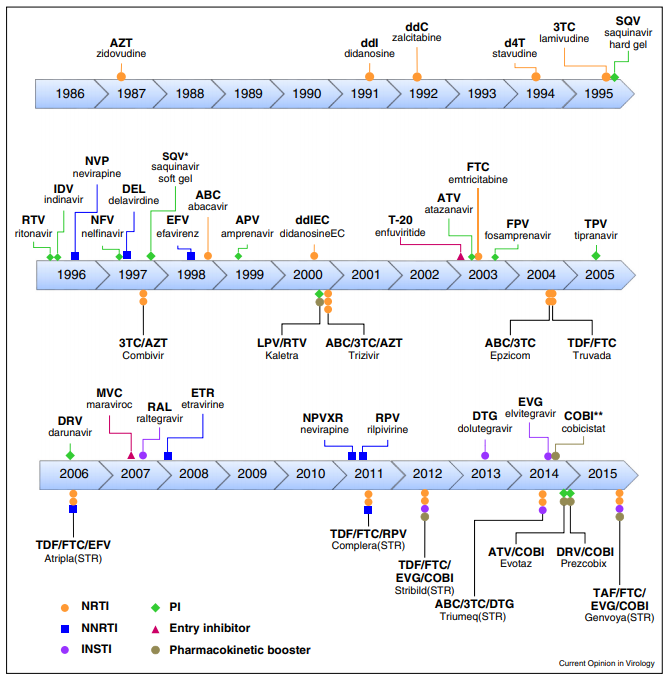
VIH: Ciclo biológico, patogenia y tratamiento (ART y sistema CRISPR-Cas9)
Jorge Maldonado Torres, Carlos Medina Sánchez y Daniel Martín Ruíz
Biología Sanitaria, Universidad de Alcalá
El VIH o virus de la inmunodeficiencia humana es muy conocido por la mayoría de la población debido a que ocasiona el síndrome de la inmunodeficiencia humana (SIDA). En un primer momento, el SIDA fue relacionado con drogadictos y gays, gente que en aquella época era tratada como los desechos de la sociedad. Esta situación es recreada muy bien en el siguiente video, donde se cuenta la historia de cómo un linfocito infectado por VIH es repudiado por el resto pero finalmente los antirretrovirales hacen efecto y permite que este siga bailando junto a otro por el torrente.
Origen
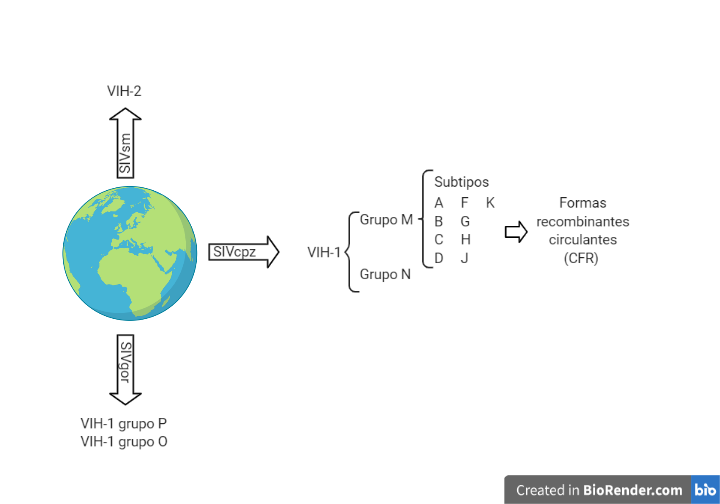
El VIH es un retrovirus perteneciente a la familia de los lentivirus. Este tipo de virus proviene de un salto inter-especie del virus de la inmunodeficiencia del simio (SIV). El VIH-2 (menos patogénico y transmisible) proviene de la variedad SIVsm (Sooty mangabey) y el VIH-1 (causante de la pandemia) a su vez lo podemos dividir en diferentes grupos: el VIH-1 de los grupos M y N proviene del SIVcpz (chimpancé) (1), mientras que el VIH-1 de los grupos O y P proviene del SIVgor (gorila) (2).
Dentro del VIH-1 grupo M encontramos diferentes subtipos a partir de los cuales mediante recombinación entre ellos se puede formar formas recombinantes circulantes. En Europa el subtipo que predomina es el subtipo B (3).
Morfología y ciclo biológico
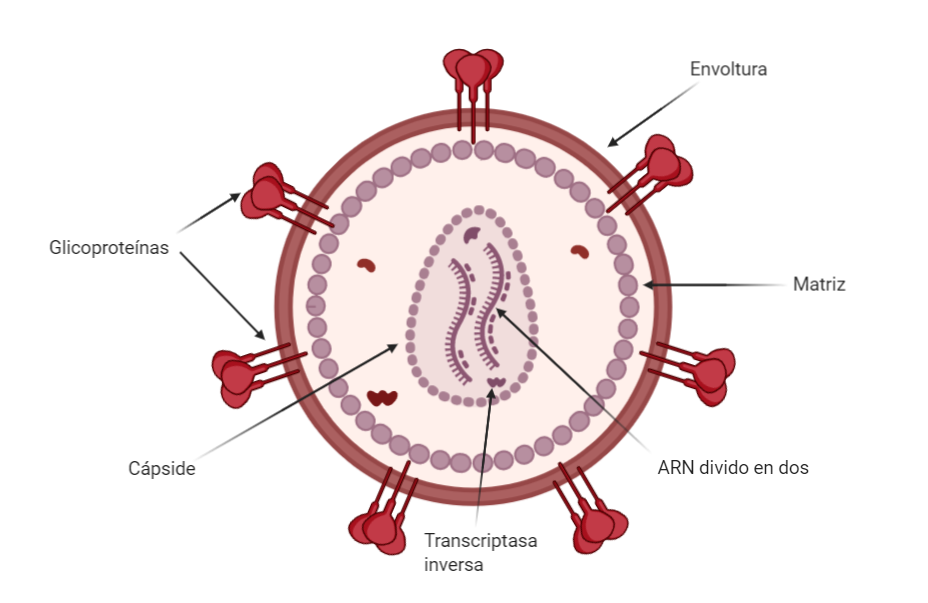
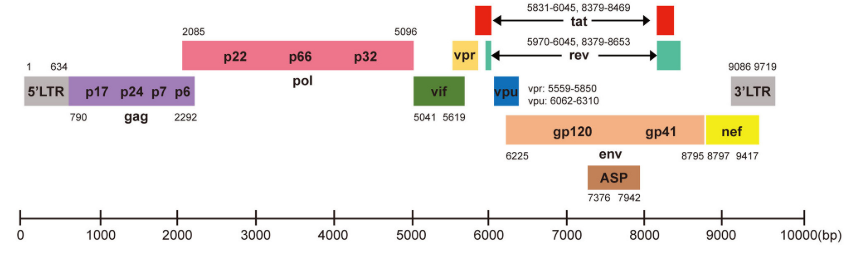
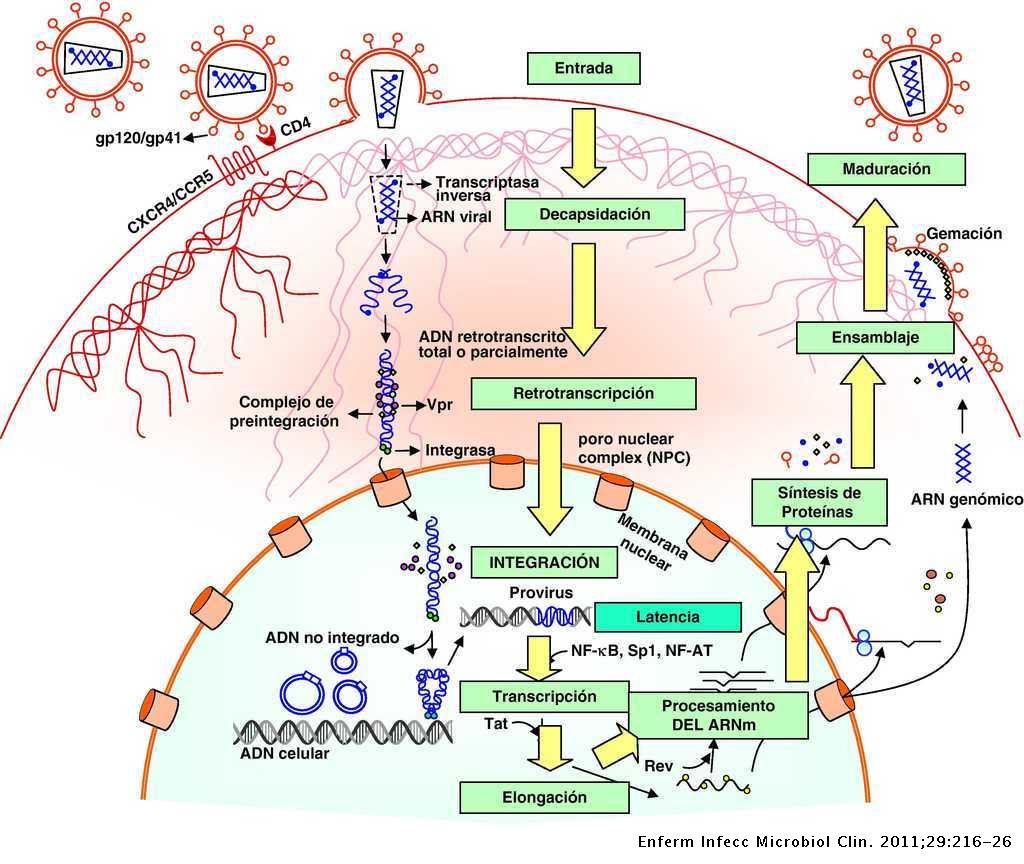
El VIH es un virus esférico con una envoltura lipídica, donde se localizan las espículas, matriz y nucleocápside, donde encontramos dos cadenas de RNA. El genoma viral está compuesto por 9 genes (Gag, Pol, Env, Tat, Rev, Nef, Vif, Vpr, Vpu), cuya estructura aparece en la Figura 2 y cuyas funciones aparecen en la Tabla 1.
