Vivir así es morir de amor: Síndrome de Takotsubo
Realizado por Sofía Alván Benito
«Morir de amor»… Esa frase que hemos escuchado y leído miles de veces en poesías, libros o en la letra de famosas canciones como la de Camilo Sesto. Suena dramático e irreal, pero a veces la realidad supera a la ficción.
Introducción
El síndrome de Takotsubo (ST) es una patología que afecta al corazón. Suele aparecer en pacientes que han estado sometidos a un gran estrés físico o emocional (Lyon et al., 2016). Estos detonantes pueden ser, por ejemplo, la pérdida de un ser querido, un gran cambio inesperado en nuestra vida o incluso una ruptura con nuestra pareja.
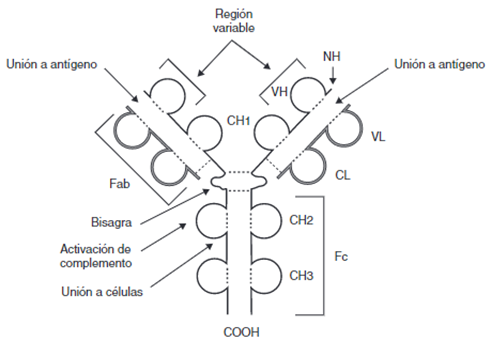
Fue diagnosticado por primera vez por un médico japonés, que bautizó a esta enfermedad con el nombre que tiene actualmente. La palabra «Takotsubo», en japonés, es el nombre de una vasija que se utiliza para cazar pulpos y que, curiosamente, tiene una forma muy similar a la que adquiere el corazón al sufrir esta enfermedad (Sato et al., 1990). (Figura 1)
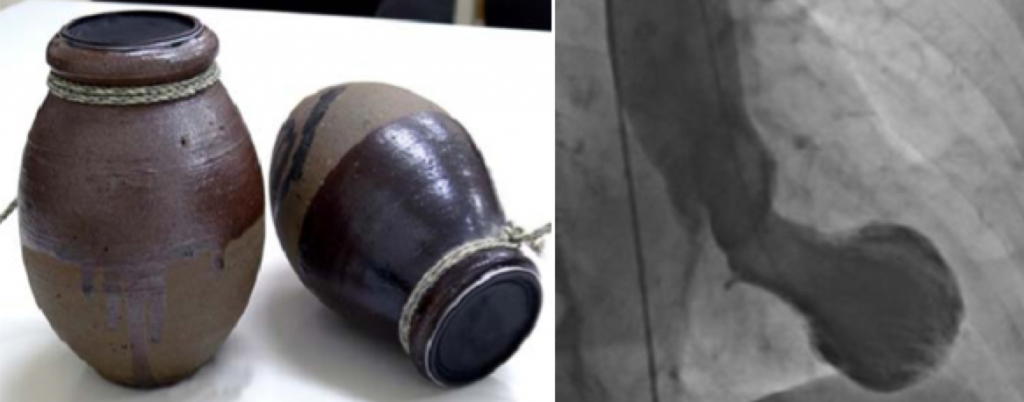
(Foto adaptada de la original tomada por: Profesor Christian Templin, Hospital Universitario de Zurich)
Síndrome de Takotsubo vs Infarto de Miocardio
Este síndrome, también llamado síndrome del corazón roto, se asemeja bastante a un infarto de miocardio (IM). Sin embargo, ambas patologías tienen grandes diferencias. (Tabla 1) (Falola, Fonbah, & McGwin, 2013; Gupta & Gupta, 2018)
| Síndrome de Takotsubo | Infarto de Miocardio | |
| Antecedentes cardiovasculares | No | Sí |
| Obstrucción arterial | No | Sí |
| Factores de riesgo | Estrés emocional o físico | Tabaco, obesidad, hipertensión, diabetes |
| Partes afectadas | Ventrículo izquierdo | Corazón (general) |
(De elaboración propia)
Epidemiología
Realmente, el Síndrome de Takotsubo es una enfermedad rara. Afecta únicamente, más o menos, a un 2% de todos los pacientes que fueron inicialmente diagnosticados con Síndrome Agudo del Miocardio. (Deshmukh et al., 2012)
