AMPLIFICACIÓN ISOTÉRMICA MEDIADA POR BUCLE (LAMP)


Laura Díez Pérez & Jaime Franco Mansilla
La Universidad es discusión, es efervescencia, no es pensamiento único
Alberto Kornblihtt
REFLEXIÓN INICIAL
Para empezar esta entrada en el blog, nos gustaría abandonar esa visión del estudiante de Biología Molecular que debe publicar un trabajo de investigación sobre un tema concreto; y centrarnos en un estudiante, futuro científico, que adquiere poco a poco una visión más crítica de la realidad, una realidad llena de injusticias.
Pues bien, este trabajo no va a estar dedicado a la ciencia teórica, nos negamos rotundamente. ¿Por qué estamos acostumbrados a realizar solo este tipo de trabajos? Vale que sea importante el saber y el conocimiento, y vale que en base a ese conocimiento actúas y te planteas hipótesis, pero ¿de qué sirve plantearte una buena hipótesis si luego no conoces los medios para poder afirmar o refutar tu idea?
Pongámonos en contexto. Nuestro grado, Biología Sanitaria comprende un total de 240 créditos ECTS, de los cuales, únicamente 12 de ellos (el equivalente a cuatro asignaturas cuatrimestrales en cuatro años de carrera) se destinan a aprender técnicas de experimentación, esto es, un 5 %. Por acotar, quitemos los 18 créditos de las prácticas externas y los 12 del TFG. Nos quedan por tanto, 198 créditos donde la aptitud que más se valora es la memorística. Recita en tu mente si te acuerdas toda la glicólisis (si lo consigues es que lo tienes estudiado de forma muy reciente; ya te darás cuenta que al cabo del tiempo, la acabarás olvidando, ¿por qué? porque este sistema se basa fundamentalmente en memorizar, devolver toda la información que te sabes en un examen, y olvidar lo que has memorizado porque tienes que empezar a estudiarte el siguiente examen; memorizar y olvidar). Ahora, si yo te digo que pienses cómo estudiarías los efectos de un fármaco A en una enzima B, y que no solo estudies un experimento, sino que luego consigas interpretar los resultados, ¿a qué te cuesta más? Pero queremos ir un poco más allá, ¿de las técnicas que se te han venido a la cabeza, cuál de ellas es puntera actualmente? Esta última pregunta hace referencia a que, aunque sepamos de alguna técnica, estamos totalmente desactualizados. Nos acordamos cuando en nuestro grado nos explicaban la técnica de secuenciación Maxam-Gilbert, una técnica completamente obsoleta (vale que históricamente sea el primer método de secuenciación, y que está bien saber algo historia; pero está mucho mejor saber cuáles son las técnicas de secuenciación masivas actuales, que es lo que se usa ahora en un laboratorio y con lo que vamos a tener que vernos las caras en un futuro muy próximo).
Por todo esto, esta entrada al blog quiere centrarse en explicar el fundamento de una técnica, y también, para qué sirve y como utilizarla. Esta técnica es la amplificación isotérmica mediada por bucle (abreviada LAMP).
«¿De qué sirve aprender y memorizar contenidos sin saber luego su empleo práctico?»
Una segunda reflexión que queríamos plasmar aquí es que la literatura científica o los artículos científicos que explican ciertos temas son de escasa utilidad a los estudiantes, científicos jóvenes, e incluso a científicos ya expertos, debido a la complejidad con la que son explicados. Muchas veces la literatura científica está demasiado centrada en su especialidad y quizás esa información vaya dedicado a otro tipo de público. Por ello, es interesante, por ejemplo, abordar este problema con una entrada a un blog, donde hemos hecho ese ejercicio de desciframiento para que entendáis de una forma destripada y mascada el fundamento de la técnica.
Desde aquí proponemos a futuras entradas al blog a hacer lo mismo, que se dejen de tantas proteínas y fármacos, y que se centren en explicar técnicas y fundamentos; ya que estamos convencidos, de que algún lector que pase por este blog, le será muy útil esta información y podrá incorporar una LAMP para su hipótesis.
