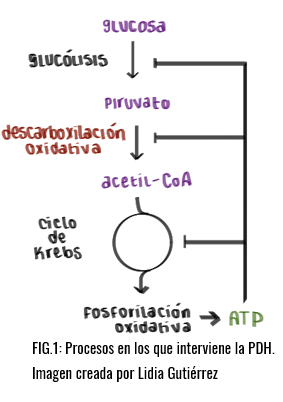
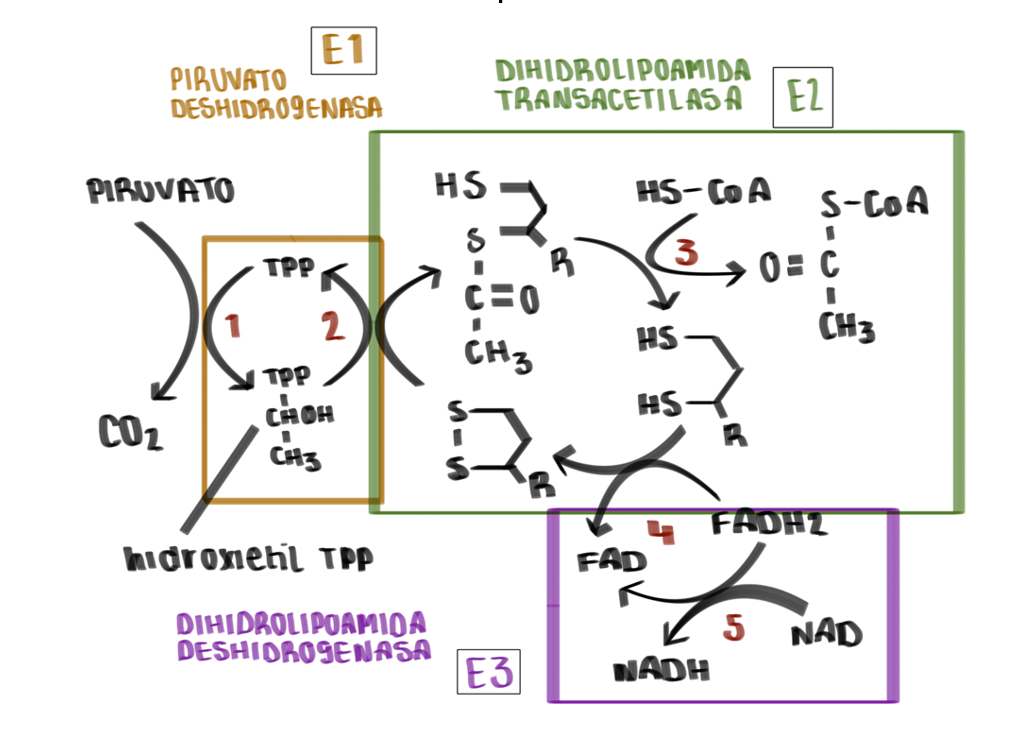
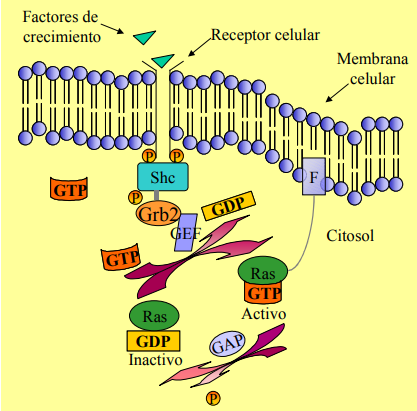
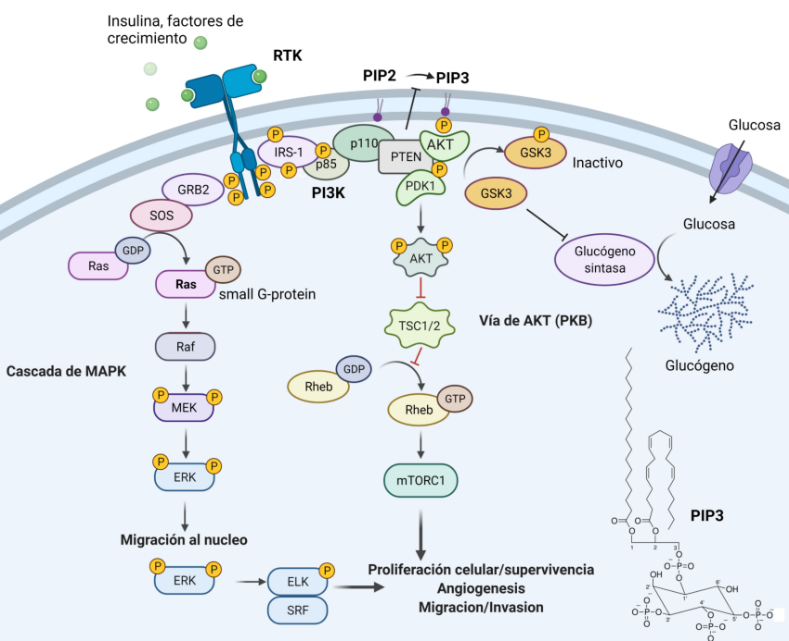
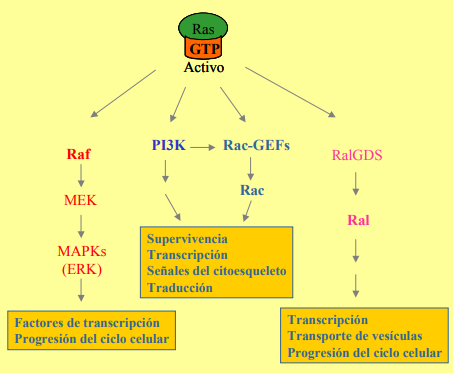
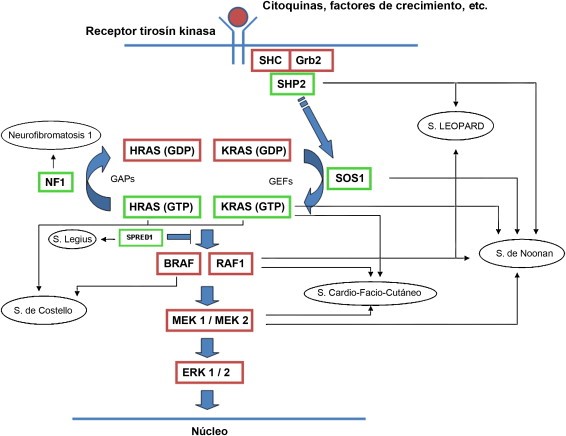
Fluorescencia
Imagina que tienes la oportunidad entrar en una habitación oscura donde hay minerales, papeles, productos químicos… y tu única fuente de luz es una linterna de luz ultravioleta. De pronto, algunos objetos comienzan a emitir luz de colores vivos – rojos, verdes, azules – como si escondieran luces internas. Este fenómeno sorprendente es la fluorescencia, una propiedad física por la cual los objetos absorben energía de la luz incidente (la llamaremos luz de excitación) y luego la re-emiten como luz visible. En términos sencillos, los fotones UV de tu linterna excitan temporalmente electrones en defectos o características propias de la red cristalina, o impurezas del material, y al volver a su estado inicial esos electrones liberan la energía extra en forma de un destello luminoso, que nuestros ojos pueden ver.
Nos vamos a centrar en los minerales. No todas los minerales son fluorescentes. Para que un mineral brille bajo luz UV o azul (las mejores luces de excitación) debe contener los ingredientes adecuados: los llamados elementos activadores o centros luminiscentes. Estos son típicamente trazas de ciertos elementos o moléculas incrustadas en la red cristalina que son capaces de provocar el fenómeno. En definitiva, la fluorescencia es una obra colectiva entre la luz UV, el activador adecuado y el entorno cristalino que lo acoje.
