

Febrero 2019. Rev. 3, Nov 2023. Por César Menor-Salván.
La electroforesis en gel de acrilamida es una de las técnicas básicas mas importantes en el laboratorio de Biología Molecular. Permite la separación de una mezcla de proteínas según su peso molecular y es la técnica de entrada para la realización del Western-Blot, purificación de proteínas para su análisis por espectrometría de masas, estimación de peso molecular o identificación de variaciones en la expresión o estructura de proteínas. Su fundamento físico tiene cierta complejidad, por lo que aquí mostraremos una versión mas simplificada, de interés para los biólogos. Vamos a centrarnos en los fundamentos de la técnica mas que en la práctica, sobre la cual hay numerosos turoriales en video.
Vamos a elaborar la electroforesis según el método desarrollado y refinado por Ulrich Laemmli en los años 1960-70. La idea básica de la electroforesis es la separación de proteínas en un campo eléctrico. Para ello necesitamos dos fuerzas opuestas: el campo eléctrico que va a provocar la movilidad de la proteína en base a su carga, y un soporte sólido que va a retener la proteína en base a su interacción debido al tamaño. La base de la técnica SDS-PAGE es hacer que el movimiento de la proteína en el soporte sólido (gel) por acción del campo eléctrico sea exclusivamente proporcional a su peso molecular.
Construcción del gel: la polimerización
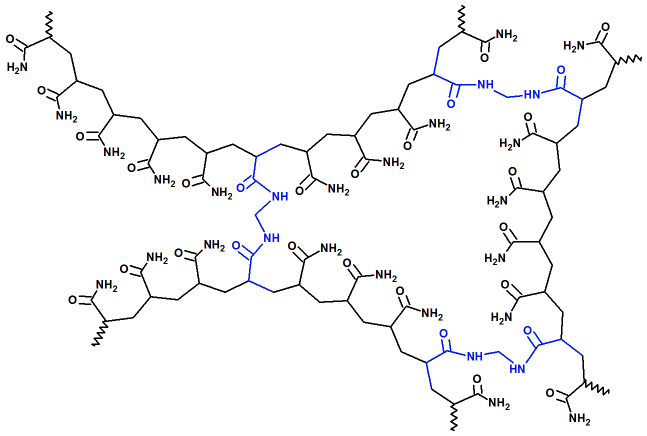
La electroforesis en gel de poliacrilamida requiere la construcción de un hidrogel (gran cantidad de agua retenida en un soporte molecular altamente poroso y reticulado, resultando un material viscoso, semisólido o un sólido blando). El hidrogel de polímero de acrilamida es el soporte donde tiene lugar la separación de proteínas, gracias a la fricción de las moléculas de proteína con la estructura del gel. Actualmente se puede obtener ya preparado (precast gel), pero sigue siendo usual que se prepare in-situ en el momento de su uso. La preparación del gel se basa en la siguiente reacción:
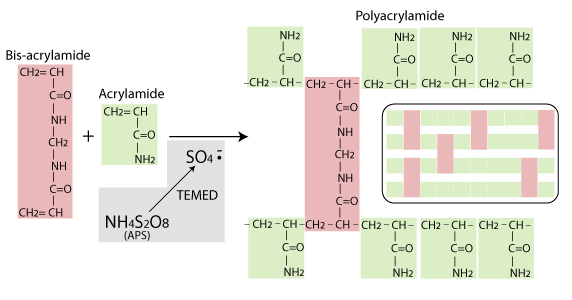
Los componentes implicados en la reacción son:
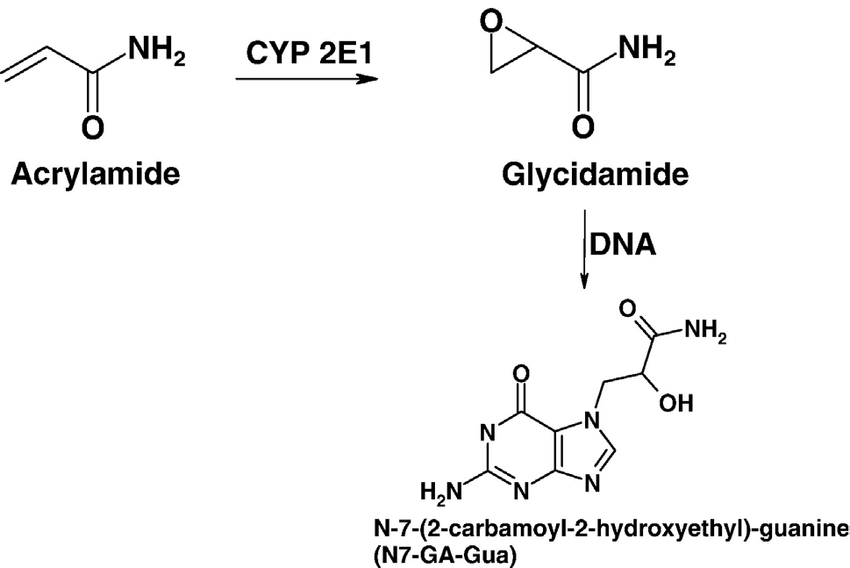
1- Acrilamida. El monómero fundamental que va a formar el gel. La acrilamida es un compuesto vinílico y, como tal, es susceptible de polimerización por via radicálida. La acrilamida es soluble en agua, no es un compuesto con gran toxicidad aguda, pero en una exposición laboral o exposición continua tiene un efecto acumulativo, es cancerígena (en especial puede inducir cáncer de piel), teratógena y es un neurotóxico. Por ello debe manejarse con precaución. Una vez polimerizada, la poliacrilamida resultante es inocua, ya que ha perdido los grupos vinilo altamente reactivos. De hecho, numerosos cosméticos, como geles fijadores de pelo, llevan polímeros acrílicos similares.