Suele aparecer con mayor frecuencia en mujeres mayores de 50 años. La mayoría de casos se producen por un estímulo, que lleva a la aparición de la enfermedad. (Lyon et al., 2016). Sin embargo, en un 30% de los casos no hay detonante físico ni emocional. (Khera, Light-Mcgroary, Zahr, Horwitz, & Girotra, 2016). Dentro de los casos que sí se deben a un evento desencadenante, un 90% corresponden a eventos negativos, son los casos del síndrome del corazón roto propiamente dicho. (Templin et al., 2015). El 10% restante se debe a eventos positivos, como por ejemplo ganar la lotería, y constituyen una variante de esta patología que recibe el nombre de síndrome del corazón feliz. (Ghadri et al., 2016)
La tasa de mortalidad es muy baja, de un 4.5%. (Singh et al., 2014). En general, esta patología tiene un buen pronóstico y la mayoría de pacientes se recuperan en unos meses. (Elesber et al., 2007). Suele ser una enfermedad transitoria, aunque en ocasiones puede llegar a ser recurrente.
En hombres, aunque la enfermedad es menos frecuente, la mortalidad es superior a la de las mujeres. (Khera, Light-Mcgroary, Zahr, Horwitz, & Girotra, 2016)
Síntomas y manifestaciones clínicas
Como ya ha sido mencionado anteriormente, los síntomas de un paciente con Síndrome de Takotsubo son muy parecidos a los de un paciente que sufre un infarto de miocardio.
Los principales síntomas son dolor de pecho, disnea o dificultad para respirar, palpitaciones, insuficiencia cardíaca, paro cardíaco… (Templin et al., 2015)
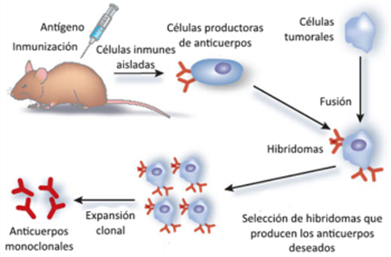
Los pacientes con este síndrome no presentan ningún otro problema cardiovascular, ni obstrucción en las arterias. A pesar de ello, se ve una pérdida de la función del ventrículo izquierdo del corazón, que es la parte más afectada. (Figura 2)
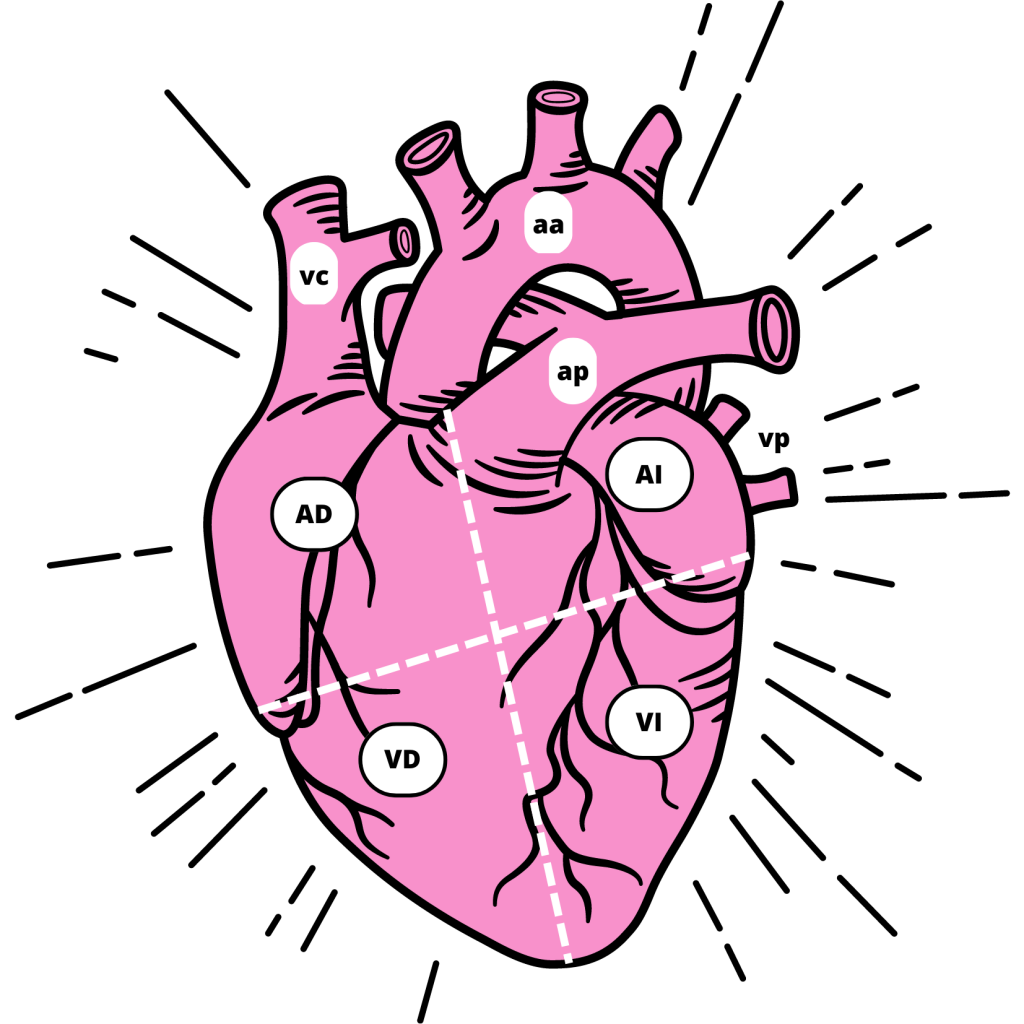
AD: Aurícula Derecha; AI: Aurícula Izquierda; VD: Ventrículo Derecho; VI: Ventrículo Izquierdo; vc: vena cava; aa: arteria aorta; ap: arteria pulmonar; vp: vena pulmonar