No hay que confundir fluorescencia y fosforescencia, aunque ambos son fenómenos muy relacionados. En la fluorescencia, la emisión de luz cesa al apagar la luz de excitación. En el caso de la fosforescencia, la emisión de luz perdura, a veces unos segundos, a veces minutos u horas, apagándose lentamente.
El rubí y el cromo: destellos rojos bajo la luz UV
El rubí es un material muy especial, no sólo por formar las conocidas gemas de intenso color rojo. Además, tiene unas propiedades ópticas únicas y, entre ellas, su espectro de fluorescencia es tan limpio y único que el primer láser de la historia (el láser de rubí de Maiman, 1960) aprovecha esta característica.

El rubí es una variedad roja del corindón, que es simplemente óxido de aluminio (Al2O3). El óxido de aluminio puro es blanco o incoloro, pero el rubí debe su color y su fluorescencia a pequeñas cantidades de cromo trivalente (Cr³⁺) en su interior. El cromo trivalente se parece químicamente al aluminio trivalente; entonces, el Cr³⁺ sustituye a unos pocos Al³⁺ en la red de óxido de aluminio, quedando enclavado en sitios octaédricos rodeados de oxígenos. Este “invitado” de transición tiene electrones 3d cuyo nivel de energía se ve partido por el campo cristalino del corindón. Bajo excitación (por ejemplo con luz verde-azulada o UV), los electrones del Cr³⁺ suben a niveles excitados y rápidamente relajan a un estado metastable desde el cual revierten a su estado fundamental emitiendo fotones rojos. El resultado es un espectro de emisión característico, dominado por dos picos estrechos en la zona del rojo profundo (alrededor de 694 nm, en el rojo oscuro) conocidos como las líneas R del rubí.

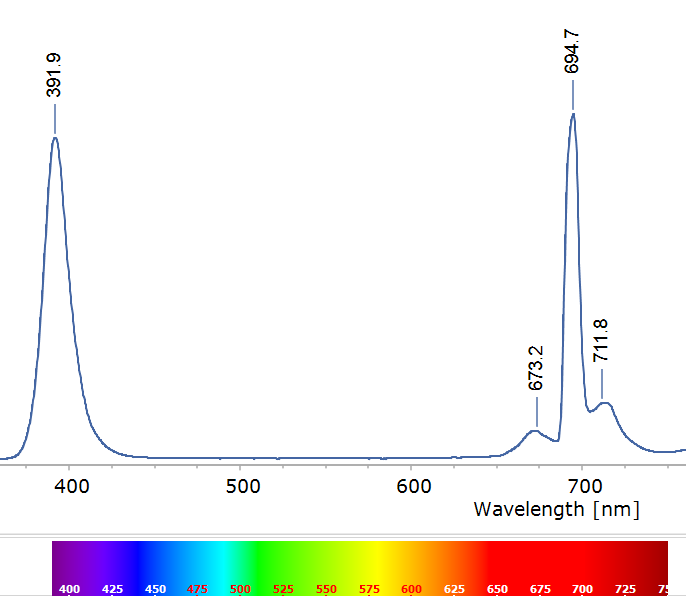
Estas características espectrales nos permiten introducir dos conceptos fundamentales en luminiscencia de sólidos: la línea cero-fonón (ZPL) y las bandas laterales vibrónicas (phonon sidebands). En el rubí podemos identificar:
- Línea cero-fonón (ZPL): corresponde a la transición electrónica “pura” del Cr³⁺ desde el estado excitado al fundamental sin cambio vibracional en la red. Es decir, ningún fonón (cuanto de vibración del cristal) es creado ni destruido durante la emisión del fotón. Las líneas R del rubí, tan intensas y definidas, son esencialmente ZPL: transiciones electrónicas internas del Cr³⁺ no acopladas a fonones. Por eso aparecen como picos bien definidos en ~694 nm. Constituyen la firma del Cr³⁺ en corindón.