La toxicidad de la acrilamida se debe en parte a la formación de su epóxido, la glicidamida, que reacciona con bases nitrogenadas de los ácidos nucleicos y proteínas, alterando su función o induciendo mutaciones.
2- TEMED y persulfato. Son la clave del mecanismo de polimerización via radicales libres.

https://doi.org/10.1016/j.reactfunctpolym.2004.06.004
El TEMED y el persulfato generan los radicales necesarios para iniciar la reacción, que prosigue en cadena hasta formar el hidrogel.
3- N,N’-metilenbisacrilamida: Agente responsable del entrecruzamiento de las cadenas de poliacrilamida y la reticulación del gel. Es esencial para regular las propiedades mecánicas y la capacidad de separación del gel.
Cómo el gel de poliacrilamida puede separar las proteínas.
El gel, una vez preparado en su soporte, nos permite separar una mezcla de proteínas en forma de bandas según su peso molecular: la electroforesis. El proceso global de separación se observa en éste estupendo video, preparado por María Jarabo Arranz (Estudiante, en ese momento, de Biología Sanitaria UAH). En el video vemos un gel preparado en su montaje, dentro de una cubeta electroforética donde se establece el campo eléctrico que induce el movimiento de las proteínas en el gel.
Los componentes implicados en el proceso del video son:
- 1- El propio gel, que consta de dos partes: gel concentrador y gel separador. Nos aporta la fuerza de retención, que va a frenar más las proteínas de mayor peso molecular
- 2- Los electrodos en la cubeta, que establecen el campo eléctrico que aportará la movilidad electroforética
- 3- El tampón de electroforesis, que permite la conducción eléctrica y contiene componentes fundamentales en la separación: la glicina y el Tris-HCl
- 4- La propia muestra, que se prepara utilizando otro componente fundamental: el tampón de carga o de ruptura que a su vez contiene cuatro componentes fundamentales: SDS, mercaptoetanol, glicerol y azul de bromofenol
Vamos a ver detalladamente que ha ocurrido en el video:
Gel concentrador y gel separador. Papel del SDS
La electroforesis SDS-PAGE se diseña de modo que podamos separar las proteínas según su peso molecular. Para ello debemos utilizar una serie de componentes que se incorporan a los tampones utilizados, tanto en la preparación del gel como en la propia electroforesis y en la preparación de la muestra. El más relevante es el SDS.
SDS (lauril sulfato sódico o dodecil sulfato sódico)
Es un detergente muy utilizado, tanto en el laboratorio como en la vida diaria. Lo podemos encontrar en champús y geles o jabones líquidos.