| Gen | Proteína | Función |
|---|---|---|
| gag | p24 | Proteína de la cápside. |
| gag | p17 | Proteína de la matriz, favorece el anclaje en la membrana y dirige el complejo de pre-integración hacia el núcleo. |
| gag | p7 | Proteína de la nucleocápside, responsable del reconocimiento y la incorporación del RNA al virión. |
| gag | p6 | Interviene en la incorporación de la proteína Vpr al virión en formación. |
| pol | Transcriptasa inversa (TI) | Sintetiza DNA usando como molde RNA. |
| pol | Integrasa (IN) | Integra el material genómico viral en el genoma del huésped. |
| pol | Proteasa (PR) | Escinde precursores como los del gen gag, interviniendo en la maduración. |
| env | gp120 | Proteína externa de la espícula de la membrana, relacionada con la gp41. Interviene en la fijación a células CD4. |
| env | gp41 | Proteínas transmembrana de la espícula, relacionada con gp120. Interviene en la fijación a células CD4. |
| tat | Tat | Proteína reguladora Activador de la transcripción. |
| rev | Rev | Proteína reguladora Factor de exportación de los RNAm al citosol para la expresión de proteínas. |
| nef | Nef | Proteína accesoria Interfiere en la activación de linfocitos T, induce la regulación negativa de células CD4 y del complejo de histocompatibilidad clase I (MHC I). Interviene en la evasión ante la respuesta inmune (4). |
| vif | Vif | Proteína accesoria Interfiere anulando el efecto antiviral de la proteína A3G (5), favoreciendo la maduración y la infectividad de la partícula vírica. |
| vpr | Vpr | Proteína accesoria Interviene en la entrada al núcleo del complejo de pre-integración. |
| vpu | Vpu | Proteína accesoria Interfiere anulando el efecto antiviral de la proteína Tetherina (6), favoreciendo la liberación de viriones y promoviendo la degradación de CD4 en el retículo endoplásmico. |
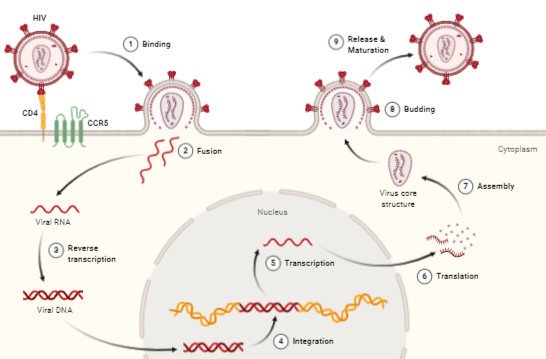
Para que se pueda llevar a cabo la infección y transmisión del VIH es necesario su integración en la célula del huésped. Primero es necesario la interacción con receptores CD4 (localizados en linfocitos T, linfocitos B, monocitos …) y posteriormente con correceptores CCR5 y CXCR4 (receptores con 7 dominios transmembrana), de tal forma que las partículas virales pueden tener tropismo por receptores CCR5, CXCR4 o ambos.
En un primer lugar, la proteína gp120, localizada en la zona exterior de las espículas de la membrana del virión interacciona con el receptor CD4, provocando un cambio de conformación en gp120 que deja expuesto a su dominio V3, para que se una a los correceptores (receptores de quimiocinas). Estos cambios a su vez provocan un cambio conformacional en la proteína gp41, que deja expuesto el péptido de fusión (N-terminal), el cual se ancla a la membrana plasmática provocando el acercamiento y finalmente la fusión de ambas membranas.
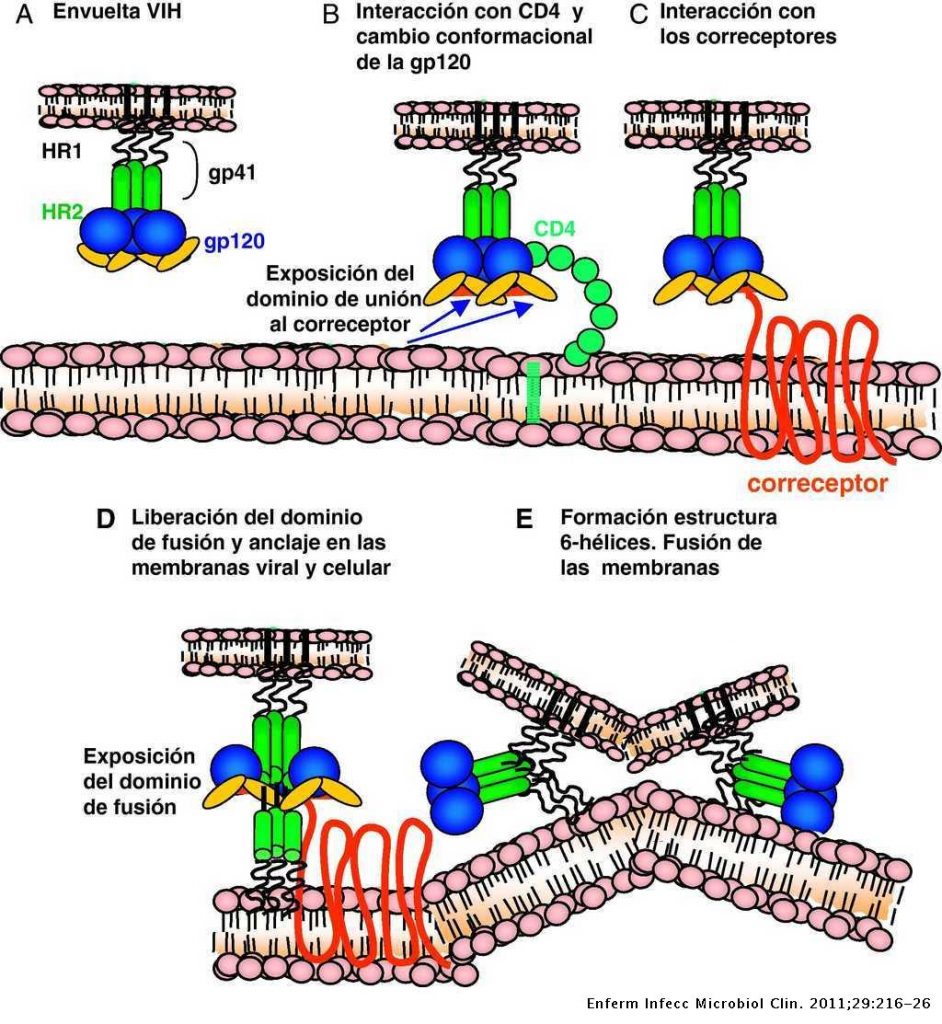
En el siguiente video representa el final del proceso mediado por gp41.
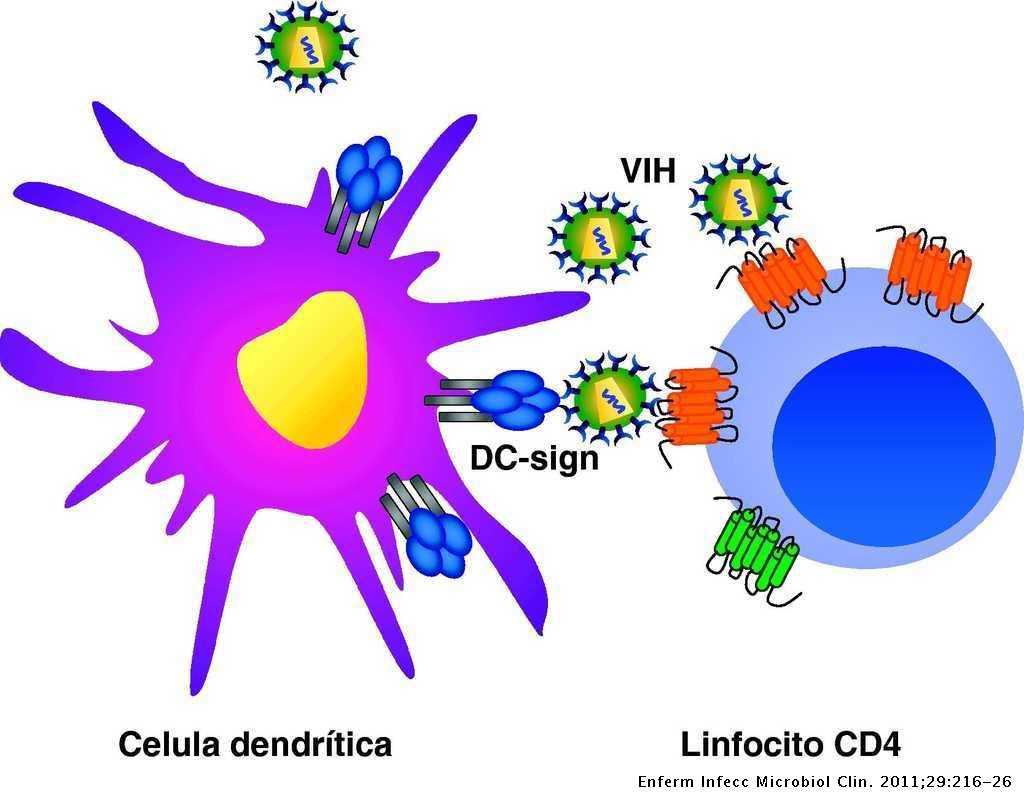
Este proceso está favorecido por las interacciones que se producen entre el VIH y las lectinas DC-SIGN y L-SIGN, localizadas en la superficie de las células dendríticas, provocando el aumento de la infección de los linfocitos circulantes. De esta forma, en las prolongaciones de las células dendríticas se facilita la infección de linfocitos CD4 (10).
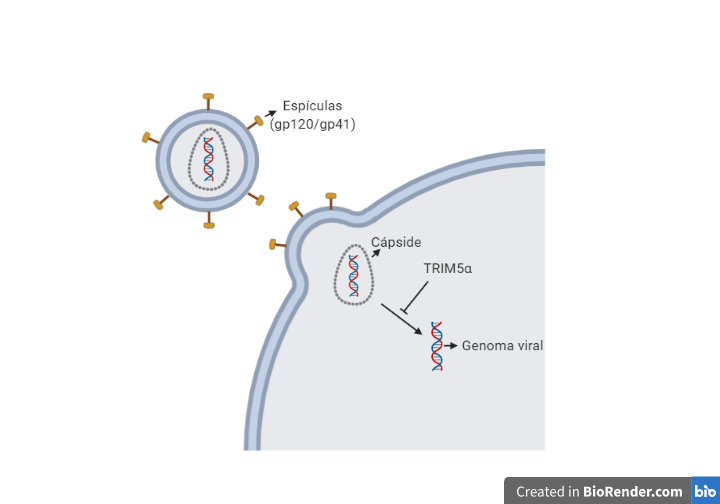
Tras la fusión de las membranas se produce la entrada de la nucleocápside viral, para a continuación descapsidar el genoma viral. Este proceso es inhibido por la proteína celular TRIM5α (específica de cada especie), que provoca que cada retrovirus deba generar variantes de la proteína de la cápside para evitar la acción de estas proteínas celulares (11)(12).