- Bandas laterales (vibrónicas): alrededor de las líneas R se observan picos o “hombros” más débiles a longitudes de onda ligeramente distintas (por ejemplo, en ciertos rubíes sintéticos se reportan pequeños picos cerca de 688 nm o 701 nm, etc.). Estas son emisiones donde la transición electrónica sí va acompañada de la creación o absorción de uno o más fonones en el cristal. En otras palabras, parte de la energía se va en forma devibraciones de la red cristalina. Si durante la emisión se crea un fonón (vibración), el fotón sale con un poco menos de energía (es decir, de longitud de onda mayor, más desplazado al rojo), porque parte de la energía se ha ‘quedado’ en forma de vibración de la red cristalina. Esta se llama desplazamiento de Stokes; si en cambio se aniquila un fonón existente, el fotón sale con un poco más de energía (longitud de onda menor, más desplazado al azul) y tenemos sideband anti-Stokes. Este fonón existente es un fonón térmico, es decir, son vibraciones de la red cristalina debidas a la propia temperatura ambiente. Este acoplamiento electrón-fonón resultan en picos satélite simétricos, de menor intensidad flanqueando la ZPL, que vemos en la imagen. Además, como es esperable, el pico Stokes es de mayor intensidad que el anti-Stokes.
Ahora bien, ¿por qué el rubí exhibe una ZPL tan intensa y apenas unas bandas laterales débiles, mientras otros materiales fluorescentes presentan espectros mucho más anchos? La respuesta nos la proporciona el principio de Franck–Condon, originalmente formulado para transiciones moleculares pero totalmente aplicable a centros luminiscentes en sólidos. Este principio dice, esencialmente, que las transiciones electrónicas son muy rápidas comparadas con el movimiento nuclear. En cristales, esto implica que cuando el sitio del Cr³⁺ emite un fotón, los iones del entorno no tienen tiempo de reajustarse instantáneamente a la nueva configuración electrónica. Si la posición de equilibrio del Cr³⁺ en el estado excitado difiere de su posición en el estado fundamental, la emisión “atrapará” al sistema en un estado vibracional excitado. Dicho de otra forma: el fotón se emite antes de que la red termine de relajarse, dejando uno o varios fonones excitados en el cristal. La intensidad relativa de la ZPL y las bandas vibónicas viene determinada por el traslape entre las funciones de onda vibracionales inicial y final; cuanto mayor sea el desplazamiento entre los mínimos de energía, menor será la probabilidad de regresar al nivel vibracional base sin excitaciones. En el rubí, este desplazamiento es pequeño, indicando un acoplamiento moderado entre el Cr³⁺ y las vibraciones del corindón. Por eso la mayor parte de las emisiones ocurren sin cambio vibracional (ZPL intensa) y solo una fracción pequeña involucra fonones (sidebands tenues). Es un equilibrio delicado, donde el rubí opta por “devolver” la energía casi íntegra en forma de luz roja pura, en vez de repartirla con el retículo cristalino. Esto se traduce en el bello color del rubí.
El destellos verde de la adamita: la firma luminosa del uranio
Nos trasladamos a Mapimí, en el semidesierto de Durango (México), donde la antigua mina de Ojuela ha dado ejemplares mundialmente famosos de adamita fluorescente. La adamita es un arseniato de zinc (Zn₂AsO₄OH) que en sí misma no es especialmente luminiscente. El secreto de su intensa fluorescencia verde está en diminutas impurezas de uranio incorporadas en su estructura, en forma de iones uranilo (UO₂²⁺) sustituyendo parcialmente al zinc o presentes en inclusiones microscópicas. Bajo la luz UV de onda larga o violeta azulada, estas muestras emiten una luminosidad verde inconfundible.