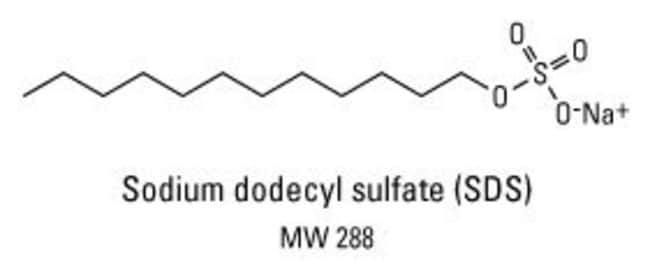
Su papel es la desnaturalización y el establecimiento de una relación carga-tamaño constante en la proteína, de modo que la movilidad de la proteína durante la electroforesis sea exclusivamente proporcional a su peso molecular. En condiciones nativas, la proteína está plegada según su estructura terciaria y tiene una carga global (positiva, neutra o negativa) que va en función de la composición de aminoácidos, el punto isoeléctrico de la proteína y el pH del tampón en el que se encuentra. Si hiciéramos la electroforesis sin SDS, la proteína tendría una carga global que dependería del pH y el punto isoeléctrico. Al añadir SDS, tiene lugar la siguiente reacción:
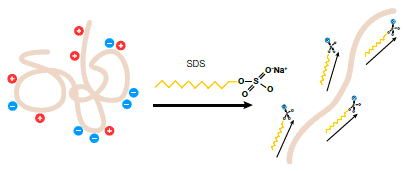
Como resultado, la proteína adquiere una carga global negativa y directamente proporcional a su tamaño, de modo que la cantidad de SDS que interacciona con la proteína es constante y de unos 1.4 gramos de SDS por cada gramo de proteína. La interacción de ésta con el polímero de acrilamida será mayor cuanto mayor sea el peso molecular. La velocidad de desplazamiento de la proteína en el campo eléctrico es inversamente proporcional al peso molecular.
Gel separador y concentrador
La separación tiene lugar en el gel separador. Éste gel realiza la separación a un pH 8.8. Antes de que las proteínas entren en el gel separador, pasan por un gel concentrador, a pH 6.8, que evita que las proteínas se dispersen en el gel y hace que todas las proteínas de las muestras se alineen en una fina banda que entra al mismo tiempo y a la misma velocidad en el gel concentrador. Este proceso de concentración es esencial para aumentar la resolución de la separación.
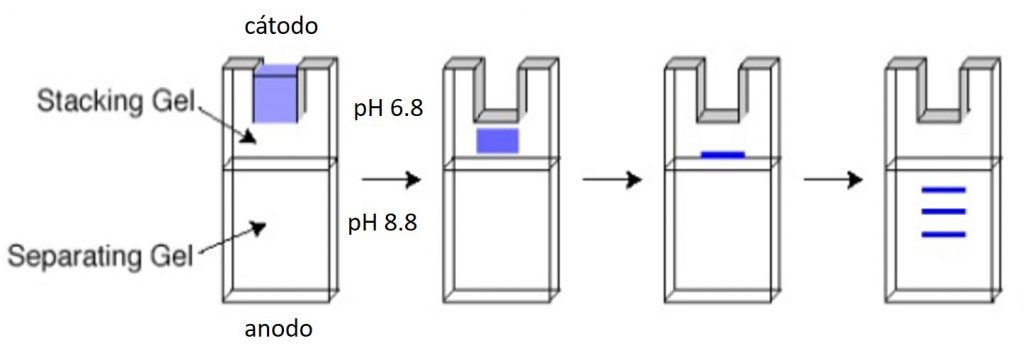
El papel de la glicina y el Tris-Cl. Cambios en el gel durante la electroforesis
En la separación juega un papel fundamental la glicina y los iones Cl- del tampón, que van entrando en el gel durante la electroforesis. Para entender el papel de la glicina hay que conocer el punto isoeléctrico y los pK de la glicina:
- pI=6
- pK1=2.34
- pK2=9.6
Esto significa que a pH 6, la glicina se encuentra en forma de zwitterion (eléctricamente neutro). A pH mayor de 9.6, la glicina está cargada negativamente y a pH por debajo de 2.34 está cargada positivamente. En los pH intermedios, prevalece la forma zwitterionica, pero a pH<6 va aumentando la concentración de gly+ y a pH>6 de gly–.