Una vez que tenemos libre el RNA viral en el citoplasma, se produce la retrotranscripción, proceso en el cual interviene la transcriptasa inversa (TI). Para la retrotranscripción será necesario unos niveles de nucleótidos y factores celulares, lo cual implica que el linfocito esté activo, porque en caso de que se encontrase en reposo se produciría una retrotranscripción incompleta dando lugar a un DNA retrotranscrito parcial que se degradaría por nucleasas celulares. Por el contrario, en linfocitos activados, una vez que tenemos el DNA transcrito este se va a unir a factores virales (Vpr) y celulares formando el complejo de pre-integración localizado en el citoplasma.
Este complejo será transportado al núcleo para ser integrado mediante la acción de la integrasa en cualquier parte del genoma de la célula huésped (es más frecuente en secuencias intrónicas de genes que estén activados). Sin embargo, no todo el DNA viral será integrado, quedando un porcentaje que será susceptible de integración.
Una vez que ya se haya integrado el genoma viral, se pueden dar diferentes escenarios, en los que el VIH puede permanecer latente, replicarse de forma masiva o de forma controlada.
Para que se inicie la replicación es necesario que se produzca la transcripción, proceso que únicamente depende de factores celulares, entre los que podemos destacar el NF-κB (13) el cual no se encuentra activado en linfocitos en reposo y sí en linfocitos activados.
Una vez que se inicia la síntesis, la proteína Tat aumenta la tasa de transcripción del RNA viral (es necesario para transcripción completa del RNA viral), el mRNA viral es transportado al citosol para ser procesado en transcritos de menor tamaño. Ambos procesos (procesamiento y transporte) están regulados por la proteína Rev (localizada en el núcleo), y si no hay Rev el mRNA viral se acumula en el núcleo. De esta forma obtenemos diferentes proteínas virales que serán procesadas para su posterior ensamblaje dando lugar a las partículas maduras. En este proceso es importante remarcar el papel del las proteínas Vif y Vpu.
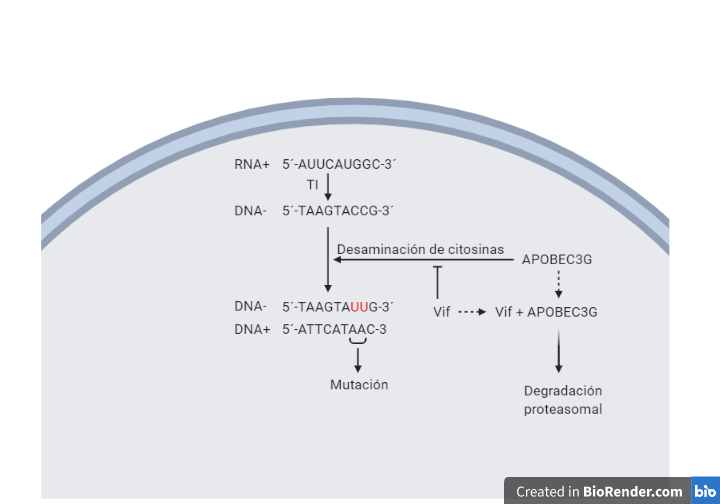
Vif va a producir la degradación proteosomal de APOBEC3G (A3G) impidiendo su entrada en los viriones en formación. La acción que tiene APOBEC3G es producir la desaminación de citosina en la primera hebra retrotranscrita por la transcriptasa inversa, provocando mutaciones en el genoma viral que provocan su degradación o una hipermutación y posterior degradación. De esta forma los efectos de su acción se ven sobre los viriones (14).
Vpu actúa inhibiendo la expresión de Tetherina, de tal forma que permite y facilita la liberación de viriones de la célula al espacio extracelular, ya que la Tetherina actúa secuestrando los viriones recién formados en la membrana celular.
Finalmente se llevará a cabo la maduración final en el proceso de gemación mediante la acción de la proteasa viral sobre poliproteínas. Cabe destacar que el precursor gp160 (da lugar a gp120 y gp41) es procesado por la proteasa celular.
La transmisión del VIH principalmente se produce vía parenteral, vía sexual o vertical (transmisión madre-hijo).
Patogenia
El VIH una vez que se introduce en un organismo va a provocar principalmente un debilitamiento del sistema inmunitario, el cual estará muy relacionado con la disminución en el número de linfocitos T CD4+ (10). Esta disminución de linfocitos CD4 puede deberse a diferentes motivos que aparecen representados en la Figura 8.
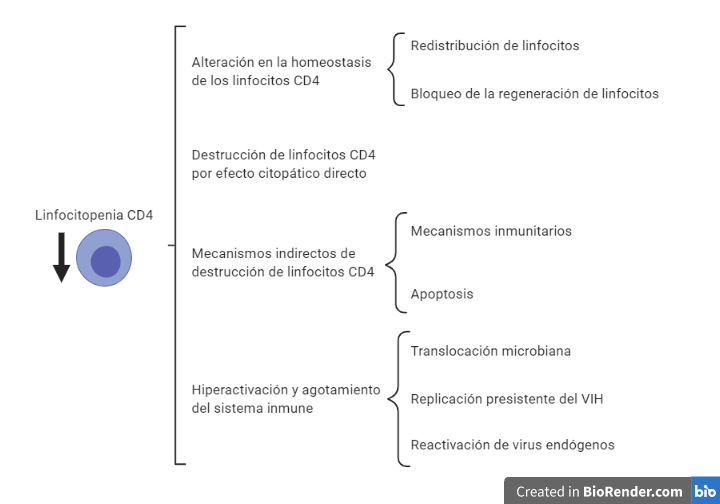
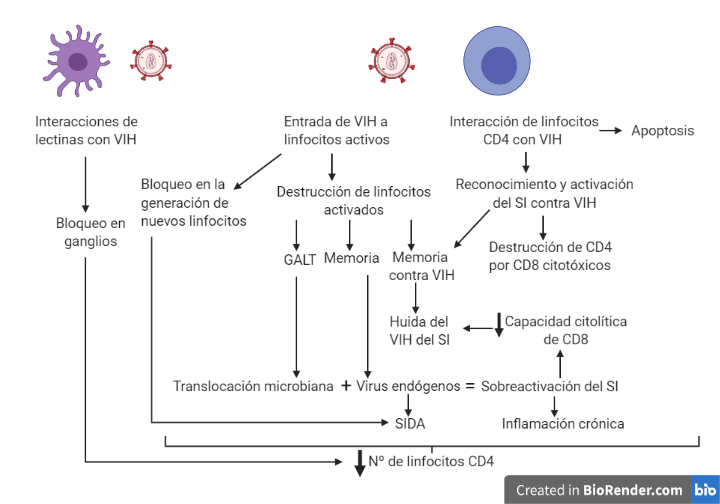
La alteración en la homeostasis esta ocasionada por una acumulación de los linfocitos alrededor de prolongaciones de células dendríticas de los órganos linfoides donde se localizan las partículas víricas. A su vez la replicación vírica provoca un bloqueo en la generación de nuevos linfocitos por parte del timo y la médula ósea, dificultando la sustitución de los linfocitos destruidos por el VIH.
En la destrucción directa por efecto citopático, es importante destacar que la destrucción es preferentemente en aquellos linfocitos activados (tienen altos niveles de CCR5, altos niveles de nucleótidos, ATP… En definitiva, factores que son necesario para la replicación del virus).
En el sistema GALT (tejido linfoide asociado al intestino) hay un predominio de linfocitos CD4 activos (debido a que están en continuo contacto con partículas antigénicas). Esto provoca una gran destrucción del sistema GALT que es irreversible. A su vez, la destrucción de linfocitos activados también explica que se destruyan linfocitos de memoria, lo cual agrava la situación inmunológica. Finalmente, el proceso de activación y reconocimiento del VIH provoca la activación de linfocitos que reconocen y atacan las células infectadas, esto ocasiona que las células que iban a matar al VIH se activen siendo infectadas y destruidas. De esta forma, el VIH elimina de manera específica los linfocitos de memoria contra él, facilitando el escape viral ante la respuesta inmune.
Por otro lado, la destrucción indirecta de los linfocitos esta mediada principalmente por dos mecanismos. En primer lugar los linfocitos infectados por el VIH presentan péptidos virales en sus moléculas de HLA clase I siendo objetivos de linfocitos T CD8 citotóxicos.
En segundo lugar, se produce apoptosis, esta se puede dar por dos vías: extrínseca (unión a la membrana plasmática de citocinas de la familia del factor de necrosis tumoral alfa (TNF-alfa)) e intrínseca (alterando la permeabilidad mitocondrial). El VIH puede inducir dicha apoptosis mediante síntesis de citocinas por macrófagos y linfocitos, aumento de la expresión de ligandos citotóxicos o mediante el efecto tóxico de proteína virales. Por otro lado, se ha visto que la proteína Tat puede tener un papel antiapoptótico en linfocitos infectados.
Finalmente, la hiperactivación y el agotamiento vienen ocasionados por una sobreproducción de linfocitos que se puede ver en los linfocitos CD8 citotóxicos producidos frente al VIH, ya que carecen de la capacidad citolítica requerida, debido al agotamiento por la sobrecarga antigénica. A su vez, es fácil pensar que la activación constante del sistema inmune viene ocasionada por la carga viral. Sin embargo, se ha visto que en pacientes con carga viral indetectable y con tratamiento antirretroviral se sigue produciendo esa activación, esto es debido, a la activación de virus endógenos (SIDA) y al aumento de la translocación de productos bacterianos debido al daño producido en el sistema GALT.
Sumado al descenso de linfocitos CD4 ocasionado por la infección del virus es necesario destacar la inflamación crónica que se produce debido a la secreción de citocinas proinflamatorias secretadas por la activación desmesurada que sufre el sistema inmune ocasionando daño endotelial y tisular sistémico.