(De elaboración propia)
En condiciones normales, el corazón utiliza como fuente principal de energía la que procede del metabolismo de ácidos grasos en vez de la glucosa. Cuando se sufre este síndrome, el corazón cambia su metabolismo a uno en el que utiliza más glucosa y menos ácidos grasos. (Gupta & Gupta, 2018)
Además, se puede ver elevación de biomarcadores cardíacos como la troponina o el péptido natriurético. Esto puede servir para el diagnóstico de la enfermedad. (Budnik et al., 2016)
Puede haber alteraciones del electrocardiograma, en algunos casos. (Migliore, Zorzi, Perazzolo Marra, Iliceto, & Corrado, 2015)
Este síndrome también recibe el nombre de síndrome de balonamiento apical transitorio, ya que se puede observar una especie de abultamiento en forma de «balón» en la región apical. (Lyon et al., 2016)
Causas
Una de las posibles explicaciones a esta patología es que se produzca debido al exceso de estimulación del eje hipotálamo-hipófisis-adrenal. (Figura 3). En situaciones de estrés, se activa esta vía, para producir hormonas como los glucocorticoides (cortisol) y las catecolaminas (epinefrina y norepinefrina). (Figura 4)
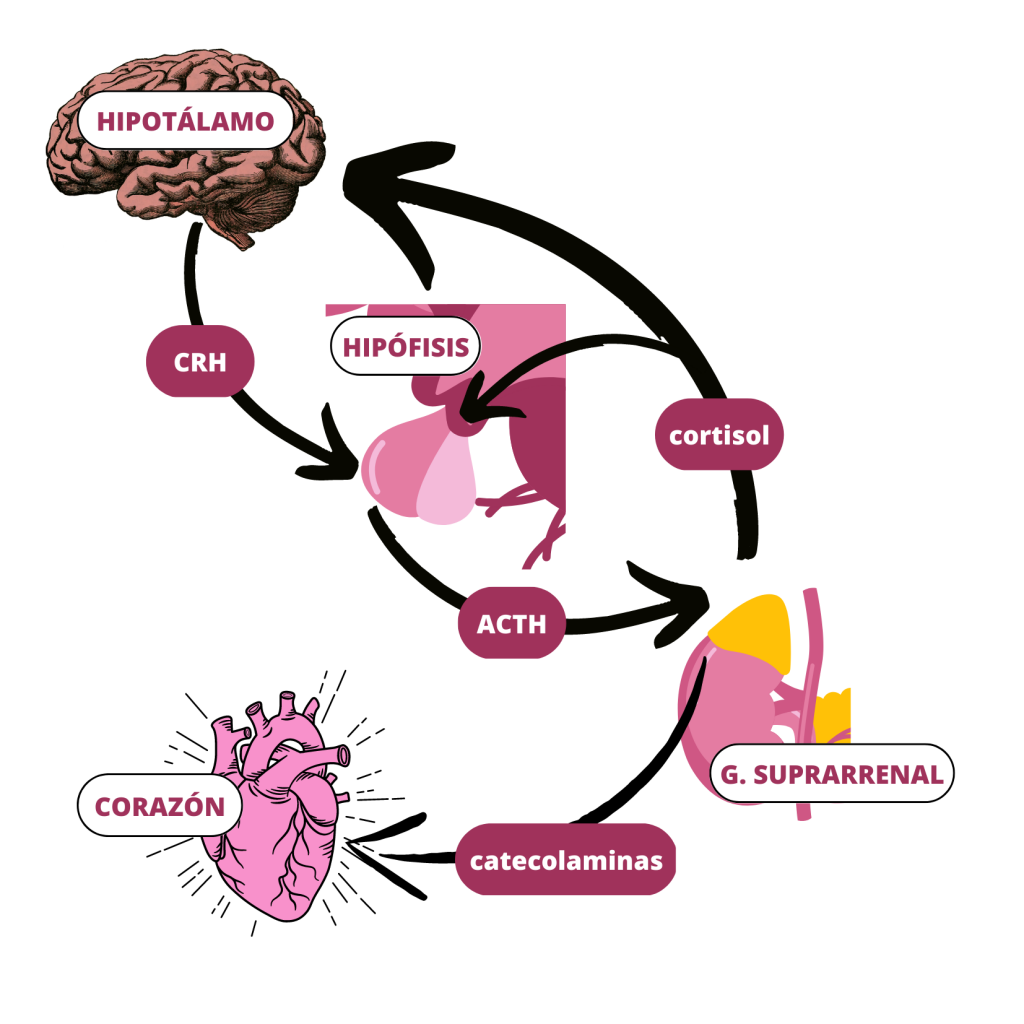
CRH: Hormona Liberadora de Corticotropina ; ACTH: Hormona Adenocorticotropa o Corticotropina
(De elaboración propia)
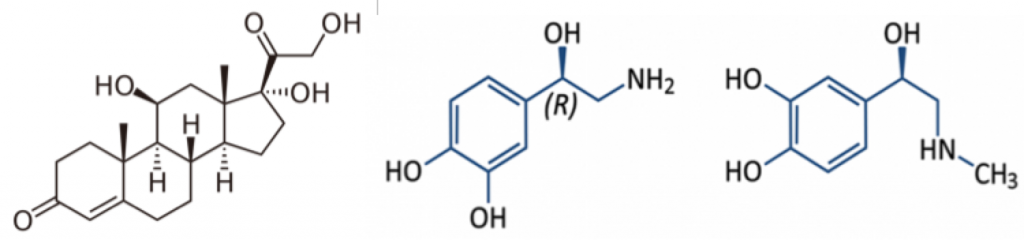
Cuando se produce esta patología, los niveles plasmáticos de epinefrina y norepinefrina son altos, lo que indica que pueden estar ejerciendo un efecto desmesurado sobre el corazón, lo que derivaría en este síndrome. (Wittstein et al., 2005)
La adrenalina y la noradrenalina actúan sobre receptores adrenérgicos para ejercer su función. En el corazón, el ventrículo izquierdo es el lugar en el que se encuentran el mayor número de receptores adrenérgicos. Esto explica que el ventrículo izquierdo del corazón sea la zona que se ve más afectada en el Síndrome de Takotsubo. (Gupta & Gupta, 2018)