Para entender bien el importantísimo papel de la glicina en la SDS-PAGE, tenemos que ver la distribución de especies en la glicina según el pH:
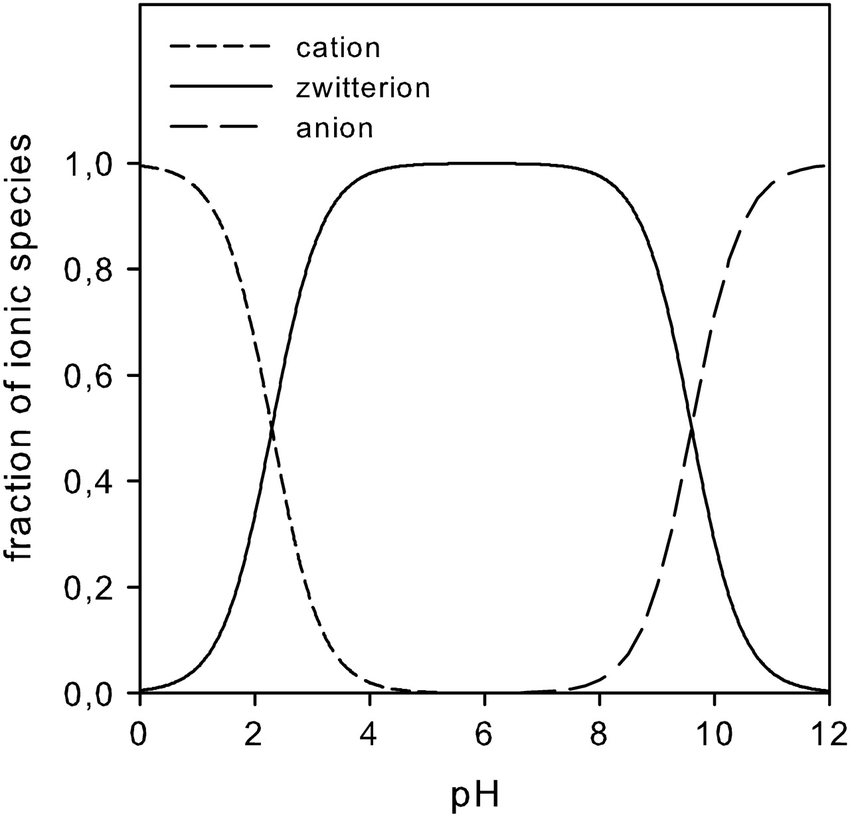
En el gel concentrador, a pH 6.8, la glicina se encuentra mayoritariamente en forma de zwitterion, pero estamos a un pH en el que no hay moléculas de glicina cargadas positivamente. Ahora, comencemos la electroforesis. El ánodo (polo positivo) se encuentra en la parte inferior y el cátodo en la parte superior. Al conectar la fuente, los iones positivos del tampón Tris+ se desplazan hacia arriba, y los iones cloruro hacia abajo, al ánodo. Se produce un cambio en las concentraciones de los compuestos y comienza a cambiar el pH del gel. Se genera una zona superior que alcanza un pH de 8.9. Esto provoca que la glicina, que estaba en forma de zwitterion, se cargue negativamente. Pero la movilidad electroforética de la glicina es inferior a la del cloruro. Por ello, se crea un frente cloruro-glicina, con las proteínas en medio, que se concentran en una fina banda, debido a la repulsión electrostática provocada por los aniones cloruro y glicina.
Aquí ahora juega un papel importante el menor porcentaje de acrilamida en el gel concentrador: las proteínas se mueven a mucha velocidad en el gradiente eléctrico, sin ser frenadas por el gel. Pero, al llegar a la zona rica en cloruro, que va desplazándose rápidamente, sufren repulsión electrostática y se frenan. El resultado es que las proteínas no pueden sobrepasar la zona enriquecida en cloruro y se alinean, atrapadas en el campo eléctrico que actúa como «rayo tractor» (guiño y codazo a los trekkies)