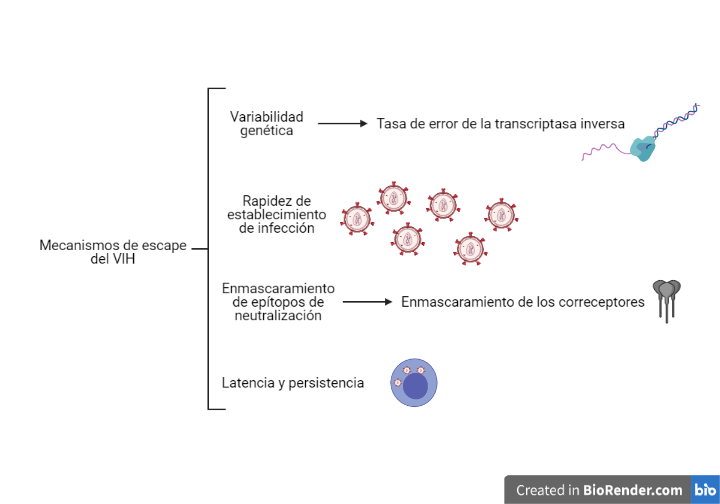
Esta acción esta favorecida por los diferentes mecanismos que tiene el VIH para evadir la respuesta inmune, los cuales están representados en la Figura 10.
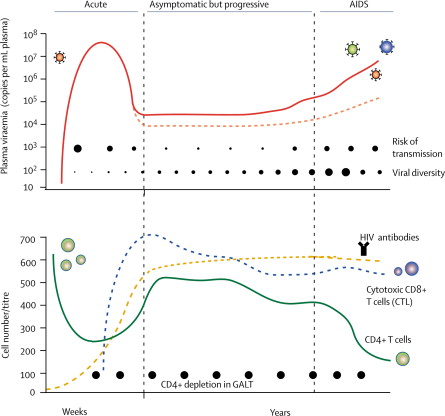
La infección por VIH a su vez se puede dividir en diferentes etapas según avanza el curso de la enfermedad (10).
Una primera etapa o infección reciente. Si se tiene en cuenta que la forma de contacto con el virus ha sido por la vía sexual, el primer contacto que tiene el virus es con las células dendríticas y los linfocitos localizados en la vagina y el recto (sistema GALT), y de ahí se va a producir una diseminación a linfocitos circulantes y ganglios (la estancia en las mucosas es relativamente corta pronto se puede observar una viremia en sangre). A partir de la infección hay un lapso de 4-12 semanas durante el cual no hay respuesta del sistema inmune, lo que ocasiona un gran avance del virus que destruye gran cantidad de linfocitos CD4, provocando la destrucción de gran parte del sistema GALT, que ya no se recuperará y que facilitará la translocación microbiana.
Una segunda etapa o infección crónica, la cual empezaría a las 12 semanas de la infección, donde se se empiezan a generar anticuerpos y linfocitos CD8 citotóxicos. Esto produce que haya un control de la viremia, pero a su vez favorece que se empiecen a producir variantes para escapar de la respuesta inmune. Durante esta fase, el sistema inmune se va debilitando provocando la aparición de alteraciones en la activación y maduración linfocitos CD8 y CD4.
Finalmente, hay una tercera etapa correspondiente a un estadio avanzado, que se caracteriza por la aparición de enfermedades oportunistas a causa de una bajada en el número de linfocitos CD4 y una elevación de la carga viral. En esta fase se ha podido observar un aumento en partículas virales con tropismo por los correceptores CXCR4.
Por lo tanto esta última fase se correspondería con la aparición del síndrome de la inmunodeficiencia humana (SIDA).
Tratamiento mediante ART
Inhibidores de la transcriptasa inversa análogos de los nucleósidos y nucleótidos (ITIAN)
Son fármacos que inhiben transcriptasa inversa del VIH mediante una inhibición competitiva actuando como análogos. Su target farmacológico se localizará en el centro catalítico (situado en el subdominio de la palma en la subunidad p66). Se dividen en dos clases:
- Análogos de nucleósidos: Abacavir, emtricitabina, lamivudina y zidovudina.
- Análogos de nucleótidos: Fumarato de disoproxilo de tenofovir (16).
Análogos de nucleósidos
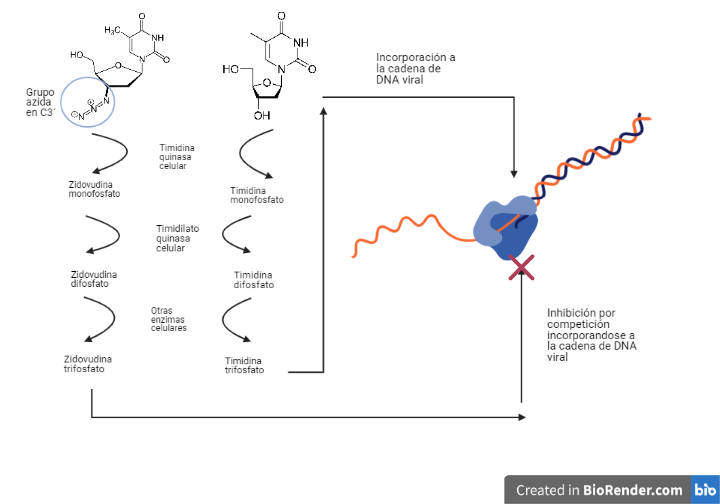
Actuarán como análogos estructurales de diferentes nucleósidos, pero diferirán de ellos, sobre todo en el radical del C 3´ de la desoxirribosa. Serán fosforilados por las mismas enzimas celulares que los nucleósidos con los que competirán.
Estos análogos de nucleósidos trifosforilados serán incorporados en el DNA viral perdiendo 2 fosfatos y quedando incorporados como análogos de nucleósidos monofosfato. Por tanto, se dará una competición entre estos fármacos y los nucleósidos trifosforilados para incorporarse en el DNA viral.
Las diferencias que poseen estos fármacos en el C 3´de la desoxirribosa impedirán la formación del enlace fosfodiéster con el siguiente nucleótido, deteniendo la elongación de la cadena prematuramente lo cual provoca la inhibición de la transcriptasa inversa (17).
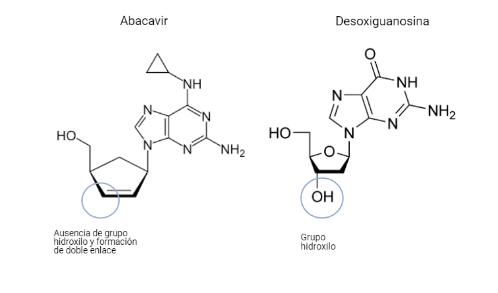
Abacavir (sulfato de abacavir, ABC)
Este fármaco será un análogo estructural del desoxirribonucleósido de guanina. Diferirá de este en que el C 3´ de la desoxirribosa no poseerá un grupo hidroxilo, por lo que formará un doble enlace, además de diferentes variaciones en la guanina.
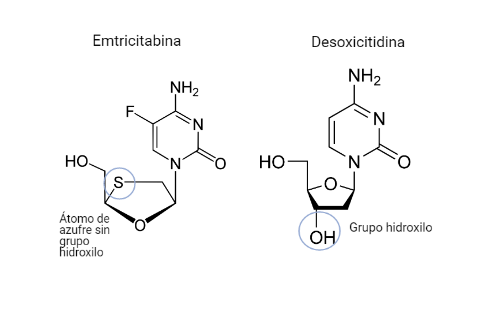
Emtricitabina (FTC)
Este fármaco será un análogo estructural de la desoxicitidina. Diferirá de este en que el C 3´ de la desoxirribosa será sustituido por un átomo de azufre, además de diferentes variaciones en la citosina como un radical de flúor.
Lamivudina (3TC)
Este fármaco será un análogo estructural del desoxicitidina. Poseerá la misma diferencia que posee la emtricitabina (átomo de azufre) pero no poseerá un átomo de flúor en la citosina.
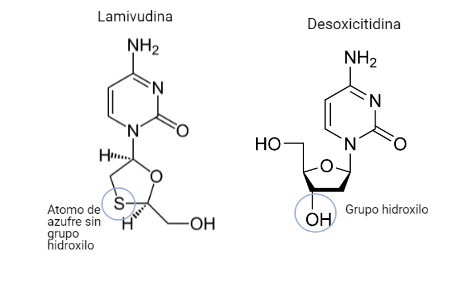
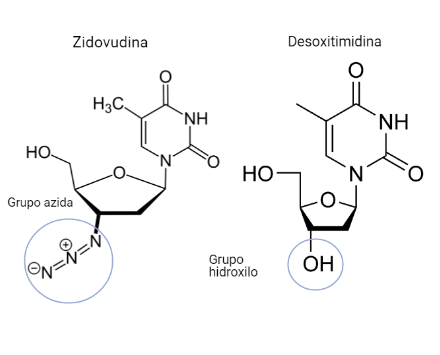
Zidovudina (azidotimidina, AZT, ZDV)
Este fármaco será un análogo estructural de la timidina (desoxirribonucleósido de timina). Diferirá de este en que el C 3´de la desoxirribosa no poseerá un grupo hidroxilo, sino un grupo azida.
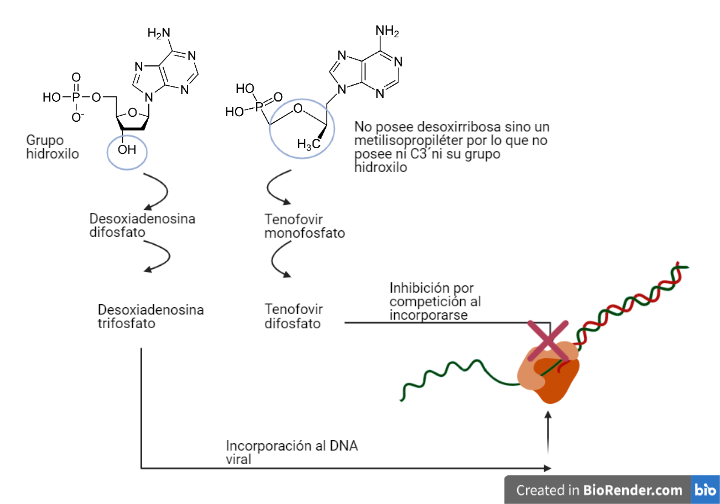
Análogos de nucleótidos: fumarato de disoproxilo de tenofovir (tenofovir DF, TDF)
Este compuesto en un profármaco que sufrirá diversas hidrolisis diestéricas hasta transformarse en tenofovir.
