombre a partir del nombre tejido donde se encuentra cada una. La PKM (actualmente denotada como PKM1) se encuentra en los tejidos musculares (muscle, M) del corazón y en el cerebro, la PKK (actualmente denotada como PKM2) se encuentra en los tejidos de los riñones (kidneys, K), el intestino y las células cancerosas; la PKR en el tejido sanguíneo (red blood cells, R) y la PKL en el tejido del hígado (liver, L) y en los riñones también. La diferenciación (que ocurre tras la fase fetal, ya que todas provienen de la isoforma PKM2) favorece la aparición de patologías mucho más especializadas debido a ligeros cambios en la conformación proteica.
Ilustración que refleja el papel de la Piruvato quinasa en el metabolismo de la glucosa. La molécula de la que partimos es la glucosa, que tiene 6 carbonos, ésta, mediante una concatenación de reacciones da lugar a 1,3-bifosfoglicerato (las bolas amarillas representan los carbonos y las P los grupos fosfato) que tras otra serie de reacciones, dan lugar a fosfoenolpiruvato (PEP) que es el precursor del piruvato. La PK es la encargada de catalizar la reacción de desfosforilación que da lugar al paso de PEP a piruvato cuyo fosfato restante se utiliza para fosforilar ATP. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
ESTRUCTURA Y CÓMO FUNCIONA
Desglose estructural
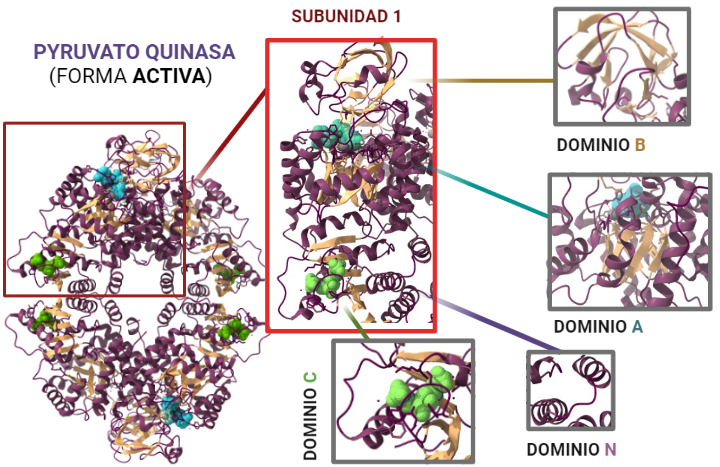
Para entender el papel que desempeña la PK es fundamental comprender su estructura, dado que de ésta dependerá el funcionamiento de la misma. ¿Qué constituye a la PK? La piruvato quinasa es una proteína conformada por 531 aminoácidos que dan lugar a un tetrámero, cuyas cuatro subunidades son iguales. Éstos están organizados en motivos de hélices alfa, láminas beta y bucles.
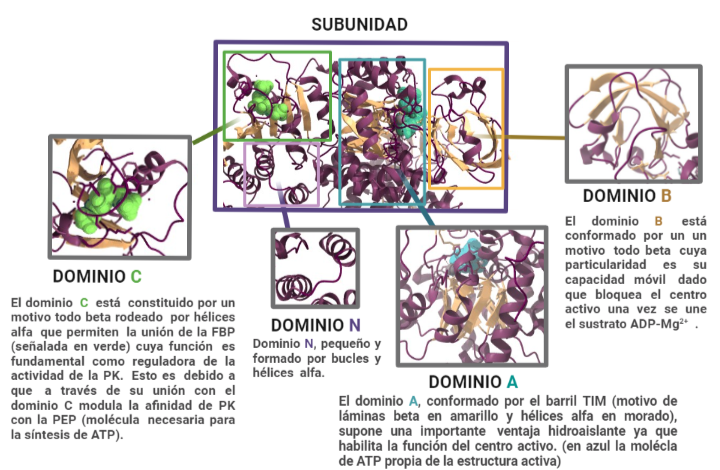
Estas subunidades se organizan a su vez en tres grandes dominios rotulados como A, B y C junto con un dominio N-terminal. El dominio A está conformado por un barril TIM α8/β8 cuyo centro activo se ubica entre el dominio A y el B, éste es además el dominio más grande de la subunidad. El dominio B sin embargo es móvil y bloquea el centro activo una vez que se le une el sustrato ADP-Mg2+. Finalmente, el dominio C contiene la fructosa-1,6-bifosfato (FBP) que es un potente activador alostérico.
Descripción gráfica del desglose estructural que presenta la Piruvato quinasa en estado activo. Los motivos de láminas beta quedan indicados en amarillo, las hélices alfa en púrpura, los bucles en violeta y los ligandos: ATP en azul y FBP en verde. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
Esto significa, que a través de la unión de la FBP al dominio C, se facilitará la unión del fosfoenolpiruvato (PEP) que es fundamental para la regulación de la actividad de la PK, ya que ésta depende de la afinidad con el PEP. En ausencia de activadores alostéricos como la FBP, la PK tiene poca afinidad con el PEP. O sea, si la PK fuera un niño pequeño y su voluntad para realizar los deberes fuera análoga a la actividad de la quinasa, éste necesitaría una motivación para realizarlos. Si se impone la condición de recibir un caramelo a cambio de la tarea, éste cumplirá. De igual forma si la PK presenta la FBP unida al dominio C, ésta aumentará su afinidad a la PEP alterando su actividad. Además, la unión de FBP estabiliza la molécula en estado activo y promueve la tetramerización. Cabe destacar, que todas las isoformas de la PK se unen con la FBP exceptuando la PKM1 que debido a una discrepancia estructural es suficientemente estable por si sola (siendo además insensible a los moduladores alostéricos) y no presenta ni la región de unión a la FBP ni el interfaz dímero-dímero debido a éstas se expresan en los exones específicos de las isoformas.
Desglose de la funcionalidad de las distintas estructuras que conforman cada subunidad de la Piruvato quinasa en estado activo. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
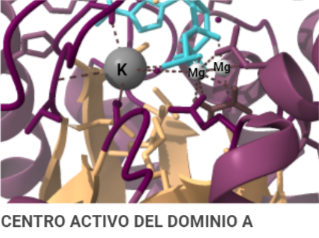
Otro detalle que cabe mencionar de la estructura de la PK es su capacidad de unión a cofactores (K+ y Mg 2+) cuya intervención en la activación de la molécula es esencial. En el caso del Mg 2+, se ha mencionado que está involucrado en bloquear el acceso al centro de activo (gracias al cambio conformacional que desplaza al dominio B) cuando éste forma un complejo de sustrato al unirse al ADP (formando el sustrato ADP-Mg2+). En el caso del K+ sin embargo, se ha observado que ante su presencia, el mecanismo cinético de la PK se mantiene desordenado (forma natural), esto supone que favorece la forma activa de la PK y permite que se unan el PEP o el complejo ADP-Mg2+ de forma independiente (mecanismo aleatorio). En ausencia de K+, por el contrario, el ADP no se pude unir al centro activo hasta que el PEP no haya terminado de formar un centro activo completamente funcional. De forma, que se deduce que el K+ es el encargado de inducir el cierre del centro activo y de que los residuos encargados de la unión al nucleótido adopten la conformación correspondiente.
Ilustración que señala la presencia de cofactores ( K+, Mg2+) en el Dominio A, fundamentales para la adopción de la conformación activa de la PK. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
Función ¿Qué hace?