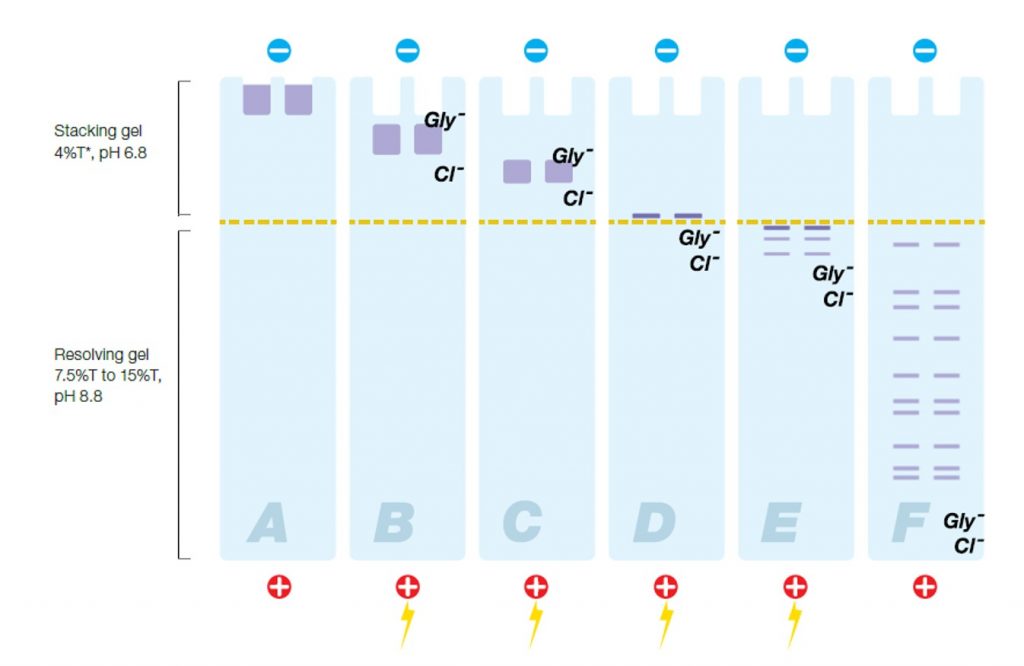
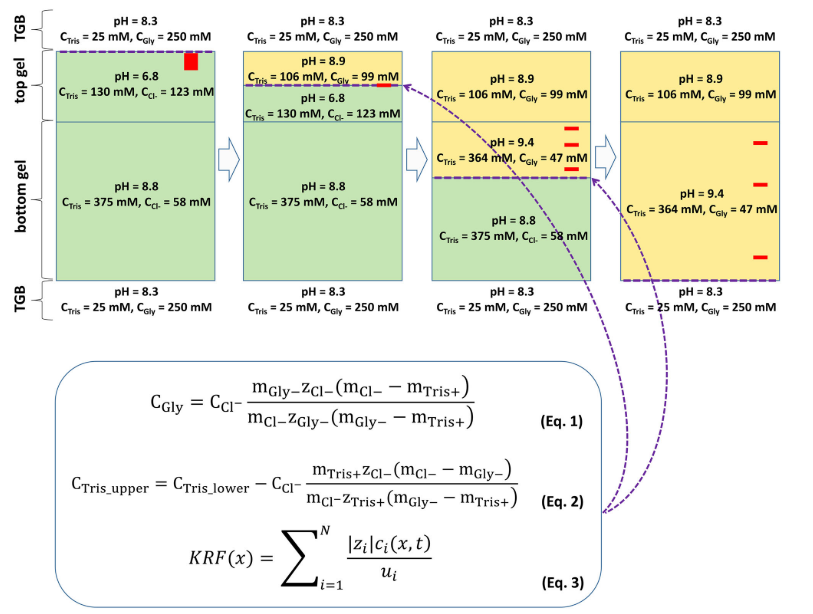
Cuando el frente rico en cloruro que retiene las proteínas alcanza el gel separador, el pH de éste comienza a aumentar hasta alcanzar un pH 9.4. Este pH aumenta la carga negativa de la glicina y su movilidad, desplazándose rápidamente hacia el frente, junto con el cloruro. Las proteínas, a su vez, se encuentran con un gel más concentrado y «liberadas» del «campo de fuerza» o el «rayo tractor» interaccionan con el gel, realizándose la separación.
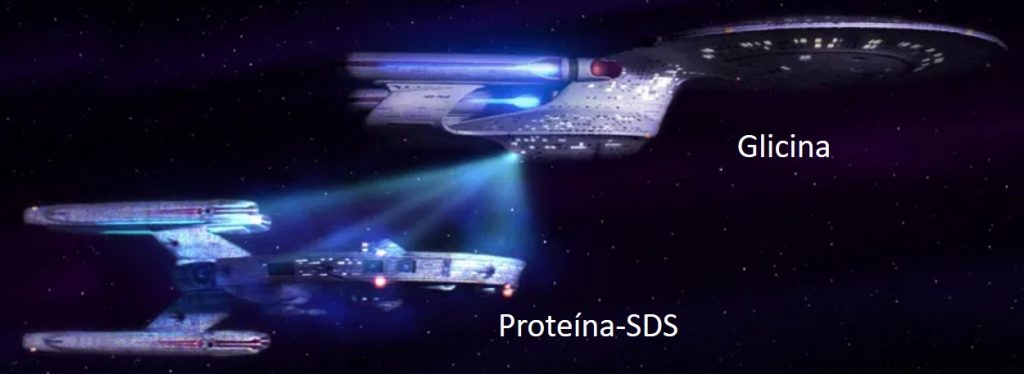
La glicina sigue jugando un papel importante durante la separación. En el gel separador, la glicina y el cloruro no desaparecen a toda velocidad, sino que se forma una banda que actúa como «barrera», evitando que las proteínas la sobrepasen y formando el frente de la electroforesis.

La electroforesis: movilidad electroforética y peso molecular.
La velocidad a la que se mueve una proteína en el gel se define como:
v=µ E
donde v es la velocidad de desplazamiento y E es el campo eléctrico. El parámetro µ se denomina movilidad electroforética
En términos de voltaje, podemos convertir la ecuación en:
v=µ V/D
donde V es el voltaje al que se realiza la electroforesis y D es la longitud del gel separador.