El tenofovir será un análogo estructural del desoxirribonucleótido de adenina (desoxiadenosina monofosfato). A diferencia de los análogos de nucleósidos, este compuesto solo sufrirá dos fosforilaciones por las mismas enzimas celulares que fosforilan el nucleótido de adenina.
El tenofovir difosfato será incorporado al DNA viral perdiendo 2 fosfatos y quedando incorporado simplemente como tenofovir. Por tanto, se dará una competición entre el tenofovir difosfato y la desoxiadenosina trifosfato por la incorporación.
El tenofovir poseerá una estructura química que diferirá de desoxiadenosina monofosfato ya que no poseerá una desoxirribosa como conector, sino que poseerá un metilisopropileter. Al no poseer la desoxirribosa, no poseerá su C 3´ y, por tanto, impedirá la formación del enlace fosfodiéster con el siguiente nucleótido. Esto supondrá la detención de la elongación de la cadena prematuramente provocando la inhibición de la transcriptasa inversa (16)(17).
Inhibidores de la transcriptasa inversa no análogos de nucleósidos (ITINN)
Estos fármacos inhiben la actividad polimerasa de la transcriptasa inversa del VIH-1. No necesitarán ser fosforilados ni se incorporarán a la cadena de DNA viral, como los anteriores, sino que realizarán una inhibición no competitiva uniéndose a la propia enzima.
Se dividen en dos generaciones:
- Primera generación: Efavirenz (EFV) y nevirapina (NVP): precisan pocas mutaciones para desarrollar resistencia.
- Segunda generación: Etravirina (ETR) y rilpivirina (RPV) utilizados contra cepas del VIH-1 resistentes a la primera generación (18).
Además de estas dos generaciones, en los últimos años, surge un fármaco de nueva generación llamado doravirina (DOR).
Se unirán en la transcriptasa inversa en un bolsillo hidrofóbico situado a 10-15 Å del centro catalítico de la actividad polimerasa y a 60 Å del sitio activo de la actividad ribonucleasa H.
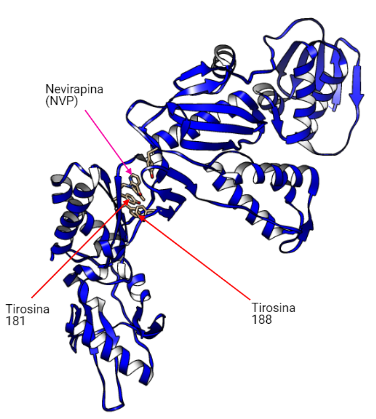
Estos fármacos actúan contra el VIH-1 y no contra el VIH-2 debido a que los residuos 181 y 188 en la RT-VIH-1 son tirosina mientras que en la RT-VIH-2 son isoleucina y leucina, respectivamente. Esto provoca que no se pueda dar la unión del fármaco (19).
Los fármacos de primera generación (EFV y NVP) se unen a los residuos de tirosina (Y181 y Y188) localizados en la región de la palma. Por otro lado, los fármacos de segunda generación (ETR y RPV) se unen a otros puntos como un residuo de triptófano (W229), sitio en el que se producen menos mutaciones. Debido a esto, los fármacos de primera generación sufrirán más resistencia que los de segunda generación (18).
La unión a estos puntos provoca cambios en la conformación que provocan la inhibición de la catálisis del RNA viral y la formación de DNA viral disminuyendo la replicación del VIH-1.
Inhibidores de la proteasa (IP)
La proteasa se encarga de realizar la hidrolisis del enlace peptídico de las poliproteínas virales gag y gag-pol. En concreto, se tiene como consenso que en estas poliproteínas se da la hidrolisis del enlace peptídico entre fenialanina (posición 1) y prolina (posición 1´).
Estos fármacos inhibirán la proteasa del VIH y los clasificaremos según su modo de acción:
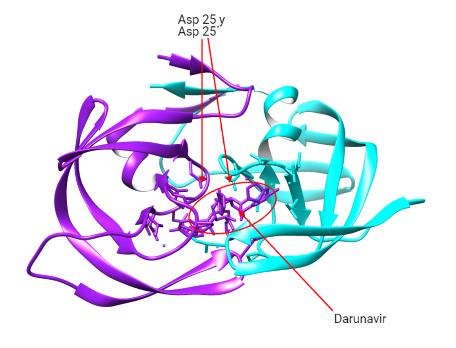
- Actuando como peptidomiméticos y compitiendo con las poliproteínas virales en la unión al centro activo (Asp25-Asp25´): Saquinavir (SQV), ritonavir (RTV), fosamprenavir (FPV), atazanavir (ATV) y darunavir (DRV).
- Uniéndose a otra zona del centro activo (Ile50-Ile50´): Tipranavir (TPV).
Los diferentes sustituyentes que tendrán formarán interacciones con aminoácidos de la estructura de la proteasa produciendo más afinidad.
Cabe destacar que el ritonavir actualmente se utiliza como potenciador farmacocinético ya que inhibe el citocromo CYP3A4, el cual se encarga del metabolismo de estos fármacos. Por ello, este se administrará junto con otro IP (20).
Inhibidores de la integrasa
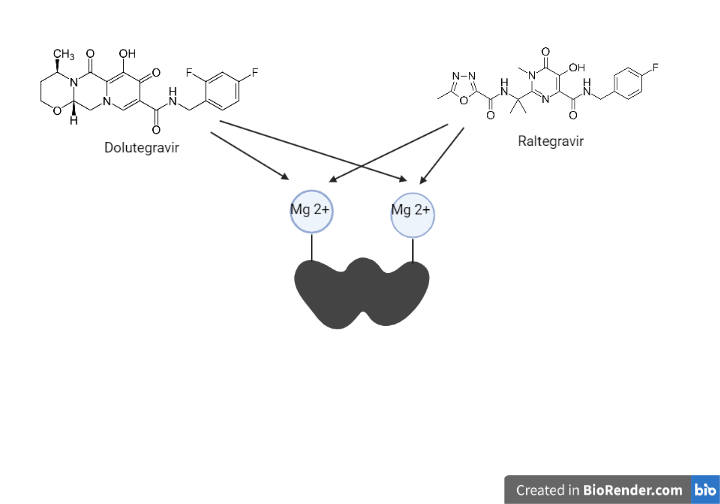
El dolutegravir (DTG) y el raltegravir (RAL) son fármacos antirretrovirales que actúan inhibiendo la integrasa. Pertenecen al grupo de los INSTI (inhibidores de la transferencia de cadenas de la integrasa).
La integrasa posee dos cationes metálicos divalentes (Mg 2+) en su sitio activo, en el cual se unirá al DNA del hospedador. Estos dos átomos metálicos serán el target farmacológico.
Se unen a los dos cationes divalentes en el sitio activo cuando la integrasa forma el complejo de preintegración. Esto provoca que la 3-desoxiadenina, que se encuentra en el extremo 3´del DNA viral, se desplace impidiendo la transferencia e integración del DNA viral al DNA del hospedador (21).
Inhibidores de la fusión
La proteína de fusión gp41 de los retrovirus posee dos regiones de repetición: HR1 y HR2. La HR1 forma una estructura de enrollamiento trimérica y HR2 se pliega dentro de los surcos hidrofóbicos de HR1 para formar una horquilla. En esto consiste el zapping de gp41. Esto provoca el acercamiento de las dos membranas y la formación de un poro de fusión.
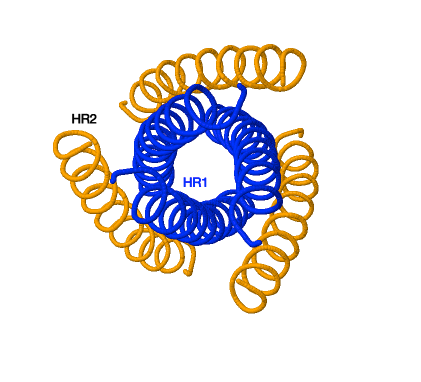
Poseemos un único fármaco que inhiba este proceso el cual es la enfuvirtida. Este previene este proceso uniéndose a la región HR1 previniendo el zapping de gp41. La secuencia de aminoácidos de este fármaco es similar a la secuencia de HR2 provocando que realice el pliegue que realizaría HR2.
Se cree que esta acción se realiza después de que se una la proteína gp41 en la membrana de la célula y antes de que se realice el pliegue de HR2. Esto provoca la inhibición del zapping de gp41 y los sucesivos cambios conformacionales para que se dé la fusión (22).
Antagonistas de CCR5
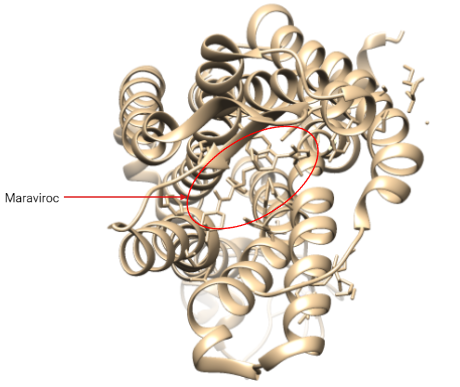
Poseemos un único fármaco, el maraviroc (MVC). Este es un inhibidor alostérico del correceptor CCR5, que inhibe la entrada de retrovirus a la célula.
Es una imidazopiridina que se une a una región transmembrana de CCR5 (dominios transmembrana 2, 3, 6 y 7). Esto provoca una serie de cambios conformacionales, especialmente en el dominio ELC2, lo cual impide que la región V3 de la glucoproteína gp120 de la envuelta interaccione con el receptor (23). Esto impide que se dieran cambios conformacionales posteriores para que se insertara gp41 en la membrana celular provocando la fusión.