INTRODUCCIÓN
Realmente, esta técnica no es tan nueva como puede parecer, ya que el artículo original fue publicado en el año 2000 por científicos japoneses de las universidades de Tokio y Osaka. Entonces, podrías pensar que es un poco contradictorio lo que expusimos en la reflexión inicial: «mejor aprender técnicas más actuales»; y razón no te falta, pero, ¿acaso habías oído algo de esta técnica? (en nuestro caso, nos ponemos en el pellejo de alumnos que cursan nuestro grado, pero estamos convencidos de que la mayoría de estudiantes que pasen por aquí, esta técnica no les sonará de nada). No obstante, para que sea también de tu agrado, proponemos al final de la entrada, una revisión de las variantes de esta técnica, y alguno de sus usos actuales (que los tiene, y más cercanos de los que piensas).
¿Y por qué esta técnica para dedicarle una entrada a este blog y no otra? Simple. Esta técnica es fiel competidora de la tan conocida, sagrada y divina PCR (Polymerase Chain Reaction); y si es así, ¿por qué no había escuchado nada (o muy poco) de esta técnica? Pues porque la PCR funciona muy bien y está muy estandarizada ¿podríamos decir que existe una cara oculta de la ciencia (realmente, de los científicos) de aferrarse a lo conservador? ¿No es curioso? ¿Y qué pasó cuando los colegas médicos de Semmelweis llegaron incluso hasta amenazarle por simplemente querer introducir un hábito antiséptico como lavarse las manos? Nosotros creemos que es pura condición humana: el rechazo a lo novedoso.
Eppur si muove! («Y sin embargo, se mueve»)
Frase (mal atribuida) de Galileo Galilei al retratarse de su idea heliocéntrica frente a la Inquisición
Queremos preguntar en este contexto otra cosa ¿es el prestigio y la fama el que determina que algo se pueda implementar en la comunidad científica? ¿Tiene la misma relevancia los descubrimientos de un científico que no es conocido, que uno que lleva años nutriéndose de fama y premios? ¿No le pasó algo así al fraile Gregor Mendel con sus estudios de la herencia que no fueron valorados hasta su redescubrimiento por Hugo de Vries, Carl Correns, Erich von Tschermak y William Bateson años después? O al olvidado Michael Creeth en el descubrimiento de que la doble hélice de DNA se estabilizaba por enlaces de hidrógenos ¿a qué no habías escuchado su nombre en la historia del descubrimiento de la estructura del DNA? Además, ¿no hizo además el prestigio de Linus Pauling retrasar el descubrimiento de esta estructura debido a su idea de la triple hélice? ¿Quién os dice que dentro de unos años, no se redescubra el LAMP, ganando esa importancia que quizás pueda tener, y pase a ser la nueva panacea?
«El prestigio en ciencia, es un lastre para la ciencia»
Frase dicha por César Ángel Menor Salvan, que pasó desapercibida, pero que deja mucho que pensar
Además, si está técnica hubiera sido ideada por científicos europeos o estadounidenses, ¿crees que seguirías sin saber nada de ella? Dejamos esa pregunta en el aire para que te respondas tú mismo.

Realmente, hay que diferenciar entre los descubrimientos científicos y la aparición de nuevas técnicas (aunque realmente, crear una nueva técnica de algún modo es un descubrimiento). Pues en el primer caso, los científicos son muy reaccionarios contra nuevos descubrimientos, en especial si cuestionan ideas de científicos prestigiosos. Con las técnicas pasa algo diferente, si funcionan bien se imponen rápidamente, si no se demuestra realmente útil (en las demandas científicas de la época), se queda en los archivos y queda desapercibida hasta que alguien encuentra una aplicación muy útil.
«La ciencia avanza funeral a funeral»
Max Planck. Os recomendamos pasaros por el siguiente enlace.