Se producen cambios en el metabolismo celular, que llevan a una disminución de la contractibilidad del corazón, principalmente en el ventrículo izquierdo. (Gupta & Gupta, 2018)
Como curiosidad, algunas publicaciones científicas apuntan a una posible relación entre esta patología cardíaca y el cáncer. Se dice que es posible que las catecolaminas que se liberan en esta enfermedad actúen sobre células tumorales, induciendo su crecimiento. (Sattler et al., 2017)
Además, se ha postulado que el hecho de que afecte más frecuentemente a mujeres puede deberse a la influencia de ciertas hormonas sexuales. (Alkhoury et al., 2016)
Otros posibles factores que podría jugar algún papel en el desarrollo de esta enfermedad son los aspectos genéticos. (Ikutomi et al., 2014)
Tratamiento
No hay un solo tratamiento claro para esta condición.
Dependiendo de las circunstancias personales de cada paciente o de otras enfermedades coexistentes, el tratamiento podría incluir beta-bloqueadores (para frenar el efecto de las catecolaminas sobre sus receptores), ventilación mecánica, levosimendan (para la insuficiencia cardíaca)… (Santoro et al., 2013; Templin et al., 2015)
Conclusión
El Síndrome de Takotsubo es un gran ejemplo de que existe una conexión entre el cerebro y diferentes órganos de nuestro cuerpo, en este caso, entre el cerebro y el corazón. No solo influyen las cuestiones físicas sobre la salud del cuerpo, sino que las cuestiones emocionales a veces pueden ser tan influyentes que no solo afectan a nuestro estado de ánimo, también pueden llegar a afectar al funcionamiento de órganos tan importantes como lo es el corazón.
En definitiva, si solías ser de los que dicen «de amor nadie se muere», apuesto a que ahora habrás cambiado de opinión y tendrás que decir «de amor sí se puede morir».
Referencias
Alkhoury, J., Lundgren, J., Ali, A., Mesinovic, D., Redfors, B., & Omerovic, E. (2016). Updates on publication trends in takotsubo syndrome doi:10.1016/j.ijcard.2016.07.059