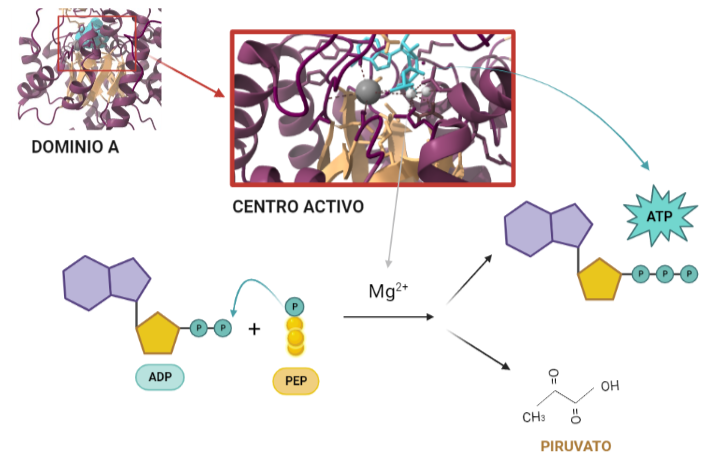
Entonces, ¿Qué función tiene? Una vez conocida la estructura, es posible dilucidar qué función lleva a cabo, y cómo. La función catalítica de la PK consiste en fosforilar moléculas, debido a su condición de quinasa; en concreto, moléculas de ADP a partir de moléculas de PEP de la etapa anterior de la glucólisis. Todo ello para dar lugar a dos productos. Por un lado, piruvatos estables a partir de sus precursores PEP y por otro, obtener energía en forma de moléculas de ATP a través de la fosforilación del ADP. De modo, que el nombre surge del producto (piruvato) + tipo de enzima (quinasa). Para ello, al centro activo del dominio A se unen el PEP y el complejo ADP-Mg2+ dado que los cationes de Mg2+ median y facilitan la transferencia del grupo fosfato del PEP al ADP dando lugar al ATP y a los piruvatos. Todo ello es posible debido a la alta energía que libera PEP al ser hidrolizada. Al perder el fosfato, el PEP pasa a su forma de enolpiruvato que es menos estable, de modo que se llevará a cabo un proceso de tautomerización que consiste en que el enolpiruvato acepte un protón procedente de una molécula de agua convirtiéndose así en un piruvato estable y favoreciendo la fosforilación del ADP.
Representación gráfica de la reacción que tiene lugar cuando la PK funciona de forma natural.El complejo ADP-Mg2+ se une al centro activo junto con el PEP. Éste le aporta el fosfato que le hace inestable con el fin de utilizarlo para fosforilar al ADP. Como productos se obtienen ATP y piruvato. A partir de PDB 1A3W. Creado por María Arranz con ChimeraX/BioRender.com
En cuanto a la capacidad alostérica de la PK, aparte de la FBP, que favorece la unión del sustrato PEP, hay otras moléculas que alteran la actividad de la enzima. Por ejemplo, un inhibidor alostérico de esta enzima (PKM1, PKM2) sería la fenilalanina (Phe) cuya unión supone la disminución de afinidad con la PEP mediante la estabilización de la estructura inactiva de la PK. El lugar de unión de Phe también puede albergar a la alanina que actúa como inhibidor, pero solo ante la isoforma PKM2, y esto lo lleva a cabo favoreciendo la conformación dimérica, contraria a la tetramérica a la que se une FBP. A pesar de que en presencia de concentraciones normales de FBP la inhibición de la alanina queda mitigada. La serina sin embargo, también puede ocupar este centro de unión, pero con función activadora no inhibidora, en la PKM2. Dejando a un lado los aminoácidos, hormonas ,como la hormona tiroidea triyodo-L-tironina (T3), actúan también como inhibidor alostérico favoreciendo la conformación monomérica inactiva de la PK. Mientras que el oxalato puede actuar como activador de la PK mediante su interacción con el centro activo por ser análogo al enolpiruvato, en caso de que la concentración de PEP sea baja.
¿PORQUÉ PODRÍA ACABAR CONTIGO?
EL PELIGRO RESIDE EN LA ISOFORMA
Las isoformas de una proteína son proteínas que provienen del mismo gen que la proteína original, dicho gen se duplica y comienza a acumular mutaciones para dar lugar a las distintas isoformas. En el caso de la PK, tras este proceso de duplicación y modificación por mutaciones se han obtenido 4 isoformas distintas: PKM1, PKM2, PKR y PKL. La importancia de las isoformas recae en que a pesar de realizar la misma función que la proteína inicial, cada una presenta ligeramente distintas: propiedades cinéticas, estructurales, de regulación o de localización en la célula. Estas ligeras diferencias atienden a las necesidades metabólicas del tejido al que pertenecen. O sea, las modificaciones que sufra PKL (L hace referencia a liver, hígado en inglés) afectarán en principio al hígado dado que la estructura de la PKL ha resultado ser la más eficaz a la hora de catalizar las reacciones que precisa este órgano. A pesar de esto, existe una isoforma que destaca en su implicación en numerosas patologías inflamatorias (como la Sepsis, IBD o Arterosclerosis) o enfermedades como el Cáncer o el Alzheimer. Ésta es la PKM2.
PKM2 Y UN COMPENDIO DE LO QUE PUEDE SALIR MAL
PKM2 en el Cáncer
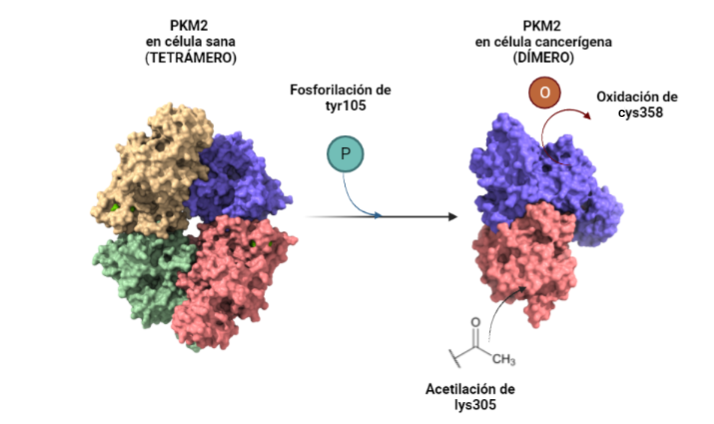
Ante el desarrollo de células neoplásicas, la PKM2 tiene un comportamiento que podría clasificarse como moonlighting. Esto se debe a que en condiciones normales la PK transforma PEP en piruvato y este sigue la ruta metabólica normal hacia el ciclo de Krebs, mientras que, ante una situación de estrés, como puede ser el desarrollo de células cancerígenas, ésta altera su forma tetramérica natural y pasa a su forma dimérica. Al dimerizarse mediante la fosforilación de su tirosina 105 la proteína deja de realizar su función natural y divierte el proceso de la glucólisis hacia la síntesis de metabolitos necesarios para la síntesis de serina. Esto se debe a que dicho aminácido regula a mTORC1 ( mammalian target of rapamacyn complex 1), que es fundamental para favorecer la proliferación celular, característica de las células cancerígenas. Otra de las modificaciones que sufre, es la acetilación de su lisina 305, junto con la oxidación de su cisteína 358 que provoca una alteración en la ruta de la glucólisis haca la PPP (pentose phosphate pathway, vía de la pentosa fosfato) que favorece la síntesis de nucleótidos para sufragar los efectos de la interrupción de la ruta glucolítica.
Descripción gráfica de la dimerización y modificación de la piruvato quinasa en presencia de cáncer. Este proceso consiste en dimerizar la proteína mediante la fosforilación de su tirosina 105 y alterar sus propiedades mediante la oxidación de su cisteína 358 y la acetilación de su lisina 305. A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
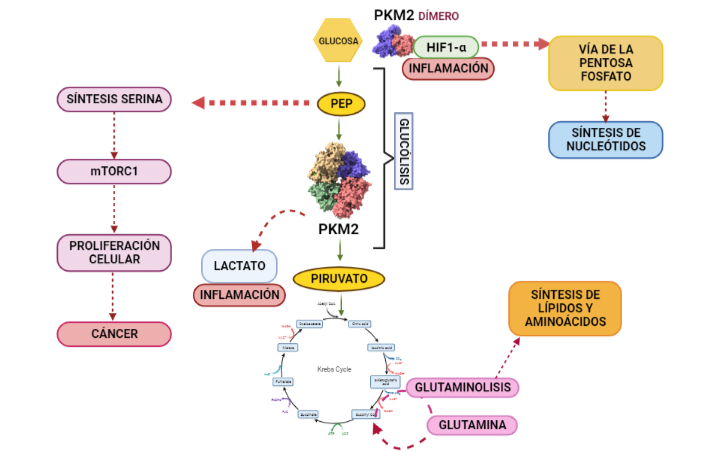
Estas modificaciones de la PK se conocen como el efecto Warburg o Glucólisis Aeróbica. El efecto Warburg supone que las células cancerígenas conviertan el piruvato en lactato, disminuyendo su producción de ATP. La disminución de síntesis de ATP se soluciona mediante los procesos mencionados anteriormente, mientras que el aumento de lactato supone la aparición de un ambiente tumorigénico dado que es excretado, reduciendo así el pH extracelular (esto favorece las condiciones para la proliferación celular) y provocando inflamación. El aumento de lactato también se utiliza para acceder a un recurso de energía alternativo, como es la glutamina. Esto es posible, dado que se disminuye la entrada de piruvato en el ciclo de Krebs, por lo que comienzan a introducirse metabolitos de glutamina en su lugar y aumenta por lo tanto la síntesis de lípidos y aminoácidos. Finalmente, la PKM2 es translocada al núcleo donde se une a HIF1-α (hypoxia-inducible factor-1 alfa) promoviendo la transactivación de HIF1 que favorece la aparición de un ambiente tumorigénico y la desviación de la glucólisis hacia la vía de la pentosa fosfato.