Con todo esto no queremos estigmatizar la fama ganada (y merecida) de la PCR, pero creemos que si se explota esta técnica lo suficiente, podría llegar incluso a superarla. Es muy difícil que otra técnica se imponga a ella, si no aporta una clara ventaja, pero si sigues leyendo, verás como la técnica LAMP posee numerosas ventajas ( frente a la PCR y otras técnicas de amplificación de DNA).
En la imagen siguiente, podemos observar el incremento de las citas en los artículos científicos frente al LAMP, desde que se creó la técnica. En estos últimos años, ha habido un ligero aumento con respecto a lo normal, debido a que con la pandemia de la COVID19, mucha gente se ha puesto a probarla y sacarla a algún partido. Quién sabe si en algún momento la imposición de esta técnica en un aspecto concreto pueda hacer que se disparen sus citas dentro de unos años.
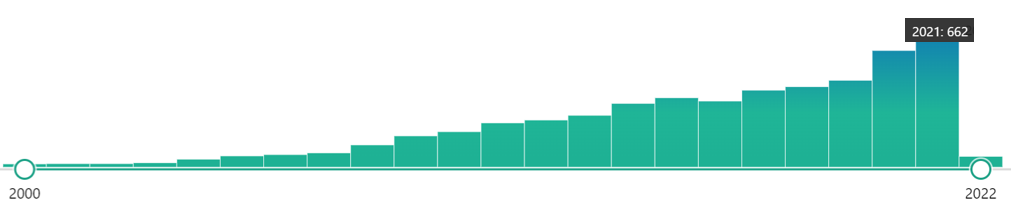
Con todo, creemos que es importante que maticemos que en este caso, tanto de la LAMP como de la PCR, estamos hablando de su papel CUALITATIVO. Os adelantamos que la técnica la LAMP no es empleada para obtener una secuencia única y pura de una secuencia de nucleótidos (al menos hasta ahora), sino de amplificar una secuencia para demostrar o si existe o no esa secuencia.
¿Por qué decimos que la técnica LAMP podría superar a la PCR? Por las siguientes ventajas:
- No requiere ningún aparato específico para llevarla a cabo, salvo un baño termostático o un termostato de bloque seco que casi todos los laboratorios suelen tener.
- El tiempo de detección de una determinada secuencia es inferior a una hora porque se amplifica en condiciones isotérmicas (no se hace con diferentes ciclos de temperatura como la PCR). Únicamente se necesita un baño a una temperatura constante (de ahí que sea isotérmica) de unos 60-65ºC. Además, no necesita una desnaturalización previa de la cadena de DNA molde, porque la polimerasa que se emplea tiene actividad de desplazamiento de cadena (actividad parecida a la helicasa).
- La detección es rápida y se puede hacer visualmente por turbidez, fluorescencia o usando reactivos con color.
- Por todo lo anterior, es una técnica más simple y menos laboriosa que una PCR.
- Es más específica, ya que emplea cuatro (o incluso seis) cebadores, frente a los dos que emplea la PCR.
- Es más sensible que la PCR convencional y sus variaciones.
- Es más barata.
Aunque esta técnica también tiene sus desventajas, como son:
- El diseño de cebadores es mucho más complejo que en una PCR.
- Pese a que la polimerasa empleada funciona en un rango amplio de temperaturas, no es tan termoestable como la Taq polimerasa usada en la PCR, por lo que a temperaturas por debajo o por encima de sus temperaturas de actuación, la polimerasa se inactivará.
- La técnica no presenta un 100 % de eficiencia, por lo que se puede generar ruido de fondo ante amplificaciones no específicas o, por ejemplo, la formación de dímeros de cebadores.
- No es tan conocida por lo que no se usa en muchos laboratorios como técnica habitual de diagnóstico.
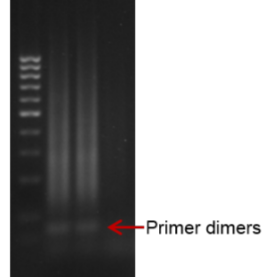
No sabemos si hay reticencia a usar LAMP, pero cuando tu tienes una técnica estandarizada en tu laboratorio y que funciona (como la PCR), necesitas tener una motivación para implementar otra técnica, lo que normalmente es bastante costoso en tiempo y fondos. No sabemos qué ocurre con LAMP, pues hay gente que la está usando y les funciona muy bien y otros se quejan de dificultades en la reproducibilidad, eficiencia, y otras dificultades técnicas.