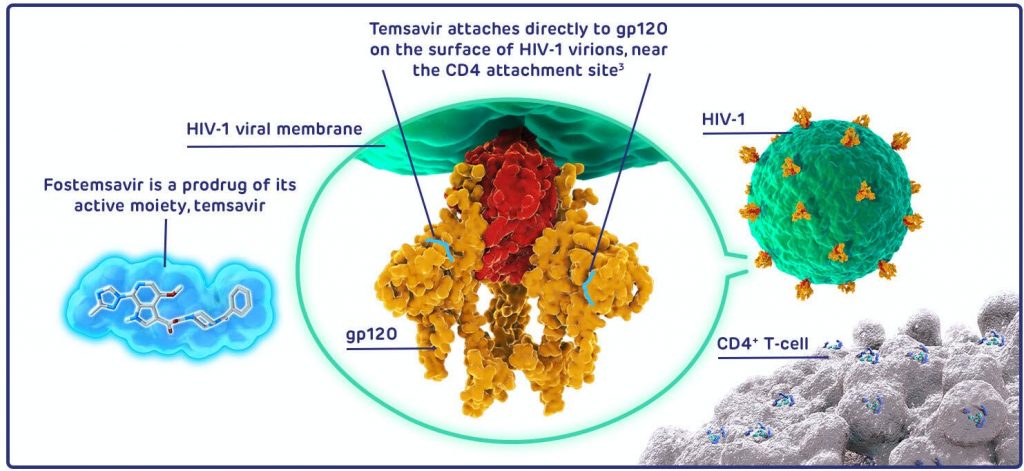
Inhibidores de la fijación: Fostemsavir
Fostemsavir es un profármaco del temsavir, el cual impedirá la fijación del VIH. Se unirá directamente a la proteína gp120 e inhibe la interacción entre el receptor CD4 celular y el virus. Por tanto, también inhibirá los procesos posteriores a la fijación impidiendo la entrada a linfocitos T CD4+ (24).
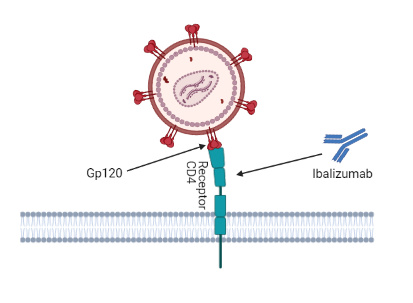
Inhibidores posfijación: Ibalizumab-uiyk
Ibalizumab es un anticuerpo IgG monoclonal humanizado el cual bloquea la entrada del virus a los linfocitos T4 sin deteriorar el sistema inmune. Es el primer anticuerpo monoclonal para el tratamiento del VIH y el primer inhibidor posfijación.
Se une al dominio 2 del receptor CD4 en la superficie opuesta tanto en el sitio de unión del complejo mayor de histocompatibilidad de clase II como en el sitio de unión de g120 (25).
Tratamiento mediante CRISPR-Cas9
¿Qué es el sistema CRISPR-Cas9?
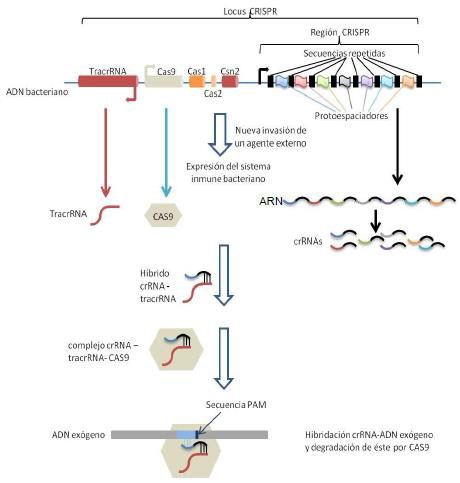
El sistema CRISPR-Cas9 ha evolucionado en procariotas como un sistema inmune de memoria que confiere a estos microorganismos resistencia a una segunda entrada de material genético exógeno, como aquel que puede ser introducido a través de fagos o plásmidos.
En el genoma bacteriano existe una región, el locus CRISPR (siglas de “Clustered regularly interspaced short palindromes repeats”, o “Agrupaciones de repeticiones palindrómicas cortas regularmente interespaciadas”), que porta porciones de RNA exógeno (de unos 20pb), perteneciente a fagos u otros elementos genéticos, insertadas por la propia bacteria cuando esta sobrevive a una infección. Estas porciones de RNA, denominadas “protoespaciadores”, se encuentran intercaladas entre otras secuencias cortas que se repiten.
Además, en este locus están también las secuencias que codifican para otros elementos formadores del sistema CRISPR-Cas9, como la enzima Cas9 (“CRISPR asociated protein 9”, una nucleasa) o el RNA transactivador de CRISPR (TracrRNA). La defensa frente un nuevo DNA “conocido” entrante por parte de la bacteria se basa en la complementariedad entre este y el protoespaciador específico (el fragmento de RNA que constituye ese protoespaciador recibe el nombre de crRNA) que se corresponde con ese material genético, además de la interacción de estos con el TracrRNA y la nucleasa Cas9, que desarrolla la acción de degradación del nuevo material genético infectante.
Este sistema, nativo de bacterias, ha servido de base para desarrollar en los últimos años un sistema de edición genética que puede ser usado para manipular y escindir partes concretas del DNA humano causantes de graves enfermedades, así como para prevenir y tratar diferentes enfermedades infecciosas como las causadas por el VIH, entre muchos otros agentes infectantes (26).
La maquinaria que permite desarrollar este método de edición génica consiste en una enzima Cas9 unida a una porción de RNA denominado RNA guía (gRNA), formado por la conjunción de un TracsRNA y un crRNA que cuenta con un protoespaciador sintetizado artificialmente y de manera intencionada para que sea complementario de la región del DNA celular (secuencia diana) que se quiere modificar o escindir.
Además de la secuencia diana, para la unión de la proteína Cas9 (y para el correcto emparejamiento con el protoespaciador) es necesaria una secuencia PAM, de apenas 3 nucleótidos, situada junto a ella. Los dominios HNH (Histidina-Asparagina-Histidina) y RuvC de la nucleasa Cas9 participan en la unión de la enzima a la secuencia diana del DNA celular (aquellas con la que interacciona el protoespaciador) y a la hebra complementaria a la secuencia diana, respectivamente (9)(27).
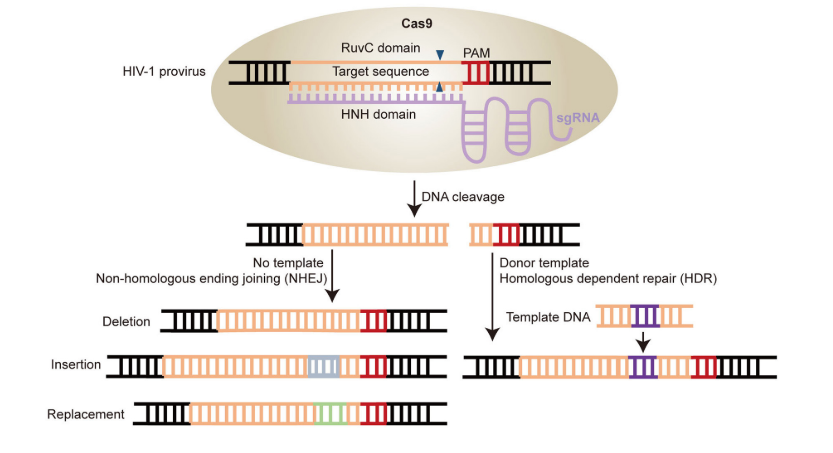
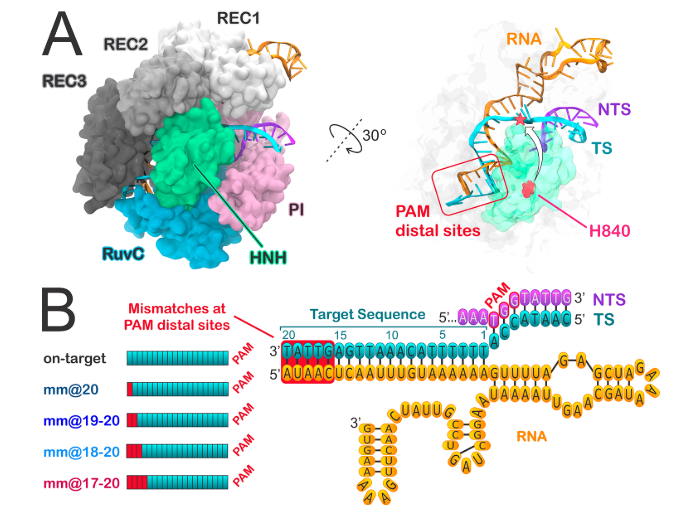
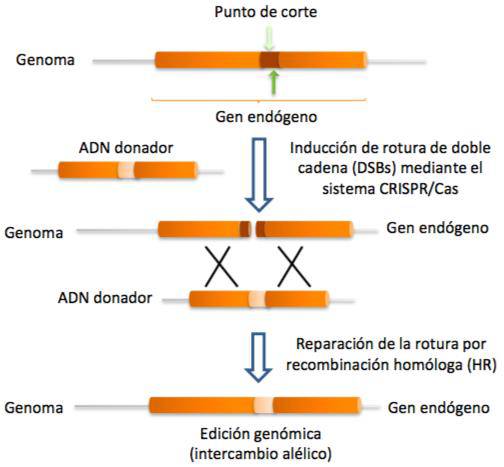
Una vez se ha producido el corte en el DNA celular, el proceso puede continuar con la reparación de las hebras mediante recombinación homóloga o no homóloga. A través de la primera, gracias a un DNA donador, se puede introducir nueva información genética.
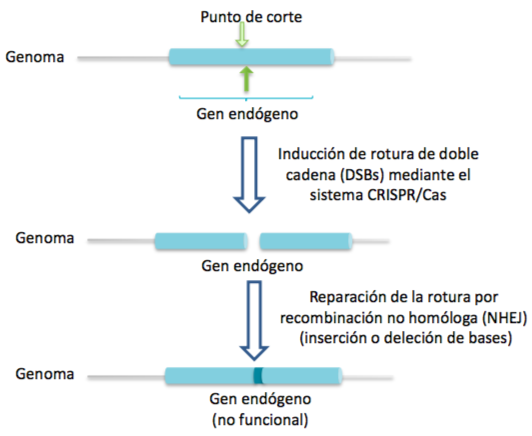
Sin embargo, si el proceso desarrollado por el sistema CRISPR-Cas9 no se acompaña por ningún DNA donador y se produce la recombinación no homóloga (unión de los extremos no homólogos formados en el DNA tras el corte), el resultado será un knock-out del gen afectado, un gen no funcional, pues simplemente se ha anulado la función del gen y no se ha introducido nueva información genética.