Diagrama que dilucida la modificación de la vía metabólica regulada por la piruvato quinasa en presencia de cáncer (efecto Warburg). Las flechas rosas indican los procesos que surgen bajo condiciones de dicho efecto, mientras que las verdes indican el curso habitual de la vía. A partir de PDB 1a3w y 6wp3(en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
PKM2 en el Alzheimer
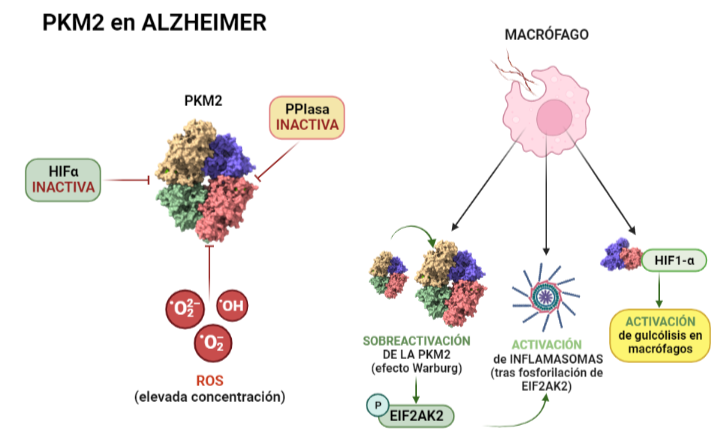
Debido a que el origen y funcionamiento del Alzheimer todavía no se ha esclarecido, se están explorando nuevas vías de investigación. Entre ellas, destacan factores como el estrés oxidativo, fallos en el transporte y metabolismo de la glucosa, y la inflamación. Dado que la piruvato quinasa juega un papel central en el metabolismo de la glucosa, su función se ve afectada por la enfermedad. Como consecuencia de la alteración de la ruta metabólica de la glucosa (debida a la acumulación de β-amiloides), se da la inactivación HIF1-α lo que supone que la PKM2 en dímero no puede unirse a ella, quedando inhibida. Otra forma en la que la PKM2 queda inhibida (en presencia de esta enfermedad) es mediante la inactivación o la regulación insuficiente de PPIasa (peptidil-prolil cis-trans-isomerasa) o la elevada concentración de ROS (especies reactivas de oxígeno), ya que ambas impiden la translocación de la PKM2 al núcleo, y consecuentemente esta no se une a HIF1-α, dando lugar en este caso a un desequilibrio en el metabolismo mediante una cascada de PEP. Respecto al papel de la PKM2 en la respuesta inflamatoria, la PKM2 regula la respuesta de los macrófagos, que son esenciales a la hora de retirar los amiloides para evitar la formación de placas. Esto se debe a que ante la inflamación, los macrófagos adoptan un fenotipo que favorece la síntesis de ATP y se ha observado que algunos incluso activan la Glucólisis Aeróbica (efecto Warburg), que se da cuando hay una sobreactivación de la PKM2. La Glucólisis Aeróbica mediada por PKM2 también promueve la activación de inflamasomas mediante la fosforilación de EIF2AK2 (factor 2 de iniciación de traducción de la α quinasa 2). Por otro lado, mediante la interacción con HIF1-α se promueve la transcripción de genes que activan la glucólisis en macrófagos. Finalmente, la PKM2 también es responsable de la glucólisis de la α-sinucleína que es una proteína presináptica asociada a la fisiopatología del Alzheimer.
Esquema que ilustra las consecuencias de la enfermedad del Alzheimer sobre la actividad de la piruvato quinasa. A la izquierda se indican las principales formas mediante las que se inhibe a la PK en condiciones patológicas (inactivación de las moléculas HIF1-α, PPIasa y la elevada concentración de radicales libres). A la derecha sin embrago, se exponen los procesos que se activan como consecuencia de la patología. A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
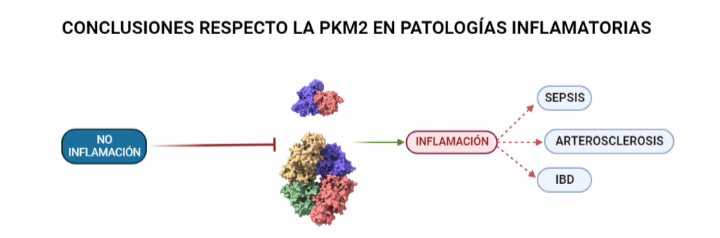
PKM2 en diversas enfermedades inflamatorias
La respuesta inflamatoria, como se ha expuesto en los dos casos anteriores, requiere de un alto consumo de energía. Células del sistema inmune, como los macrófagos, activan mecanismos similares al denominado efecto Warburg en el cáncer para aumentar la síntesis de energía y poder responder de manera eficaz. Esto está íntimamente relacionado con la glucólisis por lo que la implicación de la PK es crucial. En el caso de la Sepsis la implicación de la PKM2 en la glucólisis anaeróbica que adoptan las células bajo la patología supone un perjuicio para la supervivencia del individuo. Esto se ha observado tras inhibir su actividad comprobando que de este modo también quedaban inhibidas moléculas como el inflamasoma NLRP3 y HMGB1 (proteína de alta movilidad del grupo 1) cuya represión favorece la supervivencia del individuo. En el caso de la Arterosclerosis se ha observado que la inhibición de PKM2 en linfocitos B y T favorece la atenuación de la inflamación. Esto está unido a que parte de los productos que se sintetizan durante la glucólisis aeróbica (adoptada por las células del sistema inmune bajo condiciones de estrés) producen inflamación como se expone en la segunda imagen de la PKM2 en el cáncer. Incluso en la IBD (enfermedad inflamatoria de Bowel) la PKM2 se está valorando como posible bioindicador de la enfermedad según estudios recientes. Una vez más, la supresión de la PKM2 favoreció el control sobre la inflamación ya que en su versión nuclear dimérica contribuye a la adaptación de las necesidades metabólicas de los linfocitos. Esto significa que la presencia (en muestras) de esta proteína en estado dimérico podría ayudar a identificar la enfermedad. También cabe mencionar, que se ha observado que la PKM2 regula a Bcl-xL impidiendo la apoptosis de las células del epitelio intestinal.
Ilustración que refleja concisamente la influencia que tiene la PK en enfermedades inflamatorias como son: la Sepsis, la Arterosclerosis y la IBD. A partir de PDB 1a3w y 6wp3 (en el caso de la estructura dimérica). Creado por María Arranz con ChimeraX/BioRender.com
Israelsen, W. J., & Vander Heiden, M. G. (2015). Pyruvate kinase: Function, regulation and role in cancer. Seminars in cell & developmental biology, 43, 43–51. https://doi.org/10.1016/j.semcdb.2015.08.004
June 2022, Faiza Ahmed, Jonathan Ash, Thirth Patel, Auriel Sanders, David Goodsell, Shuchismita Dutta
Oria-Hernández, J., Cabrera, N., Pérez-Montfort, R., & Ramírez-Silva, L. (2005). Pyruvate kinase revisited: the activating effect of K+. The Journal of biological chemistry, 280(45), 37924–37929. https://doi.org/10.1074/jbc.M508490200
Patel, S., Das, A., Meshram, P., Sharma, A., Chowdhury, A., Jariyal, H., … & Shard, A. (2021). Pyruvate kinase M2 in chronic inflammations: a potpourri of crucial protein–protein interactions. Cell Biology and Toxicology, 37(5), 653-678.