COMPONENTES DE LA MEZCLA DE REACCIÓN
Para el desarrollo de esta técnica, necesitaremos:
- DNA molde: que se quiera amplificar.
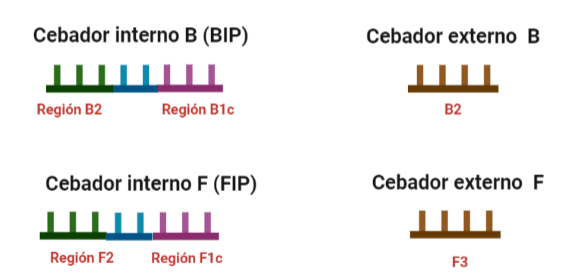
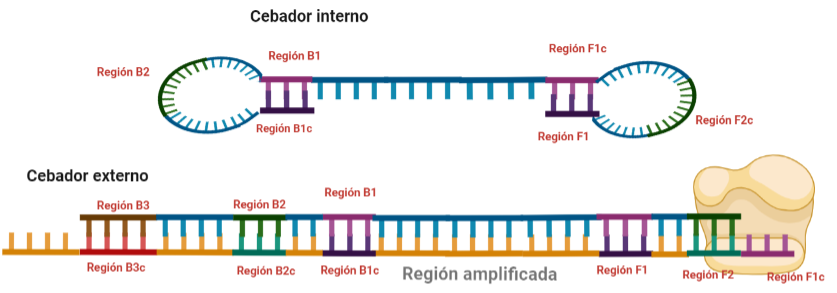
- Cebadores: cuyo diseño no es igual al de la PCR y que queda explicado en el fundamento de la técnica. Estos cebadores pueden ser de varios tipos:
- Internos: cuyo tamaño debe oscilar entre 86 y 88 nucleótidos. Dos tipos: FIP (Forward Inner Primer) y BIP (Back Inner Primer): contienen una región complementaria a la hebra en la que encontramos la secuencia a amplificar y otra región que no es complementaria a la hebra y que permitirá la formación de bucles por autohibridamiento. Entre ambas regiones encontramos otra pequeña región que tampoco deberá hibridar y que es fundamental para la formación de los bucles y para que las estructuras formadas no estén tan compactas.
- Externos: cuyo tamaño oscila entre 17 y 21 nucleótidos. También encontramos uno F y un B.
- A veces se añaden dos cebadores más que son FLP y BLP, con el objetivo de aumentar más la especificidad.
- dNTP: a iguales proporciones: dATP, dTTP, dCTP y dGTP. Para extender las cadenas.
- ADN polimerasa: la más empleada es la Bst ADN Polimerasa (Bst procede de Bacillus stearothermophylus, ya que se obtiene de esta bacteria). Esta polimerasa destaca por presentar actividad polimerasa 5′->3′ y por no tener actividad exonucleasa, pero la característica que hace que sea diferente a otras polimerasas es su actividad de desplazamiento de cadena (por eso no se requiere desnaturalización previa). La temperatura óptima de actuación es de 60-65 ºC, aunque puede operar en un rango amplio de temperaturas. A partir de la enzima original, han surgido dos generaciones con modificaciones en su estructura, la ADN polimerasa Bst 2.0 (diseñado in silico, pero con una mayor eficiencia en la amplificación) y la ADN polimerasa Bst 3.0 (mayor velocidad de amplificación, mayor tolerancia a inhibidores, mayor termoestabilidad, capacidad para incorporar dUTP y actividad retrotranscriptasa). Otras enzimas empeladas en esta técnica, con similares características, es la ADN polimerasa Φ29 (descubierta por la bioquímica española Margarita Salas) o la SD polimerasa.