DNA provirus en latencia en linfocitos T CD4+ como diana del sistema CRISPR-Cas9 para el tratamiento por VIH
Por lo general, la terapia antirretroviral (ART) para tratar el SIDA es capaz de controlar la viremia de manera efectiva, pero no consigue eliminar el virus presente en estado de latencia en los linfocitos T infectados. Durante el estado de latencia, las células infectadas por el VIH apenas generan productos víricos, lo que les permite evitar el ataque del sistema inmune del hospedador y, por tanto, permanecer en él durante larguísimos periodos de tiempo.
En estos linfocitos T CD4+ de memoria (cuando se encuentran en un estado de descanso (“a memory resting state”), algo normal tras producirse la infección de estas células por parte del virus), las copias inactivas del provirus (se denomina así al DNA viral resultante de la retrotranscripcción que se encuentra integrado en el genoma celular ) pueden reactivarse cuando lo hicieran las células en las que se encuentran y causar un nuevo rebrote si el tratamiento antirretroviral se interrumpe (28).
Debido a que se cree que estas células son los principales “almacenes” de DNA viral en estado de latencia (29) se han convertido en el objetivo de los principales tratamientos contra el SIDA basados en la utilización del sistema CRISPR-Cas9.
Escisión del provirus del genoma de linfocitos T CD4+ de memoria
En la siguiente tabla aparecen algunas secuencias diana del provirus sobre las que se puede actuar a través del sistema CRISPR-Cas9, así como los métodos de introducción de los elementos necesarios (gRNA y nucleasa Cas9) para desarrollar la técnica (9).
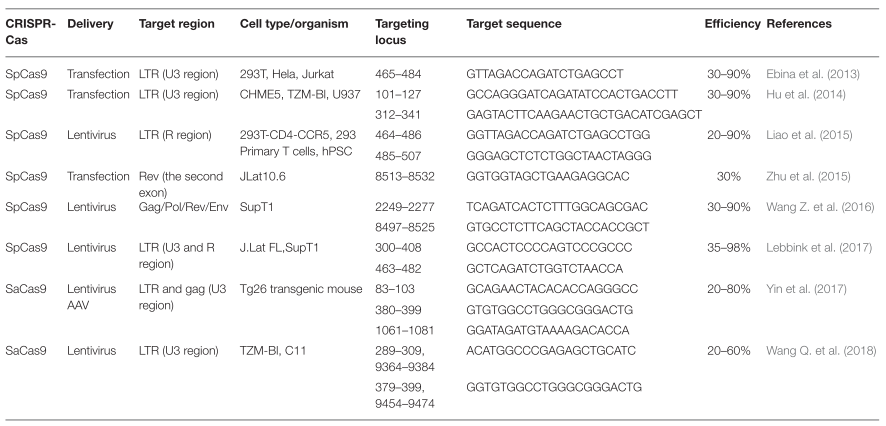
En diferentes estudios, se consideró la posibilidad de provocar la eliminación de las células hospedadoras del virus en estado de latencia a través de su reactivación, pues esto provocaría una respuesta inmune contra ellas que conllevaría también la eliminación del virus (28). Sin embargo, debido a diversos inconvenientes, consideraron mejores como cura métodos que eliminasen directamente el genoma del virus de las células hospedadoras, como es el caso del método de edición genética CRISPR-Cas9.
En su estudio, Kaminski et al. (28) utilizaron el algoritmo de secuenciación CREST (clipping reveals structure) (38) para la localización de alteraciones estructurales en el genoma de células de la línea de células Jurkat 2D10. Se encontraron cuatro puntos de rotura en el DNA de la célula hospedadora en los cromosomas 1 y 16, concretamente en las regiones p13.2 y p13.3, respectivamente.
Mediante una amplificación de corto alcance se encontró un fragmento de 504 nucleótidos correspondiente a la unión de las regiones 5’ LTR (Long Terminal Repeat) y 3’ LTR del genoma viral tras la acción del sistema CRISPR-Cas9 con un gRNA denominado B. Esta secuencia era 7 nucleótidos más larga que la secuencia de 497 nucleótidos amplificada en las células de control, pues se había producido, además, una inserción de un fragmento de 7 nucleótidos.
La acción del sistema CRISPR-Cas9 / gRNA B permitió eliminar el genoma viral, pues no se pudo secuenciar la región de 257 pares de bases correspondientes al elemento de respuesta Rev (RRE), que se encuentra en el centro de este.
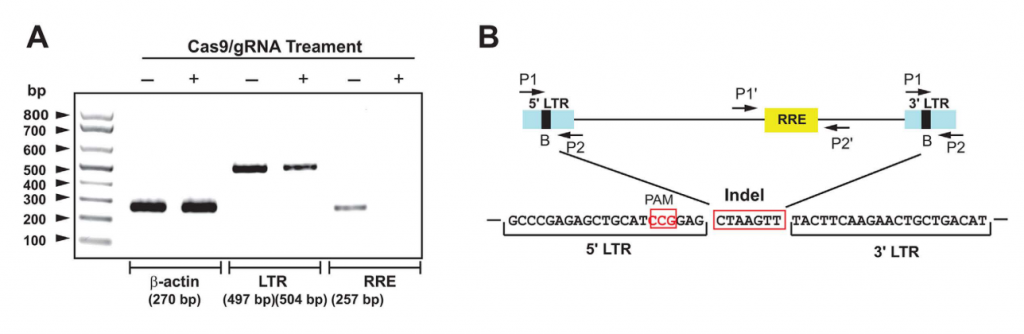
Examinaron mediante PCR de alto rango las posiciones en las que se inserta el genoma viral dentro de los cromosomas 1 y 16. En ambos, se obtuvieron tres tipos distintos de amplicones: uno de larga longitud (de 6130 pb para el cromosoma 1 y de 5467 pb para el 16), correspondiente al genoma integrado del VIH-1 más su secuencia de ADN flanqueante derivado del cromosoma; uno de tamaño medio (de 909 pb y de 759 pb, respectivamente), que se corresponde con el mismo fragmento pero tras escindir por completo el genoma viral; y otro mucho menor (de 264 pb y 110 pb), que se trata del fragmento de DNA correspondiente del cromosoma homólogo, que no ha sufrido modificación alguna.
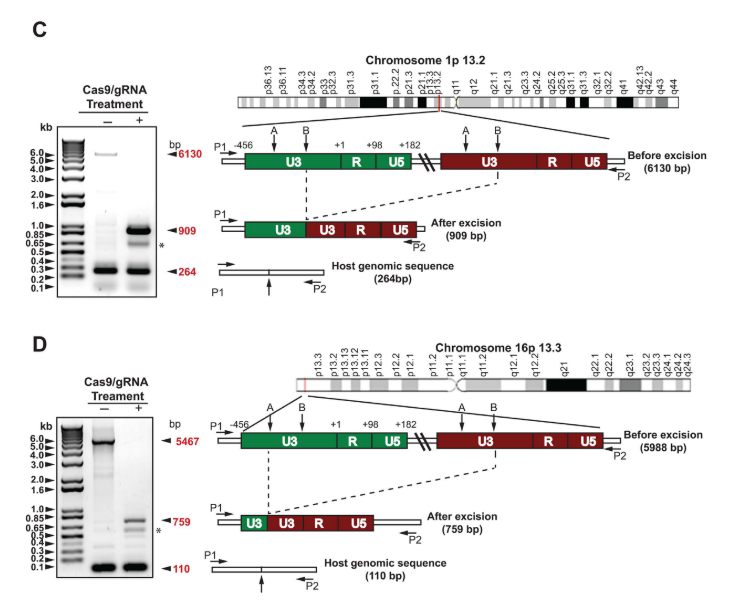
Revisiones posteriores del método determinaron que el genoma viral escindido es mayoritariamente degradado a continuación, por lo que el riesgo de transposición o reintegración en el genoma del huésped es bajo (28).
Escape viral al tratamiento y como consecuencia del tratamiento
Sin embargo, según otros estudios, como se recoge en la publicación de Das, Binda and Berkhout (39), es posible que el virus escape a la inhibición mediante mutaciones efectuadas en la región del genoma viral que es diana del corte efectuado por Cas9 que impidan su acción pero permitan la replicación viral. Entre estas mutaciones se encontraron indels (inserciones o deleciones) en regiones poco conservadas (promotores de la región LTR) y sustituciones e inserciones en regiones conservadas, como dominios que codifican para proteínas, lo que constituye un patrón de mutaciones diferentes al que ocurre para escapar de los fármacos ART.
El origen de esas mutaciones puede encontrarse, además de en el proceso de retrotranscripción desarrollado para integrar el genoma viral en el genoma de la célula hospedadora, en el proceso de modificación del genoma proviral desarrollado por parte del propio sistema CRISPR-Cas9.
Infectividad por VIH de células T tratadas con el sistema CRISPR-Cas9
En el mismo estudio (28) se probó cual era la reacción a la reinfección de clones de células T de las que se erradicó el genoma viral mediante el sistema con CRISPR-Cas9. Estas células contaban con diferentes componentes del sistema (unas con la enzima Cas9 en distintos niveles de concentración, otras con el gran y otras con ambos).
Los resultados fueron la resistencia a la reinfección por parte de aquellas células que contaban con ambos elementos (mayor resistencia en las que la expresión de Cas9 era mayor), pero no en las que solamente poseían uno de los dos elementos del sistema.
Otras dianas para el tratamiento del SIDA mediante terapia génica: Correceptores CCR5 y CXCR4
Además de tener el genoma proviral como diana para el tratamiento genético del VIH, el sistema CRISPR-Cas9 puede ser utilizado para bloquear la entrada del virus en los linfocitos T CD4+ a través de la modificación génica de los correceptores de membrana que empleados por el VIH para acceder al interior celular (9).
Simulando la resistencia al VIH que aparecen de manera natural en personas que presentan deleciones en gen que codifica para el correceptor CCR5 (40), el método CRISPR-Cas9 puede ser utilizado (como corroboran múltiples estudios, recogidos en el artículo de Xiao et al.) con la intención de editar los genes de los correceptores CCR5 y CXCR4 para evitar la infección por VIH sin que se pierda la función propia.
Ventajas y limitaciones de la utilización del sistema CRISPR-Cas9 como tratamiento contra el SIDA
La utilización del sistema de edición genética CRISPR-Cas9 se ha antepuesto a otros, como el uso de nucleasas con dedos de zinc (ZFNs) o de nucleasas que actúan como activadores transcripcionales (TALENs), debido al alto coste y tiempo requerido por los mismos. Entre los motivos que llevan al sistema CRISPR-Cas a ser el más ampliamente utilizado están su precisión, su sencillez o su alta eficacia (9).