Budnik, M., Kochanowski, J., Piatkowski, R., Wojtera, K., Peller, M., Gaska, M., . . . Opolski, G. (2016). Simple markers can distinguish takotsubo cardiomyopathy from ST segment elevation myocardial infarction. International Journal of Cardiology, 219 doi:10.1016/j.ijcard.2016.06.015
Deshmukh, A., Kumar, G., Pant, S., Rihal, C., Murugiah, K., & Mehta, J. L. (2012). Prevalence of takotsubo cardiomyopathy in the united states. American Heart Journal, 164(1) doi:10.1016/j.ahj.2012.03.020
Elesber, A. A., Prasad, A., Lennon, R. J., Wright, R. S., Lerman, A., & Rihal, C. S. (2007). Four-year recurrence rate and prognosis of the apical ballooning syndrome. Journal of the American College of Cardiology, 50(5) doi:10.1016/j.jacc.2007.03.050
Falola, M., Fonbah, W., & McGwin, G. (2013). Takotsubo cardiomyopathy versus ST-elevation myocardial infarction in a large case-control study: Proposing a new mechanism. International Journal of Cardiology, 167(3) doi:10.1016/j.ijcard.2012.10.059
Ghadri, J. R., Sarcon, A., Diekmann, J., Bataiosu, D. R., Cammann, V. L., Jurisic, S., . . . Prasad, A. (2016). Happy heart syndrome: Role of positive emotional stress in takotsubo syndrome. European Heart Journal, 37(37) doi:10.1093/eurheartj/ehv757
Gupta, S., & Gupta, M. M. (2018). Takotsubo syndrome doi:10.1016/j.ihj.2017.09.005
Ikutomi, M., Yamasaki, M., Matsusita, M., Watari, Y., Arashi, H., Endo, G., . . . Ohnishi, S. (2014). Takotsubo cardiomyopathy in siblings. Heart and Vessels, 29(1) doi:10.1007/s00380-013-0345-y
Khera, R., Light-Mcgroary, K., Zahr, F., Horwitz, P. A., & Girotra, S. (2016). Trends in hospitalization for takotsubo cardiomyopathy in the united states. American Heart Journal, 172 doi:10.1016/j.ahj.2015.10.022
Lyon, A. R., Bossone, E., Schneider, B., Sechtem, U., Citro, R., Underwood, S. R., . . . Omerovic, E. (2016). Current state of knowledge on takotsubo syndrome: A position statement from the taskforce on takotsubo syndrome of the heart failure association of the european society of cardiology doi:10.1002/ejhf.424