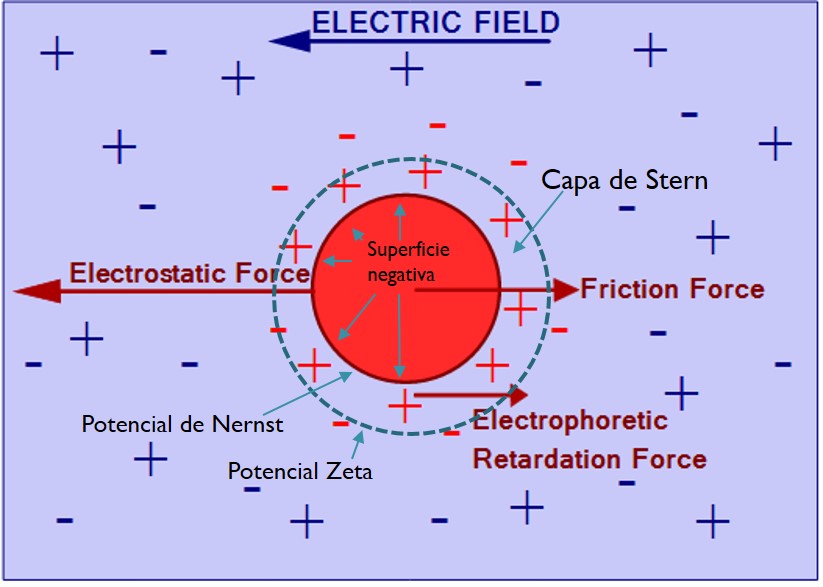
Durante la electroforesis, la partícula proteica está sometida a tres fuerzas principales: la debida al campo eléctrico aplicado (que tira de la proteína hacia el ánodo), la fuerza de retención electrostática, creada debido a la capa de Stern, capa de iones unidos por atracción electrostática a la superficie de la partícula, y la fuerza de fricción de la partícula en el polímero del gel. En el formalismo clásico de Hückel, la movilidad electroforética considera el efecto contrapuesto de la fuerza inducida por el campo eléctrico que «tira» de la partícula y la fuerza debida a la fricción de Stokes, que «retiene» la partícula en el medio viscoso del gel,
Q E=f v; f = 1/ 6 πhR
Donde Q es la carga de la partícula, E el campo eléctrico, f la fricción de Stokes (fricción de una partícula de radio R que se desplaza en un medio viscoso de viscosidad h) y v la velocidad. La movilidad será el cociente v/E que es igual a f/v, y puede definirse entonces la movilidad electroforética como:
µ=Q/6 πhR
donde Q es la carga neta, h la viscosidad del medio y R el radio de la partícula proteica que se desplaza. Según esta ecuación, la movilidad depende de la carga neta (a mayor carga, mayor movilidad) y de la fricción de Stokes de la partícula en el gel (a mayor tamaño de la partícula, mayor fricción y menor movilidad y a mayor viscosidad del gel, mayor fricción y menor movilidad). El radio R es el radio de Stokes que es la suma del radio de la partícula y la región de fluido que la engloba. El cociente Q/R es la densidad de carga superficial. Aquí está el meollo de la técnica: si la densidad de carga es constante, la movilidad electroforética es dependiente del radio y la velocidad sólo va a depender de la fricción.
La teoría de Smoluchowski explica mucho mas detalladamente el fenómeno. De modo muy simplificado, según ésta, podemos definir la movilidad relativa de la partícula como la suma de dos contribuciones:
µF Fext + µE E
µE es la movilidad electrocinética y µF la movilidad no eléctrocinética, que se aproxima a la movilidad de una partícula en un medio viscoso según Stokes: 1/6πhR
La movilidad eléctrocinética fue definida por Smoluchowski como el cociente entre el potencial zeta, o potencial electrocinético, y la constante dieléctrica del medio (e), dividido entre la viscosidad del gel (h).
µE = e Z/h
Es decir, a mayor potencial zeta, mayor movilidad electroforética. El potencial eléctrico de superficie de la partícula en sí mismo se denomina potencial de Nernst. El potencial zeta es el potencial en el límite de la capa de Stern con la zona de fricción de la partícula en el medio. La teoría de Smoluchowski ya nos dice que la movilidad eléctrica no va a depender exactamente de la carga neta de la partícula, sino de su densidad de carga superficial que generará un potencial zeta determinado. Aquí está el meollo: el SDS provoca que el potencial zeta de las partículas proteicas sea el mismo, debido a que genera una densidad de carga superficial constante para todas la proteínas. Por tanto, la movilidad relativa va a depender únicamente de la fuerza de fricción, y, por tanto del tamaño de partícula y su peso molecular y de la viscosidad (o porcentaje de acrilamida) del gel. Viendo la movilidad de Stokes anterior, a mayor peso molecular o mayor viscosidad del gel, menor movilidad relativa.