- Betaína: sirve para potenciar la técnica y mejorar su rendimiento, ya que disminuye la cantidad de estructuras secundarias que se forman y que dificultan el desarrollo de la técnica. Además, aumenta la sensibilidad hacia las dianas y evita uniones inespecíficas; y desestabiliza los enlaces guanina-cisteína lo que ayuda a los cebadores a unirse a sus secuencias diana (ejerce cierta función desnaturalizante).
- MgSO4: el magnesio sirve de cofactor a la polimerasa.
- Buffer o tampón: Tris-HCl + (NH4)2SO4 + KCl + Triton X-100 + MgSO4. Estos componentes proporcionan el entorno adecuado para la reacción.
- Triton X100: detergente no iónico que rompe conglomerados de ácidos nucleicos y proteínas.
- Agua destilada.
Creemos que no es necesario poner en esta entrada un protocolo específico para el desarrollo de la técnica, pues dependiendo del kit que hayas comprado, este se deberá ajustar a sus condiciones impuestas.

FUNDAMENTO
Lo que ocurre en una LAMP es bastante complejo. En la siguiente imagen, tomada del artículo original, vemos todos las hibridaciones y estructuras secundarias que se forman pero si te pones a seguir el esquema, es algo difícil ¿verdad?. Además, el texto del artículo tampoco es que ayude mucho.
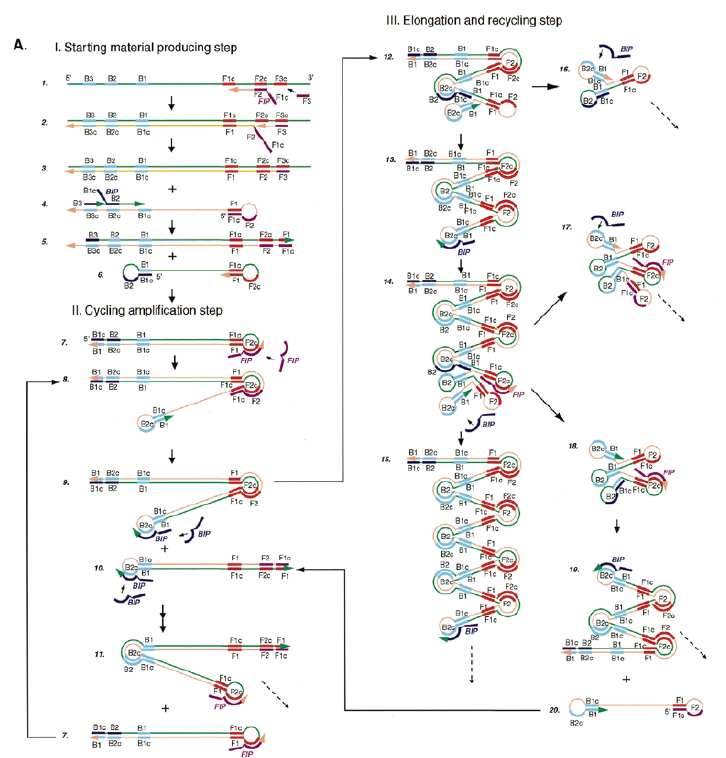
Por ello, vamos intentar explicar de una forma clara lo que ocurre en esta técnica. Te recomiendo que tengas paciencia y una mente bien abierta y atenta, ya que vas a ver que lo que ocurre, es complejo de pillar a la primera.