Por otro lado, uno de los mayores inconvenientes que aparecen a la hora de utilizar el sistema CRISPR-Cas9 es el método de “aporte” o “liberación” (delivery) de los elementos que lo componen en las células infectadas. Entre los métodos comúnmente utilizados se encuentran los vectores adenovirales, vectores de AAV (virus adenoasociados, que requieren la coinfección con adenovirus) o los vectores lentivirales (41), en función de los requerimientos de expresión o el tamaño del material genético que se quiere introducir en el interior celular. También se puede llevar a cabo la transfección directa de los componentes del sistema (procedentes de, por ejemplo, Streptococcus pyogenes o Staphylococcus aureus) tras electroporación (42) o utilizando vesículas generadas artificialmente para permitir el transporte hasta el interior celular (43).
Además, como se comentó anteriormente, otra desventaja de este método es su alta especificidad (su precisión se le puede volver en contra), pues le da al virus la posibilidad de escapar y hacerse resistente al sistema mediante simples mutaciones en su secuencia.
Bibliografía
1. Delgado R. Virological characteristics of HIV. Enferm Infecc Microbiol Clin. 2011;29(1):58–65.
2. D’Arc M, Ayouba A, Esteban A, Learn GH, Boué V, Liegeois F, et al. Origin of the HIV-1 group O epidemic in western lowland gorillas. Proc Natl Acad Sci U S A. 2015;112(11):E1343–52.
3. Albert Hernández M, San Miguel Hernández Á. Epidemiology, detection of resistance and tropism of HIV-1: update. Rev Med Lab. 2020;1(1):10–20.
4. Salmen S, Gabaldon-figueira JC, Teran-angel G. Nef- VIH-1 como principal orquestador de la disfunción celular durante la inmunopatogenia de la infección por el VIH-1 ( Nef- HIV-1 as the main orchestrator of the cellular dysfunction in the immunopathogenesis of the HIV-1 infection ) Introducción. Av en Biomed. 2016;4(3):126–37.
5. Goila-Gaur R, Strebel K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008;5:1–16.
6. Neil SJD, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451(7177):425–30.
7. Noé REA, Guadalupe HMP, Israel NG, Ricardo GPC, Denise CAF. Caracteristicas estructurales y funcionales del Virus de la Inmunodeficiencia Humana. Vol. 33, Enfermedades Infecciosas y Microbiologia. 2013. p. 163–73.
8. Alcamí J. The HIV replication cycle. Established therapeutic targets and potential targets. Enferm Infecc Microbiol Clin [Internet]. 2008;26(SUPPL. 12):3–10. Available from: http://dx.doi.org/10.1016/S0213-005X(08)76566-2
9. Xiao Q, Guo D, Chen S. Application of CRISPR/Cas9-based gene editing in HIV-1/AIDS therapy. Front Cell Infect Microbiol. 2019;9(MAR):1–15.
10. Alcamí J, Coiras M. Inmunopatogenia de la infección por el virus de la inmunodeficiencia humana. Enferm Infecc Microbiol Clin. 2011;29(3):216–26.
11. Lukic Z, Campbell EM. The cell biology of TRIM5α. Curr HIV/AIDS Rep. 2012;9(1):73–80.
12. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature [Internet]. 2004 Feb;427(6977):848–53. Available from: https://pubmed.ncbi.nlm.nih.gov/14985764/
13. Lozano De León-Naranjo F. Infección por el VIH (I). Med. 2014;11(49):2893–901.
14. Imahashi M, Nakashima M, Iwatani Y. Antiviral mechanism and biochemical basis of the human APOBEC3 family. Front Microbiol. 2012;3(JUL):1–7.
15. Simon V, Ho DD, Abdool Karim Q. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet. 2006;368(9534):489–504.
16. Ribera E, Tuset M, Martín M, Del Cacho E. Characteristics of antiretroviral drugs. Enferm Infecc Microbiol Clin. 2011;29(5):362–91.
17. McTavish D, Campoli-Richards D, Sorkin EM. Carvedilol: A Review of its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Efficacy. Drugs. 1993;45(2):232–58.
18. Portilla J, Estrada V. Mecanismo de acción y farmacocinética de rilpivirina. Enferm Infecc Microbiol Clin [Internet]. 2013;31(SUPPL.2):2–5. Available from: http://dx.doi.org/10.1016/S0213-005X(13)70136-8
19. Wang Y, De Clercq E, Li G. Current and emerging non-nucleoside reverse transcriptase inhibitors (NNRTIs) for HIV-1 treatment. Expert Opin Drug Metab Toxicol [Internet]. 2019;15(10):813–29. Available from: https://doi.org/10.1080/17425255.2019.1673367
20. Mahmoud SS, Hussain ZM, O’Shea P, Schaubach KR, Iv NJD, Rappaport TS, et al. الابتزاز الإلكتروني.. جرائم تتغذى على طفرة «التواصل الاجتماعي»No Title. CNR-ISTI Tech Rep [Internet]. 2015;3(2):356–69. Available from: https://www.metis2020.com/wp-content/uploads/METIS_D1.4_v3.pdf%0Ahttps://www.metis2020.com/documents/deliverables/index.html%0Ahttps://www.metis2020.com/metis-deliverables-d1-4-d2-4-d3-3-d4-3-d6-5-and-d7-3-were-completed-in-february-2015/index.html%0Ahttp
21. Ribera E, Podzamczer D. Mecanismo de acción, farmacología e interacciones de dolutegravir. Enferm Infecc Microbiol Clin [Internet]. 2015;33(S1):2–8. Available from: http://dx.doi.org/10.1016/S0213-005X(15)30002-1
22. Adams J. Drug evaluation. Aust Prescr. 1996;19(1):5.
23. Soriano V, Poveda E. Maraviroc: Farmacocinética, interacciones y mecanismo de acción. Enferm Infecc Microbiol Clin [Internet]. 2008;26(SUPPL. 11):12–6. Available from: http://dx.doi.org/10.1016/S0213-005X(08)76558-3
24. Markham A. Fostemsavir: First Approval. Drugs [Internet]. 2020;80(14):1485–90. Available from: https://doi.org/10.1007/s40265-020-01386-w
25. Markham A. Ibalizumab: First Global Approval. Drugs [Internet]. 2018;78(7):781–5. Available from: https://doi.org/10.1007/s40265-018-0907-5
26. McCarthy MW. Harnessing the potential of CRISPR-based platforms to advance the field of hospital medicine. Expert Rev Anti Infect Ther [Internet]. 2020;18(8):799–805. Available from: https://doi.org/10.1080/14787210.2020.1761333
27. Ricci CG, Chen JS, Miao Y, Jinek M, Doudna JA, McCammon JA, et al. Deciphering Off-Target Effects in CRISPR-Cas9 through Accelerated Molecular Dynamics. ACS Cent Sci. 2019;5(4):651–62.
28. Kaminski R, Chen Y, Fischer T, Tedaldi E, Napoli A, Zhang Y, et al. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci Rep [Internet]. 2016;6(December 2015):1–15. Available from: http://dx.doi.org/10.1038/srep22555
29. Bruner KM, Hosmane NN, Siliciano RF. Towards an HIV-1 cure: Measuring the latent reservoir. Trends Microbiol [Internet]. 2015;23(4):192–203. Available from: http://dx.doi.org/10.1016/j.tim.2015.01.013
30. Ebina H, Misawa N, Kanemura Y, Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep. 2013;3:1–7.
31. Hu W, Kaminski R, Yang F, Zhang Y, Cosentino L, Li F, et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A. 2014;111(31):11461–6.
32. Liao HK, Gu Y, Diaz A, Marlett J, Takahashi Y, Li M, et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat Commun. 2015;6:1–10.
33. Zhu W, Lei R, Le Duff Y, Li J, Guo F, Wainberg MA, et al. The CRISPR/Cas9 system inactivates latent HIV-1 proviral DNA. Retrovirology. 2015;12(1):1–7.
34. Wang Z, Pan Q, Gendron P, Zhu W, Guo F, Cen S, et al. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell Rep [Internet]. 2016;15(3):481–9. Available from: http://dx.doi.org/10.1016/j.celrep.2016.03.042
35. Lebbink RJ, De Jong DCM, Wolters F, Kruse EM, Van Ham PM, Wiertz EJHJ, et al. A combinational CRISPR/Cas9 gene-editing approach can halt HIV replication and prevent viral escape. Sci Rep [Internet]. 2017;7(February):1–10. Available from: http://dx.doi.org/10.1038/srep41968
36. Yin C, Zhang T, Qu X, Zhang Y, Putatunda R, Xiao X, et al. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol Ther [Internet]. 2017;25(5):1168–86. Available from: http://dx.doi.org/10.1016/j.ymthe.2017.03.012
37. Wang Q, Liu S, Liu Z, Ke Z, Li C, Yu X, et al. Genome scale screening identification of SaCas9/gRNAs for targeting HIV-1 provirus and suppression of HIV-1 infection. Virus Res [Internet]. 2018;250:21–30. Available from: https://doi.org/10.1016/j.virusres.2018.04.002
38. Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods. 2011;8(8):652–4.
39. Das AT, Binda CS, Berkhout B. Elimination of infectious HIV DNA by CRISPR–Cas9. Curr Opin Virol [Internet]. 2019;38:81–8. Available from: https://doi.org/10.1016/j.coviro.2019.07.001
40. Biti, R., Ffrench, R., Young, J. et al. HIV-1 infection in an individual homozygous for the CCR5 deletion allele. Nat Med 3, 252–253 (1997). Available from: https://doi.org/10.1038/nm0397-252
41. Deng Q, Chen Z, Shi L, Lin H. Developmental progress of CRISPR/Cas9 and its therapeutic applications for HIV-1 infection. Rev Med Virol. 2018;28(5):1–7.
42. Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A. 2015;112(33):10437–42.
43. Campbell LA, Coke LM, Richie CT, Fortuno L V., Park AY, Harvey BK. Gesicle-Mediated Delivery of CRISPR/Cas9 Ribonucleoprotein Complex for Inactivating the HIV Provirus. Mol Ther [Internet]. 2019;27(1):151–63. Available from: https://doi.org/10.1016/j.ymthe.2018.10.002