The RCSB PDB «Molecule of the Month»: Inspiring a Molecular View of Biology D.S. Goodsell, S. Dutta, C. Zardecki, M. Voigt, H.M. Berman, S.K. Burley (2015) PLoS Biol13(5): e1002140. doi: 10.1371/journal.pbio.1002140
Yang, L., Venneti, S., & Nagrath, D. (2017). Glutaminolysis: a hallmark of cancer metabolism. Annual review of biomedical engineering, 19, 163-194.
El plegamiento de proteínas y las mutaciones del código genético
escrito por C. Menor-Salvan | 17 diciembre, 2024
C. Menor-Salván ,oct 2022. V2 Nov 2023
En el año 1969 ya se conocía mucho sobre la estructura de las proteínas y hacía 10 años que había nacido la Biología Molecular moderna. Ya se entendía la relación entre el código genético, la secuencia de aminoácidos de un péptido y la proteína final. Poco tiempo antes, el bioquímico Christian Anfinsen había descubierto que una proteína desnaturalizada podía recuperar su estructura y actividad en cuestión de milisegundos, por lo que quedaba demostrado que la estructura estaba, de alguna manera, codificada en su secuencia.
Para Cyrus Levinthal esto era paradógico: una secuencia de aminoácidos tiene un número enorme de posibilidades estructurales, por lo que las diferentes conformaciones posibles no pueden tener la misma probabilidad y el plegamiento no es resultado de la búsqueda de la conformación nativa, sino que debía ser dirigido por un camino específico y predeterminado. La resolución de la paradoja de Levinthal, es decir, entender cómo se pliegan las proteínas, es uno de los problemas más complejos de la Bioquímica y la Biofísica, y sólo ahora, gracias a la inteligencia artificial y a los métodos computacionales modernos, estamos en condiciones de resolverla completamente.
La paradoja de Levinthal: si un péptido de 100 aminoácidos puede generar tantas conformaciones, más que átomos hay en el Universo, ¿cómo es posible que se pliegue formando proteínas bien definidas?. A la derecha, Cyrus Levinthal con unos estereomicroscopios clásicos de Bausch&Lomb.
El plegamiento proteico es un problema muy complejo, incluso para las células que generan las proteínas: los fallos o, simplemente, las diferentes soluciones al plegamiento estable de un péptido están detrás de enfermedades como el Alzheimer ,las enfermedades por priones o las amiloidosis. Pero, aunque sea tan complejo, podemos señalar algun aspecto interesante, como es el papel esencial de los aminoácidos hidrofóbicos y su relación con la evolución del código genético.
Los inicios de la aplicación de los métodos computacionales a la estructura de proteínas. Actualmente, un PC portátil es miles de veces más potente que esas enormes computadoras. Aun así, fueron claves para la comprensión de las primeras estructuras proteicas, como la de la mioglobina o la lisozima. Este sistema, llamado ‘Kluge’ fue utilizado por Levinthal.
El colapso hidrofóbico
El proceso de plegamiento proteico es gradual, formándose regiones, llamadas foldones, que van adquiriendo conformaciones similares a la forma nativa final. Estos foldones son cooperativos y, cuando empiezan a formarse, favorecen el plegamiento del resto de la proteína. Esto se denomina la hipótesis del foldón.
En la formación de las regiones plegadas es esencial la estabilización energética que proporciona el efecto hidrofóbico, es decir, la expulsión del agua y el secuestro de los grupos R de los aminoácidos hidrofóbicos en el interior de la estructura plegada, dejando los grupos polares y cargados expuestos al solvente. Este efecto es uno de los responsables de la extraordinaria capacidad de las enzimas como catalizadores: al tener un núcleo hidrofóbico, la exclusión del agua y la desolvatación de los sustratos de las enzimas promueven la actividad enzimática.
Esta fase de plegamiento de la proteína, en la que adquiere su estructura nativa en regiones y se ha producido el colapso hidrofóbico, es muy dinámica: la estructura se mueve, los foldones se forman y reconvierten de un modo fluido. Este estado se denomina glóbulo fundido. Finalmente, la proteína alcanza un estado similar a un sólido tridimensional y se estabiliza, formando el estado nativo de la proteína. En este estado, aparecen otras interacciones, como los puentes disulfuro, el stacking de residuos de aminoácidos aromáticos y la unión de cofactores, que terminan de ‘fijar’ la estructura nativa.
En estas condiciones, tenemos la proteína plegada y funcional. Así puede cristalizar, lo cual es muy importante para conocer la estructura. Pero el estado nativo no es totalmente sólido: la proteína puede desnaturalizarse, o volver al estado de glóbulo fundido y cambiar sus conformaciones en respuesta a algún estímulo.
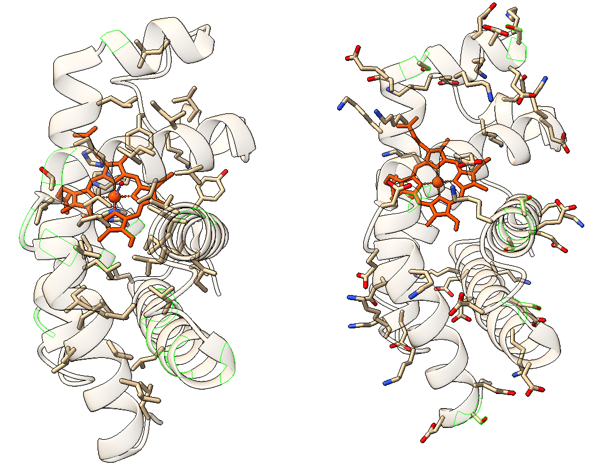
Estructura de la lisozima (derecha), mostrando su plegamiento nativo, en el que se observan dos regiones, una con hélices alfa y, en la parte inferior, un motivo de lámina beta antiparalela. En amarillo, los puentes disulfuro que estabilizan la estructura. Izquierda: Estructura mostrando los residuos hidrófobos, que ‘colapsan’ en el núcleo de la proteína, creando un centro donde el agua queda excluida. Centro: Estructura mostrando los resíduos polares y con carga, que se organizan hacia el exterior, en contacto con el agua o con otras estructuras. Cristal de lisozima, la proteína mostrada en la imagen anterior. La cristalización de las proteínas es un proceso en muchos casos necesario para poder determinar la estructura tridimensional de las proteínas. Los cristales obtenidos, como éste, generado en nuestro laboratorio, permiten obtener las coordenadas de los átomos de la estructura mediante difracción de rayos X.
La idea del colapso hidrofóbico puede parecer contraintuitiva: como es posible que una fuerza débil, como la fuerza de Van der Waals entre grupos apolares, sea lo que dirige el plegamiento de la proteína. ¿cual es el papel de los enlaces de hidrógeno?. Al principio se pensaba que los enlaces de hidrógeno, mucho mas fuertes, eran esenciales en la estabilización de la estructura proteica. Actualmente sabemos que los enlaces de hidrógeno juegan un papel director durante el proceso de plegamiento, ya que dirigen la formación de diversas estructuras secundarias, que se agrupan en motivos y van dando lugar al proceso cooperativo de plegamiento. Sin embargo, no sólo no son esenciales en la estabilización de la estructura nativa, sino que, en ocasiones, son desestabilizadores que favorecen los cambios de conformación. La clave no es la fuerza de las interacciones hidrofóbicas per se, sino el efecto termodinámico, como la variación de entropía asociada al efecto hidrofóbico, que favorece el proceso.
Dada la importancia del colapso hidrofóbico, está claro que los aminoácidos hidrofóbicos van a ser claves en la evolución de las estructuras. Ello ha condicionado la evolución del código genético.