A diferencia del rubí, aquí no vemos un par de picos aislados, sino tres picos principales en la región verde (aproximadamente a 510, 530 y 554 nm) junto con algunos hombros o picos menores adyacentes. Este patrón de múltiples líneas estrechas y casi equidistantes es característico del ión uranilo (UO₂²⁺). De hecho, los minerales que contienen uranio suelen revelar su presencia justamente por esa “firma” de fluorescencia verde multilineal. Cada uno de estos picos corresponde a una transición electrónica del uranilo acompañada de un cambio vibracional interno de la molécula UO₂²⁺. Podemos imaginar el ion uranilo como una pequeña molécula lineal (O=U=O) incrustada en el cristal: cuando sus electrones se excitan, la molécula vibra como un muelle y al relajarse emite fotones verdes. Las diferencias en longitud de onda (del orden de 20 nm entre picos) corresponden a la energía de vibración de los enlaces U–O, de modo que observamos una progresión vibromolecular, una serie de líneas (0→0, 0→1, 0→2, etc. en notación de transición vibracional) análoga a la que se ve en moléculas gaseosas, pero aquí congelada dentro del sólido.
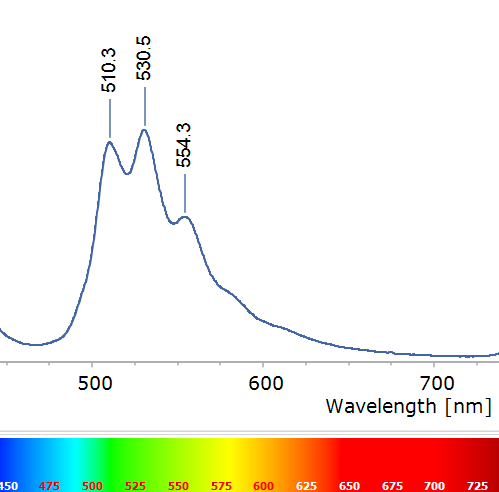
En el caso del uranilo, la transición electrónica responsable de la emisión de luz es un retorno desde un estado excitado de tipo ligando-a-metal hacia el estado fundamental, con una vibración simétrica característica acoplada a esa emisión. El resultado es este espectro de “códigos de barras” verde. Su presencia en la adamita confirma que el activador de su fluorescencia es UO₂²⁺ (uranio en estado de oxidación +6), probablemente incorporado durante la formación del mineral en cantidades traza.
Después de ver dos ejemplos de espectros bien estructurados (rubí y adamita), surge la pregunta: ¿qué pasa cuando la fluorescencia no se manifiesta en líneas definidas sino en una banda ancha difusa? Para explorarlo, viajemos a las canteras aragonesas en busca de un mineral mucho más común pero con un misterio escondido: el yeso.
El yeso de Fuentes de Ebro
El yeso (sulfato de calcio dihidratado, CaSO₄·2H₂O) es un mineral abundante y, en principio, no famoso por su luminiscencia. Sin embargo, en las canteras de alabastro de Fuentes de Ebro, cerca de Zaragoza (España), se han encontrado grandes cristales de yeso que bajo luz UV exhiben un resplandor entre cian y amarillo apreciable.

Al registrar el espectro de fluorescencia de estas muestras (Figura 3), en lugar de picos definidos se observa una banda ancha que abarca buena parte del azul, con un máximo alrededor de ~486 nm (azul verdoso) y se extiende al amarillo. ¿Qué está provocando esta fluorescencia y por qué su espectro es tan diferente?