Estimando el peso molecular con la movilidad relativa o migración
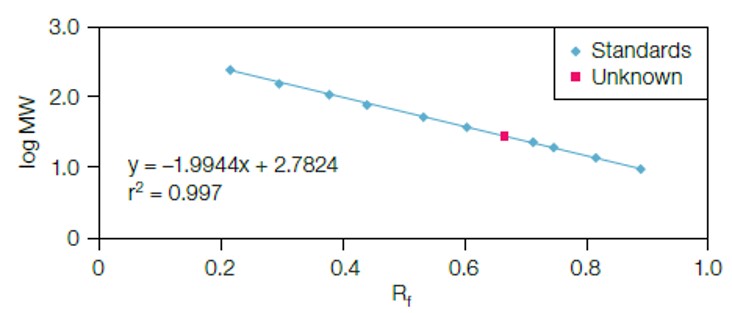
En cada electroforesis se incorpora una mezcla de proteínas de peso molecular conocido que sirven como patrón. Este patrón nos permite hacer una estimación de nuestras proteínas de interés. Para ello medimos la movilidad relativa o la migración de la banda de la proteína como:
Rf = distancia migrada por la proteína / distancia migrada por el frente.
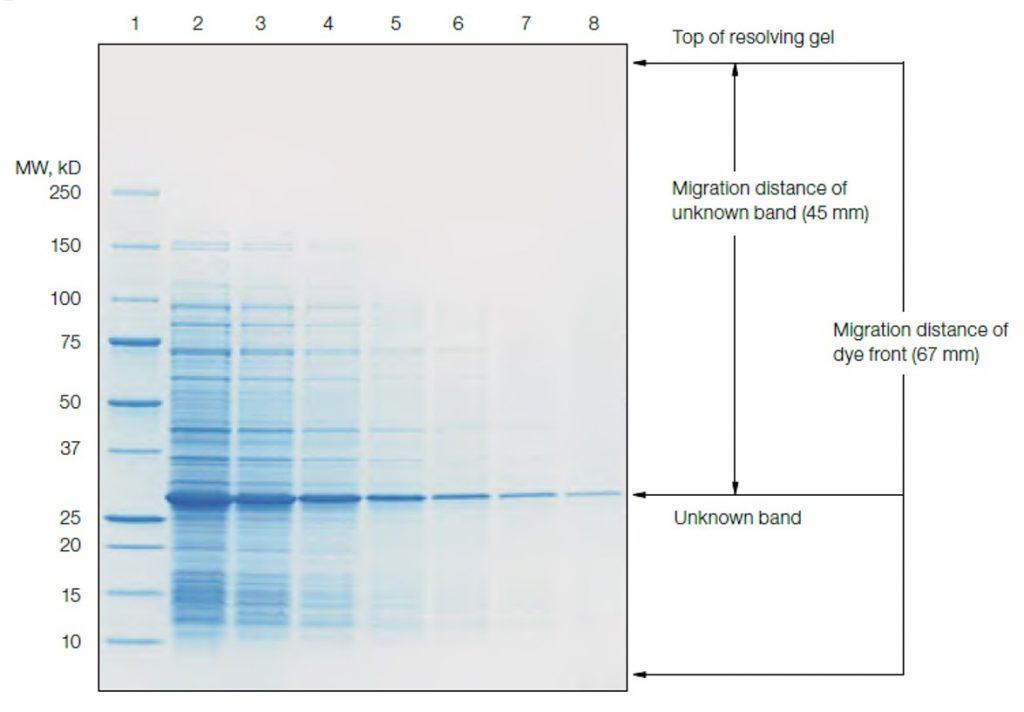
Una vez que hemos determinado los Rf los patrones, podemos representar gráficamente el logaritmo del peso molecular frente al Rf, obteniéndose una recta. Interpolando en la recta el valor de Rf de la proteína que se está analizando, podemos determinar su peso molecular.
Visualizando las proteínas: colorantes
Tras terminar la electroforesis (ver video anterior) no vemos las proteínas de nuestras muestras. Tan sólo los marcadores de peso molecular, que están pre-teñidos. Es el momento de visualizar el resultado. Para ello, uno de los métodos mas sencillos y comunes es la tinción con el colorante azul Coomassie. Este colorante nos permite detectar entre 0.1 y 0.5 µg de proteína en el gel.