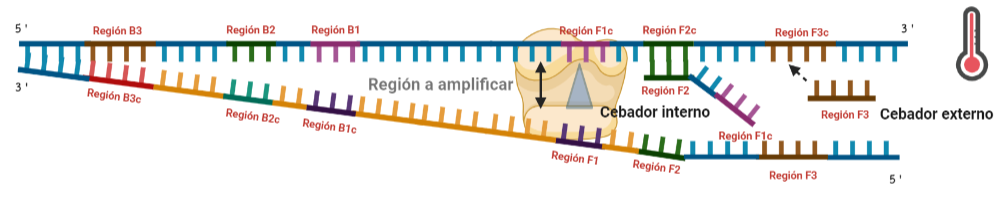
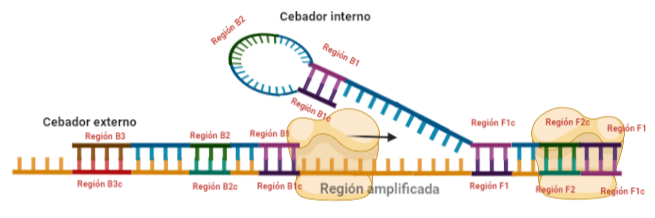
SITUACIÓN INICIAL: Partimos de una doble cadena (pese a que en la Figura 2 hemos representando una sola hebra con el objetivo de simplificar) con una región a amplificar (nuestra región de interés). Adyacentemente a esta región encontramos una serie de regiones que serán con las que hibridaremos una serie de cebadores (en las figuras, hemos decido mantener la nomenclatura empleada por artículo original de cada una de las regiones). Las regiones más cercanas a la región a amplificar se denominan regiones internas (que a su vez se subdividen en otras dos regiones), y las más alejadas, regiones externas. Las regiones que se encuentran por delante de la región a amplificar (corriente abajo, en sentido 3′) se denotan con la letra F (del inglés forward) y los que se encuentran por detrás (corriente arriba, en sentido 5′) con la letra B (backward). El subíndice c marcado en algunas regiones indica complementariedad (así por ejemplo, la región F1c será complementaria a la región F1; podrás pensar que la figura 2 es incorrecta, pero no, dentro de poco entenderás por qué).
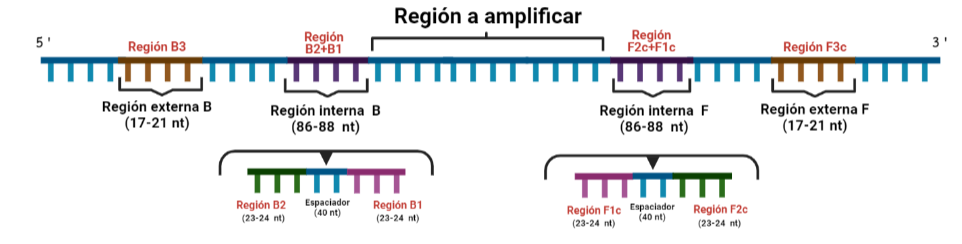
El diseño de cebadores utilizados es muy específico y muy diferente al diseño de cebadores empleados en técnicas como la PCR. En la LAMP, los cebadores internos, inicialmente solo complementan con la zona más externa de la región interna de la hebra de DNA (si tomamos como ejemplo el cebador interno F, solo la región F2 es la que complementa con la región F2c de la hebra de DNA). La otra región del cebador tendrá la misma secuencia que la zona más interna de la región interna de la hebra de DNA (en el cebador interno F, hablamos de la región F1c) por lo que no hibrida. Esto te puede parecer extraño (nosotros pensamos lo mismo cuando lo vimos por primera vez), pero es fundamental que sea así para la formación de bucles y estructuras secundarias que se forman durante el desarrollo de la técnica.
PASO 1: Una vez hemos echado todos los componentes de la mezcla de reacción, lo primero que ocurrirá es que la actividad desplazadora de cadena de la polimerasa (junto con la betaína en el caso de que la hayamos echado) permitirá la apertura de la doble hélice de DNA y con ello, permitirá la unión de los cebadores. Por como dijimos antes con el diseño de cebadores, el cebador FIP solo se unirá por su región F2 a la región F2c de la cadena. El cebador externo F3 también se acabará uniendo. Aquí ya empieza lo curioso de la LAMP, en función de donde se una la polimerasa y donde empiece a desplazar la doble hebra, favorecerá a que la síntesis del DNA se produzca a partir del cebador interno o externo, pero no solo del F, sino también del B. Nosotros ejemplificaremos a partir de aquí, suponiendo que la polimerasa ha permitido una unión primera del cebador interno F (si lo pensamos, si la síntesis hubiese empezado por el externo, daría una situación muy similar al material de partida).