Migliore, F., Zorzi, A., Perazzolo Marra, M., Iliceto, S., & Corrado, D. (2015). Myocardial edema as a substrate of electrocardiographic abnormalities and life-threatening arrhythmias in reversible ventricular dysfunction of takotsubo cardiomyopathy: Imaging evidence, presumed mechanisms, and implications for therapy. Heart Rhythm, 12(8) doi:10.1016/j.hrthm.2015.04.041
Santoro, F., Ieva, R., Ferraretti, A., Ienco, V., Carpagnano, G., Lodispoto, M., . . . Brunetti, N. D. (2013). Safety and feasibility of levosimendan administration in takotsubo cardiomyopathy: A case series. Cardiovascular Therapeutics, 31(6) doi:10.1111/1755-5922.12047
Sato, H., Tateishi, H., Uchida, T., Dote, K., Ishihara, M., Kodama, K., … & Hori, M. (1990). Clinical aspect of myocardial injury: from ischemia to heart failure. Kagaku Hyoronsha, 2, 55-64.
Sattler, K., El-Battrawy, I., Lang, S., Zhou, X., Schramm, K., Tülümen, E., . . . Akin, I. (2017). Prevalence of cancer in takotsubo cardiomyopathy: Short and long-term outcome. International Journal of Cardiology, 238 doi:10.1016/j.ijcard.2017.02.093
Singh, K., Carson, K., Shah, R., Sawhney, G., Singh, B., Parsaik, A., . . . Horowitz, J. (2014). Meta-analysis of clinical correlates of acute mortality in takotsubo cardiomyopathy doi:10.1016/j.amjcard.2014.01.419
Templin, C., Ghadri, J. R., Diekmann, J., Napp, L. C., Bataiosu, D. R., Jaguszewski, M., . . . Lüscher, T. F. (2015). Clinical features and outcomes of takotsubo (stress) cardiomyopathy. New England Journal of Medicine, 373(10) doi:10.1056/nejmoa1406761
Wittstein, I. S., Thiemann, D. R., Lima, J. A. C., Baughman, K. L., Schulman, S. P., Gerstenblith, G., . . . Champion, H. C. (2005). Neurohumoral features of myocardial stunning due to sudden emotional stress. New England Journal of Medicine, 352(6) doi:10.1056/nejmoa043046