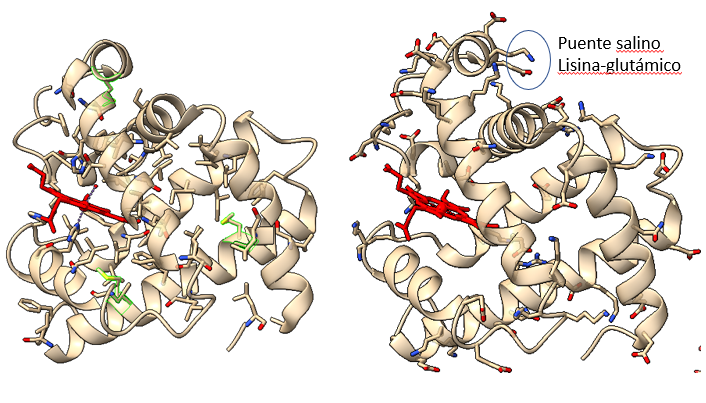
Estructura de la mioglobina, una de las primeras proteínas cuya estructura terciaria fue resuelta. A la derecha, aminoácidos hidrófobos, orientados hacia el interior de la estructura. A la izquierda, aminoácidos cargados, orientados hacia el exterior de la estructura. Algunos forman puentes salinos, que estabilizan la estructura y, en el caso de la mioglobina, juegan un papel importante relacionado con su función. En rojo, grupo hemo. Si rotamos la molécula y la observamos longitudinalmente, se ve mejor el efecto. A la izquierda, aminoácidos hidrófobos Leu, Ile y Val, orientados hacia el interior. A la derecha, aminoácidos cargados Asp, Glu y Lys orientados al exterior.
Código genético, mutaciones y la preservación de la estructura proteica
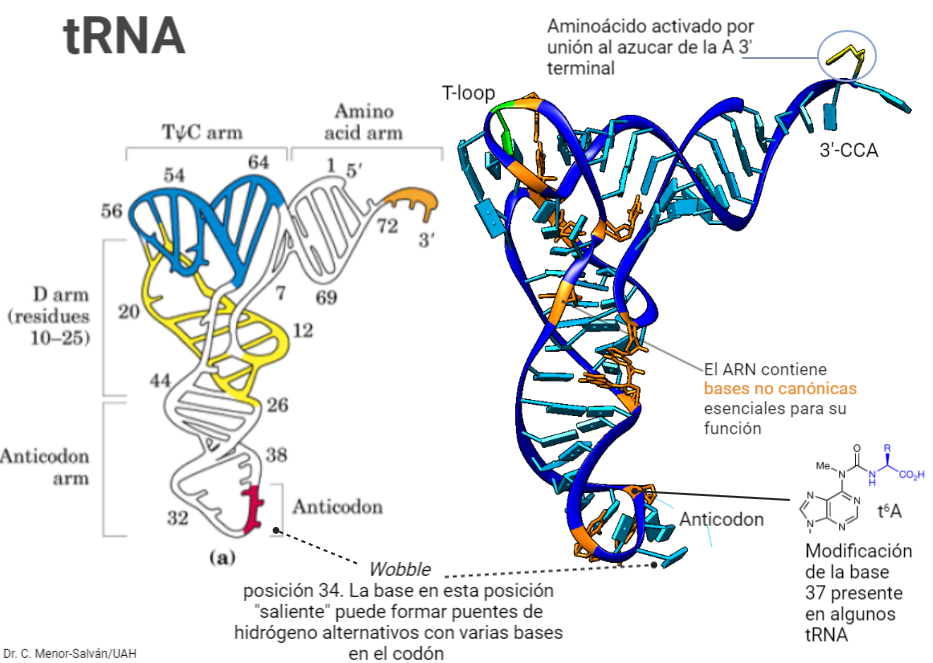
La secuencia de bases del ADN sufre constantes cambios: mutaciones que cambian bases en la secuencia y que se traducen en cambios en la secuencia de una proteína. Estos cambios pueden ser deletéreos, pero también silenciosos o incluso beneficiosos, dando lugar a nuevas funciones. El código genético ha evolucionado de tal forma que las mutaciones mas comunes, llamadas mutaciones missense, en las que un par de bases sustituye a otro, limiten sus efectos sobre las proteínas resultantes. Afortunadamente, el código genético, que se compone de unas ‘palabras’ de tres letras, llamadas codones, esta degenerado. Esto quiere decir que los 64 posibles codones (‘palabras’ de tres letras formadas con las bases A, G, C y U, es decir 43) van a codificar para tan solo 20 aminoácidos distintos. Ello implica que algunos aminoácidos van a tener hasta 6 codones sinónimos. Esto es posible gracias al balanceo o wobble del RNA de transferencia: la tercera posición del anticodon puede reconocer diferentes bases en el codón, gracias a la formación de pares de bases no Watson-Crick, o la presencia de bases no canónicas en esa posición.
Estructura terciaria de un RNA de transferencia, mostrando sus características más importantes: el patrón de minihélices que se repite, la terminación CCA en 3′ con el aminoácido unido en su posición, y el anticodón, cuya base en la posición 34 es la posición de balanceo o wobble y puede puede formar puentes de hidrógeno alternativos. Por ejemplo, una uridina en el anticodón puede reconocer tanto una adenina como una guanina en el codón.
Gracias al balanceo, hay tan solo 32 diferentes tRNA para todos los codones, y estos transportan sólo 20 aminoácidos (es decir, hay más de un tRNA por aminoácido en algunos casos). Por ejemplo, un tRNAarg de las levaduras tiene el anticodón GCI (donde I es inosina, una base que usualmente no está presente en el DNA ni el RNA). La posición I es de balanceo y puede unirse a los codones CGA, CGU y CGC. Ahora, imagina que hay una mutación y un codón CGU se transforma en un codón CGC. Esta sería una mutación silenciosa, pues, al traducirse el gen, seguiría codificando para una arginina y no se producirían cambios en la proteína. La degeneración del código genético hace posible que cambie el aminoácido sólo en un 25% de las mutaciones de este tipo. Ello aporta estabilidad al genoma: durante la evolución, solo perduraron los genomas capaces de mantener sus estructuras funcionales en un ambiente con muchos cambios. Así, la degeneración del código genético y la limitación del número de aminoácidos pudo aportar una ventaja selectiva a los organismos, reduciendo los efectos de las mutaciones. Un potencial organismo con otro código que implicara más aminoácidos debió ser inviable con las mismas tasas de mutación.
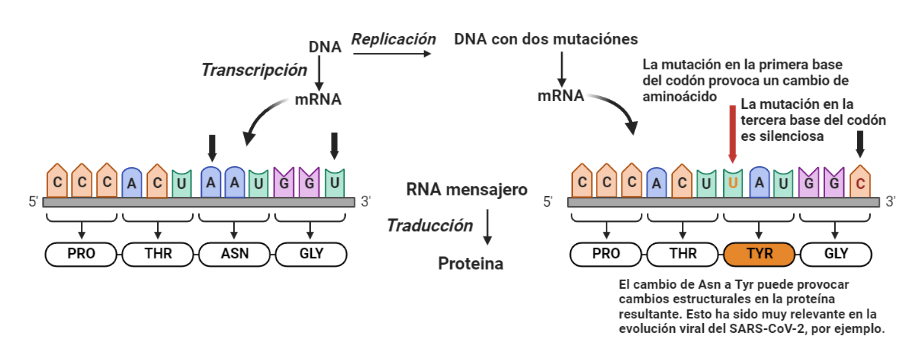
Vista simplificada del efecto de las mutaciones según la posición del codón. Las mutaciones representadas estan basadas en un modelo real, la evolución del SARS-CoV-2 hacia nuevas variantes. El cambio de asparagina a tirosina representa el caso habitual: no cambia la clase de aminoacido (ambos son aminoacidos polares, y la estructura global se va a mantener) pero las propiedades son lo suficientemente distintas como para que se produzcan cambios funcionales o de estabilidad en la proteína. En este caso, la tirosina tiene un anillo aromático, lo que le permite crear interacciones acidicionales.