El azul Coomassie, formalmente azul brillante Coomassie R-250 o G-250, es un colorante desarrollado en el siglo XIX por la compañía británica Levinstein Ltd. para la tinción textil, específicamente de lana (que está formada por proteína). El nombre de azul Coomassie se debe a que pocos meses antes, los británicos conquistaron la capital del reino Ashanti, en la actual Ghana, llamada Coomassie. El reino Ashanti era uno de los más poderosos de Africa y el único capaz de ofrecer resistencia contra el imperio británico, en una guerra que duró décadas, llamadas las guerras anglo-ashanti. La síntesis del azul de Coomassie coincidió con el final de las guerras y recibió su nombre como conmemoración de la victoria colonial británica.

En el año 1963, el microbiólogo hungaro-australiano Stephen Fazekas de St. Groth utilizó el azul Coomassie para la tinción de proteínas en electroforesis por primera vez. Desde entonces, se ha convertido en un método de rutina en el laboratorio de Biología Molecular.
El azul de Coomassie se une fuertemente a la proteína por una combinación de atracción electrostática a aminoácidos básicos (histidina, lisina y arginina) e interacciones hidrofóbicas entre fenilalanina y los grupos fenilo del colorante.
Como resultado, si todo ha salido bien, obtenemos unas bonitas bandas de color azul intenso en los sitios del gel donde se encuentra la proteína, lo cual produce una gran satisfacción, pues es una recompensa a muchas horas de trabajo.
Otro colorante importante en su uso en el análisis de proteínas es el Rojo Ponceau S.

Es un colorante azoico que tiñe reversiblemente las proteínas y no afecta a la aplicación de métodos posteriores, como el inmunoblot, por lo que es muy útil para verificar que la transferencia de proteínas del gel de electroforesis a una membrana para inmunoblot ha salido bien.
Referencias
Andrews AT (1986). Electrophoresis: theory, techniques and biochemical and clinical applications (New York: Oxford University Press).
Bio-Rad (2012) Technical Bulletin 6040. A guide to Polyacrylamide gel electrophoresis and detection.
Bjellqvist B et al. (1982). Isoelectric focusing in immobilized pH gradients: principle, methodology and some applications. J Biochem Biophys Methods 6, 317–339.
Brunelle J L and Green R (2014). Coomassie blue staining. Methods in Enzymology (1st ed., Vol. 541). Elsevier Inc.
Congdon, R. W., Muth, G. W., & Splittgerber, A. G. (1993). The binding interaction of coomassie blue with proteins. Analytical Biochemistry.
Davis BJ (1964). Disc electrophoresis. II. Method and application to human serum proteins. Ann NY Acad Sci 121, 404–427.
Debye P and Hückel E, Physikalische Zeitschrift 24, 305 (1923).
Dunn MJ (1993). Gel electrophoresis: Proteins (Oxford: BIOS Scientific Publishers Ltd.).
Fenselau C (2007). A review of quantitative methods for proteomic studies. J Chromatogr B Analyt Technol Biomed Life Sci 855, 14–20.
Garfin DE (1990). One-dimensional gel electrophoresis. Methods Enzymol 182, 425–441.
Garfin DE (2009). One-dimensional gel electrophoresis. Methods Enzymol 463, 497–513.
Georgiou, C. D., Grintzalis, K., Zervoudakis, G., & Papapostolou, I. (2008). Mechanism of Coomassie brilliant blue G-250 binding to proteins: A hydrophobic assay for nanogram quantities of proteins. Analytical and Bioanalytical Chemistry, 391(1), 391–403.
Goldenberg DP and Creighton TE (1984). Gel electrophoresis in studies of protein conformation and folding. Anal Biochem 138, 1–18.
Hames BD (1998). Gel electrophoresis of proteins: A practical approach, 3rd ed. (Oxford: Oxford University Press).
Koshkina, M. K., Shelomov, M. D., Pometun, A. A., Savin, S. S., Tishkov, V. I. & Atroshenko, D. L. (2023) ‘Speeding up SDS–PAGE: Theory and experiment’, Electrophoresis, 44(15–16), pp. 1155–1164. doi: 10.1002/elps.202300011.
Laemmli UK (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685.
Lizana L and Grosberg AY (2014) Exact expressions for the mobility and electrophoretic mobility of a weakly charged sphere in a simple electrolyte. ArXiv:1305.4060v2
McLellan T (1982). Electrophoresis buffers for polyacrylamide gels at various pH. Anal Biochem 126, 94–99.
O’Farrell PH (1975). High resolution two-dimensional electrophoresis of proteins. J Biol Chem 250, 4007–4021.
Rabilloud T (2010). Variations on a theme: changes to electrophoretic separations that can make a difference. J Proteomics 73, 1562–1572.
Steinberg TH (2009). Protein gel staining methods: an introduction and overview. Methods Enzymol 463, 541–563. Steinberg
von Smoluchowski M, Bull. Int. Acad. Sci. Cracovie (1903).