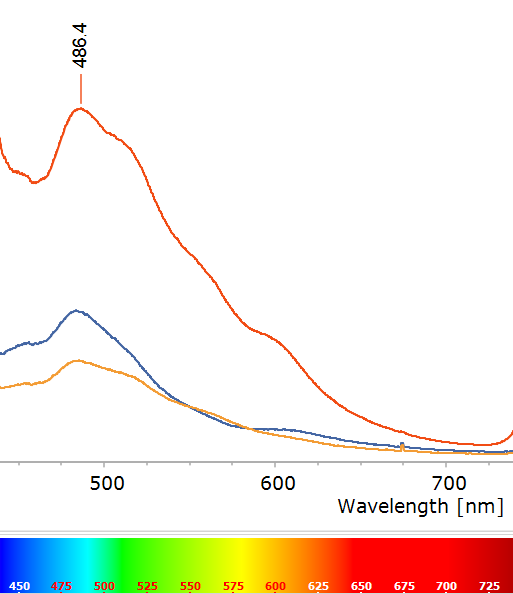
Este caso es más complejo. Los datos y estudios sobre yeso fluorescente apuntan a que el activador no es el propio CaSO₄, sino alguna impureza presente en el cristal. En el caso de Fuentes de Ebro, una hipótesis plausible es la presencia de materia orgánica incluida durante la formación del yeso sedimentario. Por ejemplo, trazas de compuestos aromáticos policíclicos (PAHs) o sustancias húmicas derivadas de materia vegetal podrían haberse incorporado microscópicamente en el cristal mientras se formaba a partir de soluciones evaporíticas. Estos compuestos orgánicos suelen ser fuertemente fluorescentes en el rango del espectro cuando se les excita con UV. De hecho, en minerales sedimentarios es conocido que inclusiones de petróleo, betunes u otros restos orgánicos producen frecuentemente fluorescencia: muchos calcitas y fluoritas exhiben colores desde azulados a blancos o amarillento-verdoso por esta causa. En el yeso de Fuentes de Ebro, todo sugiere que la fluorescencia proviene de tales impurezas orgánicas, más que de centros cristalinos inorgánicos.
¿Podrían existir alternativas a la explicación orgánica? Cabe preguntarse si algunos centros de tipo defecto en el cristal o trazas de tierras raras (elementos lantánidos) están involucrados. No tenemos datos para establecer una causa definitiva, pero el espectro de fluorescencia es consistente con la materia orgánica, en la que las transiciones se reparten entre multitud de niveles vibrónicos, y pequeñas diferencias en cada centro ensanchan y solapan las emisiones. Es como un “coro” desordenado de muchas voces: las notas se mezclan en un continuo, resultando en un brillo difuso sin picos aislados.

La próxima vez que veas una vitrina de minerales fluorescentes en un museo, o apuntes con una linterna UV a tus propias muestras, recuerda que cada color y cada espectro narran una historia: la del baile entre impurezas atómicas y la estructura cristalina. Ya sea el rojo láser de un rubí, el verde fantasmagórico debido al uranilo, o el color misterioso de las impurezas orgánicas, todas esas luces son mensajes del mineral que revelan una química escondida, que solo se deja ver bajo la magia de la luz ultravioleta.
Referencias
Adachi, S. et al. “Franck-Condon analysis of Cr³⁺ luminescence in Al₂O₃ polymorphs”. J. Luminescence 239 (2021): 118369.
Hunault, M. O. J. Y. et al. “Direct Observation of Cr³⁺ 3d States in Ruby: Toward Experimental Mechanistic Evidence of Metal Chemistry”. J. Phys. Chem. A 122 (2018): 4399–4413.
Fluomin – Barmarin, G. et al. Database of fluorescent minerals (Adamite, Gypsum entries).
Modreski, P. J. & Newsome, D. “Green uranium-activated fluorescence of adamite from the Ojuela mine, Mapimí, Mexico.” FM-MSA-TGMS Symposium, Minerals of Mexico (Tucson, 1984).
Robbins, M. Fluorescence: Gems and Minerals Under Ultraviolet Light. Geoscience Press, 1994. ISBN 0-945005-13-X.
Gorobets, B. S. & Rogojine, A. A. Luminescent Spectra of Minerals. Reference Book, Moscow, 2002