PASO 2: Mientras se va sintetizando la cadena complementaria a partir del cebador interno F, otra polimerasa podría desplazar la hebra en una posición cercana a la región F3c del DNA y permitir la unión de ese cebador externo. De esta manera, empezará la síntesis de otra cadena complementaria, y debido a esa actividad desplazadora de la polimerasa, según sintetiza la hebra a partir del cebador externo, la hebra sintetizada a partir del cebador interno se irá soltando y quedando desplazada.
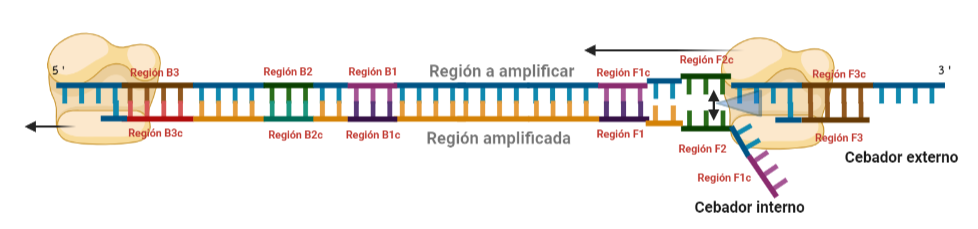
PASO 3: Aquí empieza la formación de bucles característicos del LAMP. Recordemos que dijimos que el diseño de cebadores debía de ser de una determinada manera. Con ese diseño conseguimos que la región F1c del cebador interno, que no había logrado hibridar con la cadena, autohibride ahora con la región F1 de la cadena que se había sintetizando (utilizando como molde la región F1c de la cadena inicial). ¿Y por qué se forma un bucle? Esto lo comentamos anteriormente por encima, el cebador interno hibrida con dos regiones que deben estar separadas por otra región denominada espaciador. Cuando nosotros diseñemos los cebadores internos, la región que supuestamente debería aparear con el espaciador, la modificaremos para que no pueda hibridar (en el artículo original esta región la denominaron TTTTT, debido a que colocaron solo timinas, pero realmente, podemos colocar cualquier secuencia que sepamos que no va a hibridar con el espaciador). Además, la presencia de esta secuencia también es fundamental para que las estructuras no estén tan compactas, siendo sitios por lo que es más fácil que la polimerasa pueda acceder y desplazar cadenas. A pesar de esto, el bucle principalmente se forma porque la región que comprende (en este caso la región F2) es de mayor tamaño que esos espaciadores.
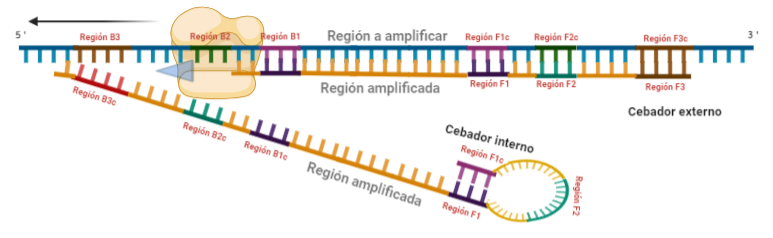
PASO 4: Cuando la polimerasa que amplificaba a partir del cebador externo acaba, genera una doble hebra que hace que partamos de la situación inicial (volver al paso 1). La hebra que había formado el bucle quedará suelta completamente y sus regiones B quedarán expuestas para su hibridación con sus cebadores. Aquí podría unirse directamente el cebador externo y dar una secuencia como la inicial (cuando la polimerasa llega al bucle, lo acaba deshaciendo y sintetiza unas bases sobre ellas. Nosotros desarrollaremos las imágenes a partir de la unión del fragmento interno, que al igual que pasaba con el F, este solo hibridará con una de sus regiones, que en este caso, es la B2, que hibrida con la región B2c de la hebra que toma como molde.