Aun así, el efecto del wobble no es suficiente; es necesario proteger la función de las proteínas, y, para ello, es clave mantener su estructura. Así, los aminoácidos hidrofóbicos están especialmente protegidos, debido a su paper fundamental en el plegamiento proteico. Cuando se produce una mutación en la primera posición del codón, se va a producir un cambio de aminoácido. Pero, en general, el cambio da lugar a la sustitución de un aminoácido por otro de propiedades similares. Por ejemplo, la valina, un aminoácido hidrófobo, esta codificada por un codón GUU. Si hay una mutación en la tercera posición y se transforma en GUC, sigue codificando para valina, y estamos ante una mutación silenciosa. Pero si hay una mutación en la primera posición y se convierte en AUU, el aminoácido cambia a isoleucina, que también es hidrófobo y tiene propiedades similares. Si la mutación lo convierte en CUU, el aminoácido cambia a leucina, que, de nuevo, es hidrófobo y tiene propiedades similares. Estos cambios no alteran significativamente el plegamiento proteico, ya que el colapso hidrófobo va a seguir sucediendo, por lo que, a pesar de las mutaciones, el plegamiento (y por tanto, la función) se va a mantener.
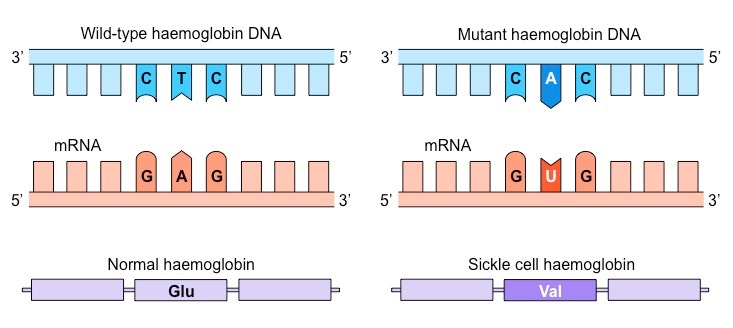
El problema surge cuando se produce un cambio de un aminoácido a otro de un tipo diferente. Esto puede provocar un cambio más o menos drástico en la estructura o propiedades de la proteína. Es el caso, por ejemplo, de la anemia falciforme, en la que hay una mutación de una base en el gen de la cadena beta de la hemoglobina. El aminoácido mutado cambia el codón para un ácido glutámico CTC por el codón para la valina CAC.
Mutación en la cadena beta de la hemoglobina, que provoca un cambio de un aminoácido con carga a un aminoácido hidrofóbico.
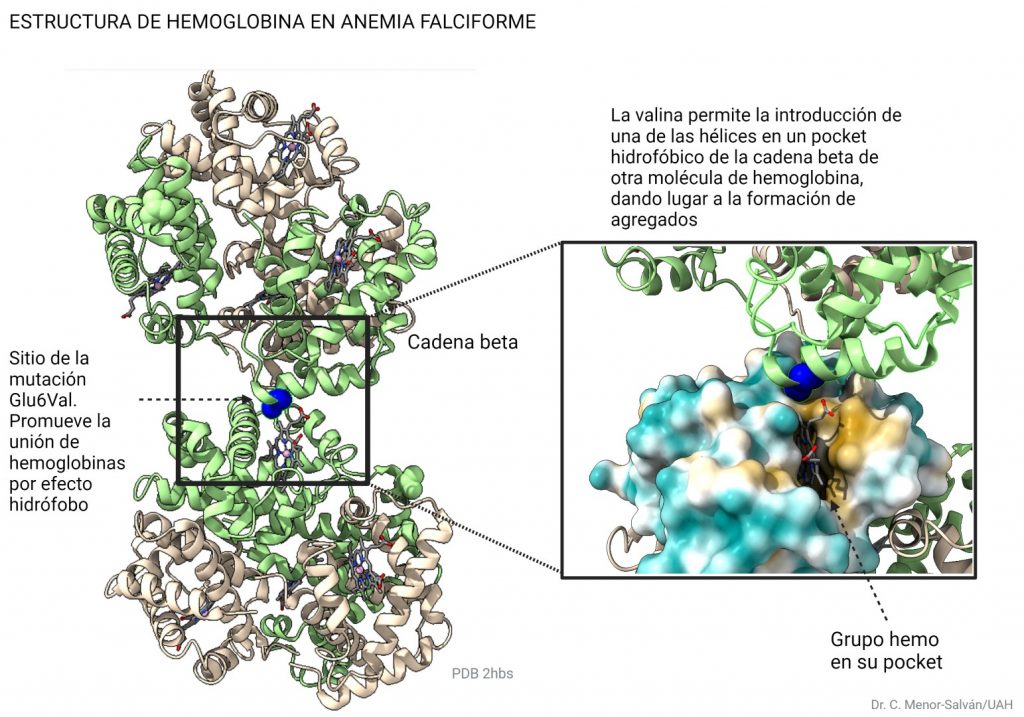
El cambio elimina un aminoácido cargado, el glutámico, por un aminoácido hidrófobo, la valina. Al desaparecer la carga, desaparece la repulsión electrostática entre unidades de hemoglobina, y se sustituye por un aminoácido hidrófobo superficial, que provoca una interacción hidrófoba entre cadenas. Como resultado, la hemoglobina precipita en forma de fibras. De nuevo, los aminoácidos hidrófobos son claves en la estructura. Aquí, la introducción del aminoácido hidrófobo valina tiene consecuencias relevantes, provocando la enfermedad de la anemia falciforme.
Aunque un cambio de aminoácido no tenga gran efecto estructural, influyen en la variabilidad genética y pueden tener importancia. Los SNP o polimorfismos de nucleótido único, son cambios en la secuencia de genes que se traducen en modificaciones de algún aminoácido en las proteínas. Estos SNP crean muchas variaciones entre individuos de la misma especie, y, normalmente, no tienen ningún efecto.
Sin embargo, en algunos casos, los cambios afectan a aminoácidos que alteran ligeramente la función de la proteína (haciendo una enzima mas activa o menos activa, o modificando la afinidad de una proteína por un sustrato). Aunque un cambio de afinidad sea pequeño, tienen una gran importancia farmacológica, explicando la diferente respuesta a algunos fármacos entre individuos. Entre diversas especies, los cambios son mayores para la misma proteína (o para la proteína ortóloga: proteína que ejerce la misma función en otro organismo y proviene del mismo ancestro común, y que normalmente son estructuralmente equivalentes). Esto permite trazar la evolución de las especies y de las propias proteínas.
Referencias
Alas-Guardado, S. de J., Rojo, A. and Merino, G. (2011) ‘La paradoja de Levinthal: cuando una contradicción se vuelve lógica’, Educación Química, 22(1), pp. 51–54. doi: 10.1016/S0187-893X(18)30114-9.
Baldwin, R. L. & Rose, G. D. (2013) ‘Molten globules, entropy-driven conformational change and protein folding’, Current Opinion in Structural Biology, 23(1), pp. 4–10. doi: 10.1016/j.sbi.2012.11.004.
Braakman, R. & Smith, E. (2013) ‘The compositional and evolutionary logic of metabolism’, Physical Biology, 10(1). doi: 10.1088/1478-3975/10/1/011001.
Englander, S. W. & Mayne, L. (2014) ‘The nature of protein folding pathways’, Proceedings of the National Academy of Sciences of the United States of America, 111(45), pp. 15873–15880. doi: 10.1073/pnas.1411798111.
Francoeur, E. (2002). Cyrus Levinthal, the Kluge and the origins of interactive molecular graphics. Endeavour, 26(4), 127–131. doi:10.1016/s0160-9327(02)01468-0
Honig, B. (1999) ‘Protein folding: From the levinthal paradox to structure prediction’, Journal of Molecular Biology, 293(2), pp. 283–293. doi: 10.1006/jmbi.1999.3006.
Karplus, M. (1997) ‘The Levinthal paradox: yesterday and today’, Folding and Design, 2, pp. S69–S75. doi: 10.1016/S1359-0278(97)00067-9.
Kurata, S., Weixlbaumer, A., Ohtsuki, T., Shimazaki, T., Wada, T., Kirino, Y., Takai, K., Watanabe, K., Ramakrishnan, V. & Suzuki, T. (2008) ‘Modified uridines with C5-methylene substituents at the first position of the tRNA anticodon stabilize U·G wobble pairing during decoding’, Journal of Biological Chemistry, 283(27), pp. 18801–18811. doi: 10.1074/jbc.M800233200.
Pak, D., Root-Bernstein, R. & Burton, Z. F. (2017) ‘tRNA structure and evolution and standardization to the three nucleotide genetic code’, Transcription, 8(4), pp. 205–219. doi: 10.1080/21541264.2017.1318811.
Root-Bernstein, R., Kim, Y., Sanjay, A. & Burton, Z. F. (2016) ‘tRNA evolution from the proto-tRNA minihelix world’, Transcription, 7(5), pp. 153–163. doi: 10.1080/21541264.2016.1235527.
¿Por qué aumenta la infectividad del coronavirus?: Un sencillo experimento virtual con la ‘variante británica’ del SARS-CoV-2
escrito por C. Menor-Salvan | 17 diciembre, 2024
por C. Menor-Salván
En los medios y redes sociales ha causado una gran alarma la aparición, reportada en diciembre de 2020 en Gran Bretaña, de una nueva variante del virus SARS-CoV-2. Esta variante, denominada B.1.1.7, contiene varias mutaciones puntuales respecto de la variante original que se extendió en febrero-marzo de 2020. Según los datos clínicos disponibles, la variante B.1.1.7 tiene mayor infectividad y velocidades de transmisión. Desde diciembre, según el COVID-19 UK Genomics Consortium, más del 50% de los nuevos contagios son de ésta variante, que presenta una transmisibilidad incrementada entre un 50 y un 70% respecto de la variante original que llamaremos WT (por ‘wild type’). Como es lógico, la aparición de variantes con mayor transmisibilidad crean alarma, pero son perfectamente esperables, dado que estamos forzando el proceso de evolución viral: mediante confinamientos y medidas anticontagio, creamos una selección artificial de las variantes más infectivas, que prevalecen sobre las variantes originales.
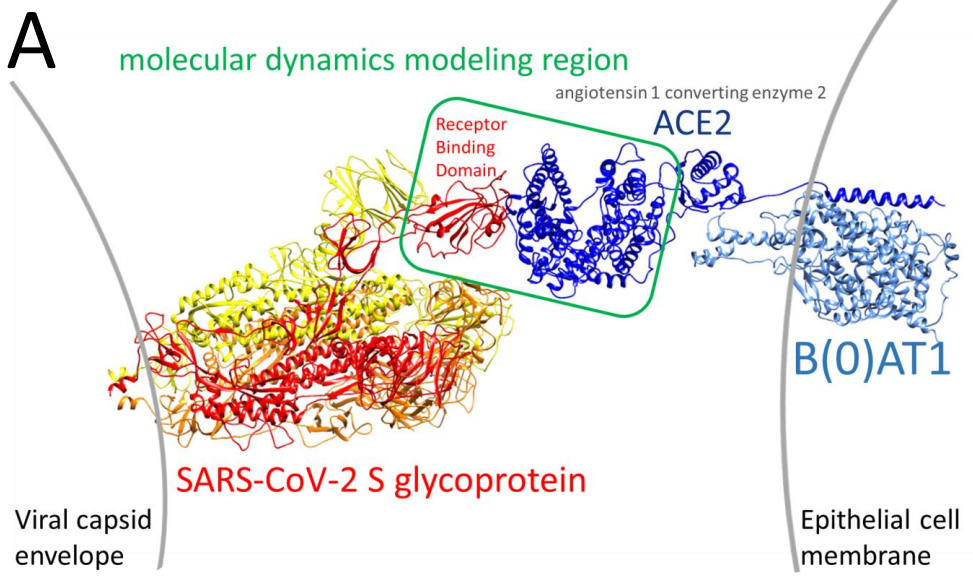
La infectividad viral es una consecuencia de varios aspectos moleculares del virus; uno de ellos es la afinidad por el receptor celular, que una proteína del virus reconoce, y al que se une, iniciando el proceso de infección. La proteína viral que reconoce el receptor en la célula que va a infectar es la proteína de la espícula. Como si de un puzzle molecular se tratara, la espícula viral ‘encaja’ en el receptor. Os remito a la Noticia Nº 59 para una introducción general al SARS-CoV-2
La proteína de la espícula, que contiene un dominio o región, que interacciona con el receptor de la célula.
Como es lógico, si modificamos la proteína de la espícula de modo que mejore la estabilidad de su interacción con el receptor, aumentará la infectividad. ¿como?. Para entenderlo, hay que tener en cuenta que un virus es un agregado supramolecular. Digamos que es una gran molécula heterogénea. No es un ser vivo. Si el virus tiene mayor afinidad por su receptor, quiere decir que la infección se producirá a una concentración menor. Dicho de otra manera, si aumentamos la afinidad del virus modificado por el receptor, se producirá una infección con una cantidad de virus menor que los necesarios con el virus sin modificar. Y, posiblemente, este proceso está detras del efecto de la variante UK B.1.1.7: una de sus mutaciones se encuentra precisamente en la región de la espícula que interacciona con el receptor de la célula a infectar.
En ésta entrada voy a mostraros como nosotros mismos podemos ver éste efecto: voy a tomar los datos de la estructura PDB 60MJ y, a partir de ellos, voy a:
Calcular las interacciones por puentes de hidrógeno entre la espícula y el receptor. Estas interacciones determinan la estabilidad el complejo espícula-receptor, y, como es lógico, cuando más puentes y más fuertes, mayor estabilidad = mayor afinidad =mayor infectividad. También observaremos otras interacciones evidentes, como el stacking pi: un tipo de enlace que se produce entre aminoácidos aromáticos.
Voy a introducir la mutación N501Y de la ‘variante británica’, que implica el cambio de la asparagina 501 de la espícula por una tirosina, justamente en la región de interacción entre la espícula y el receptor.
Después, usando el software, el ordenador calculará la estructura más estable resultante de ése cambio y veremos si la mutación introduce la aparición de nuevas interacciones. Si la mutación implica la aparición de nuevos enlaces, esto explica fácilmente el aumento de infectividad de la ‘variante británica’.
Este procedimiento lo voy a llevar a cabo usando los softwares UCSF Chimera, Pymol y Chem3D.
La mutación N501Y de la ‘variante británica’ del SARS-CoV-2 introduce nuevas interacciones y podría aumentar la afinidad por el receptor y la infectividad
Como hemos comentado, la unión entre la espícula viral y el receptor desencadena el proceso infectivo. Esta unión se lleva cabo gracias a que se establecen una serie de interacciones no covalentes entre las dos proteínas: puentes de hidrógeno e interacciones tipo apilamiento entre anillos aromáticos. Cuanto más fuerte sea la unión, mayor estabilidad tendrá el complejo y, por tanto, mayor afinidad tendrá el virus por el receptor. Al aumentar la afinidad, aumenta la infectividad del virus, dado que se requiere una inoculación de menor cantidad de virus para producir una infección efectiva. Si, por ejemplo, el virus WT necesita (número arbitrario) introducir 1000 unidades de virus en el hospedador para producir una infección, al aumentar la afinidad por el receptor ese número se puede reducir a 500-300.
Interacción entre la glicoproteína S o espícula viral y la proteína receptora ACE2 de la célula, que a su vez forma un complejo con el transportador transmembrana de aminoácidos B0AT1. En las siguientes figuras, nos centraremos en la zona recuadrada, donde se produce la interacción espícula-receptor. Imagen tomada de Rynkiewicz et al. (2021)
Centrándonos en las estructuras del dominio RBD de la espícula y de la proteína ACE2 (recuadrados en la figura anterior), vamos a ver el resultado que obtenemos usando los datos de la variante original del virus:
Vemos la estructura molecular del dominio RBD, en azul. Este dominio es la parte de la espícula que se une al receptor celular, la proteína ACE2. Las líneas azules representan los puentes de hidrógeno entre aminoácidos. Aquí tienen especial protagonismo el Gln 493 y la lisina 417, que forman puentes con el receptor. La fenilalanina 486 da lugar a un apilamiento pi con una tirosina del receptor. Esta interacción no existía en el SARS de 2003, y es una de las que explica la mayor afinidad (e infectividad) del SARS-CoV-2. En amarillo veis la Asn (asparagina) 501, que ya en su momento se señalaba como un aminoácido potencialmente interesante en la estructura. Vamos a ver qué ocurre cuando introducimos la mutación:
Fe de errata: Donde dice «E50Y» debe decir «N501Y»
La introducción de la mutación, con la nueva tirosina en la posición 501, tiene consecuencias que son sutiles pero de gran importancia. Primero, aparecen nuevas interacciones entre el nuevo aminoácido y el receptor: nuevos puentes de hidrógeno y un enlace por stacking pi entre la tirosina 501 y una tirosina del receptor. Por otro lado, interacciones que ya existían previamente disminuyen su distancia. Los puentes de hidrógeno entre las lisina 417 y la glutamina 493 se hacen más cortos (y por tanto mas fuertes, ya que es una interacción electrostática). Como resultado la proteína mutante se une con mayor estabilidad al receptor, ya que aumentan el número de puntos de atracción electrostática y disminuye la distancia de las que ya existían. Esto da lugar a una unión más estable, y, por tanto, a un aumento de la afinidad. Esto se traduce en la práctica en un aumento de la infectividad de la variante.
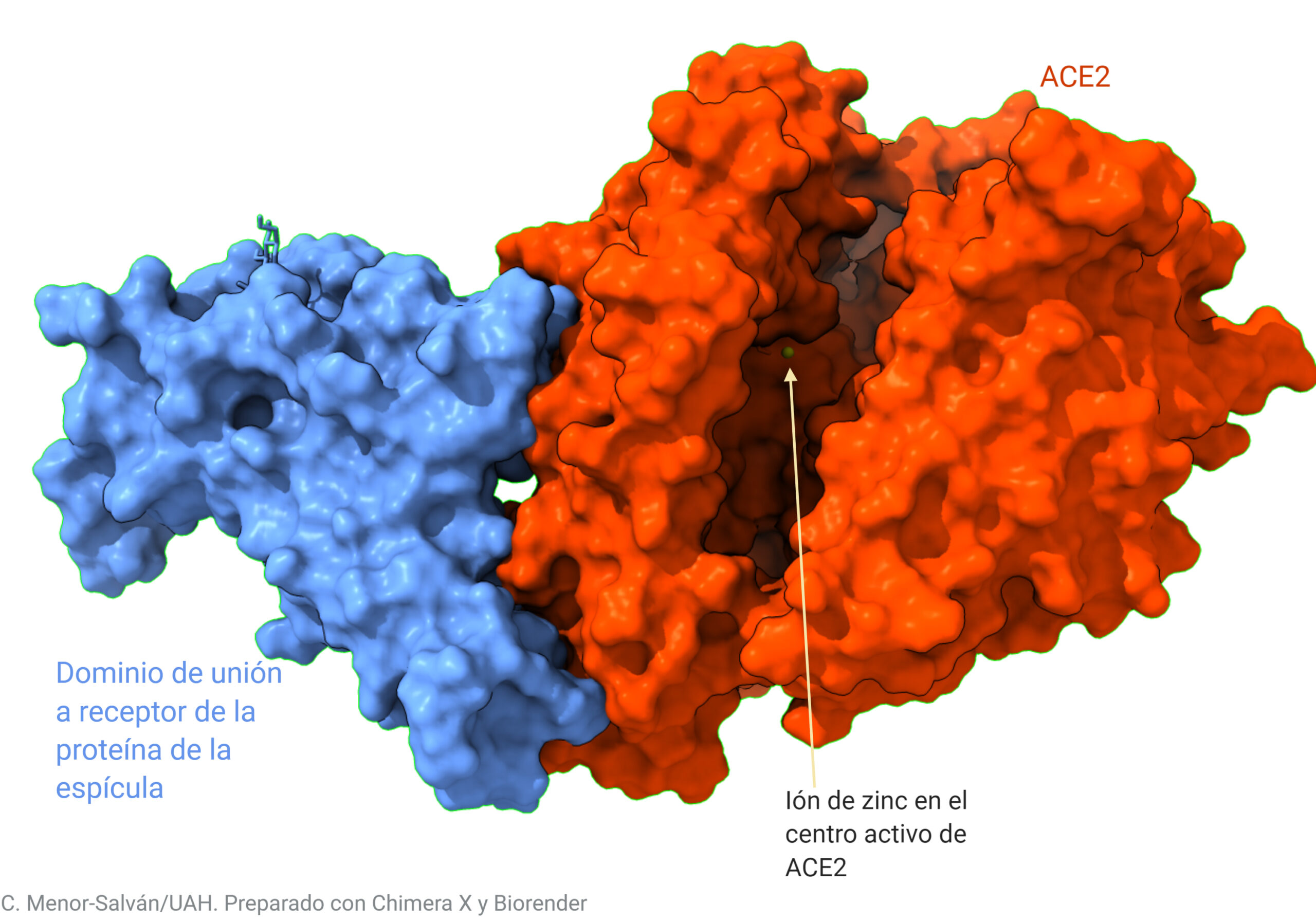
Las representaciones de proteínas son algo abstractas. Para que el lector pueda ver más intuitivamente cómo se produce la unión, vamos a ver las superficies de las moléculas de proteína. Estas interaccionan ‘encajando’ y uniéndose por las interacciones electrostáticas, de modo similar a como si uniéramos unas piezas con imanes.
Interacción entre el dominio de unión de la proteína de la espícula y el receptor. El receptor es una enzima del tipo zinc- metaloproteasa, y puede observarse el gran surco del centro activo, donde encaja la angiotensina.
En cierto modo, el virus y su receptor interaccionan de modo similar a las piezas de un conector magnético, pero con una interacción electrostática en lugar de magnética.
La fuerza y geometría de las interacciones electrostáticas determina la estabilidad de la unión entre el virus y su receptor, y, por tanto, su afinidad e infectividad. La acción de los anticuerpos se basa en un principio similar. Una pregunta habitual es si ésta nueva variante supondrá una pérdida de eficacia de las vacunas o mejorará la evasión inmune del virus. Todavía queda mucho trabajo para los científicos, pero los primeros datos indican que ésta variante no alterará la eficacia de la vacuna ni la respuesta inmune, al ser mutaciones puntuales que no afectan al reconocimiento de los anticuerpos.
Por otro lado, las modificaciones en el dominio de reconocimiento de receptor en la espícula viral, tampoco implican un aumento de la agresividad del virus, por lo que el aumento de infectividad no va asociado a un aumento de gravedad de las infecciones. Como digo, aún queda mucho que investigar y que aprender y todo puede ir cambiando, pues la pandemia evoluciona más rápido que la capacidad de los científicos para obtener resultados y avances.
Estos sencillos resultados que he mostrado, obtenidos mediante un no menos sencillo análisis computacional de datos de estructuras de proteínas, no son mas que una aproximación muy sencilla. Un estudio mas complejo y riguroso requeriría muchos mas medios y tiempo de los que dispongo, pero espero que sirva para entender la mecánica molecular que hay tras la infección del coronavirus.
Referencias
Kupferschmidt, K. Fast-spreading UK virus variant raises alarms. Science (New York, NY), 371(6524), 9-10.
Lauring, A. S., & Hodcroft, E. B. Genetic Variants of SARS-CoV-2—What Do They Mean?. JAMA.
Rynkiewicz, P., Babbitt, G. A., Cui, F., Hudson, A. O., & Lynch, M. L. A comparative survey of Betacoronavirus binding dynamics relevant to the functional evolution of the highly transmissible SARS-CoV-2 variant N501Y. bioRxiv, 2021: https://doi.org/10.1101/2020.09.11.293258
Tang, J. W., Tambyah, P. A., & Hui, D. S. (2020). Emergence of a new SARS-CoV-2 variant in the UK. Journal of Infection.
Volz, E., Mishra, S., Chand, M., Barrett, J. C., Johnson, R., Geidelberg, L., … & Ferguson, N. M. (2021). Transmission of SARS-CoV-2 Lineage B. 1.1. 7 in England: Insights from linking epidemiological and genetic data. medRxiv, 2020-12.