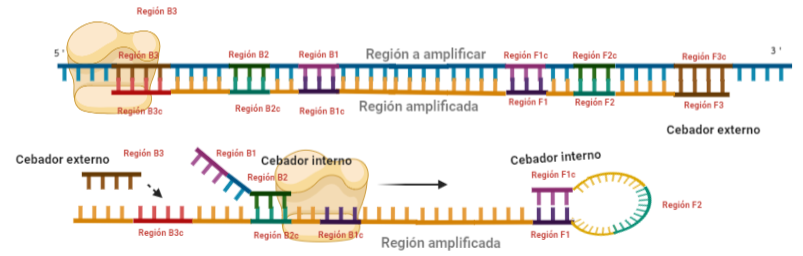
PASO 5: La polimerasa sintetizará la hebra complementaria desde el cebador interno.
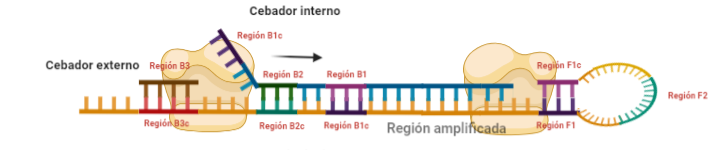
PASO 6: Al igual que pasaba antes, la polimerasa podrá unirse al cebador externo mientras se va sintetizando la hebra a partir del cebador interno, por lo que según se sintetice la nueva, se irá desplazando la otra, y como el cebador B no se había unido en su totalidad, la región que queda suelta, B1c, podrá autohibridar con B1. Es importante señalar que, cuando la polimerasa llega a la región en la que había un bucle, debido a su actividad desplazadora, conseguirá deshacer el bucle y replicar utilizando su secuencia como molde.
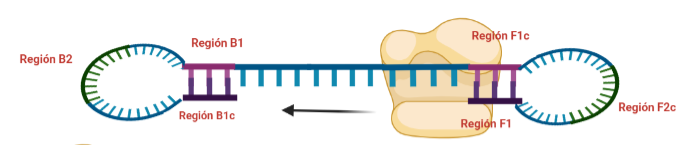
PASO 7: El resultado final del paso anterior es la formación de una doble hélice similar a la inicial (volver al paso 1) debido a la síntesis a partir del cebador externo, y una estructura conocida como dumb-bell (mancuerna en español), ya que al soltarse de nuevo la región F, se vuelve a formar el bucle que existía antes.
PASO 8: Ahora, la polimerasa se unirá a la región F1 (en este caso, no podrá unirse a la región B1c porque el extremo 3′ únicamente se lo proporciona la región F1).
PASO 9: Como pasaba antes, la actividad desplazadora de la polimerasa deshará el bucle formado. A continuación, el cebador interno especifico de la región F se unirá al bucle que queda en la estructura. Si lo pensamos, este cebador podría haberse unido también durante el paso 8; al igual que el cebador interno de la región B, que se podría haber unido en el paso 8 y generar una estructura diferente a la que estamos exponiendo. Y es que eso también ocurre. Como veis, hay muchas posibilidades, lo que hace que se generen estructuras secundarias muy complejas.
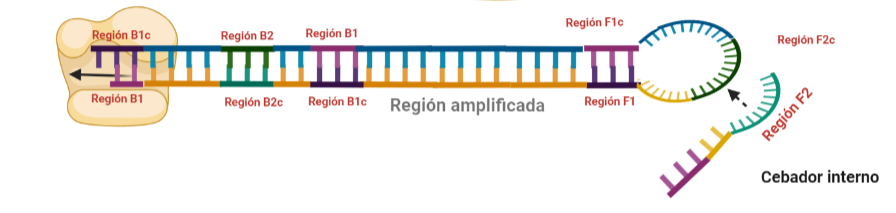
PASO 10: Una vez se ha unido el cebador interno a la región F2c del bucle, la polimerasa empezará su síntesis a partir de ahí. La región F1c del cebador no logra unirse a F1 porque esta queda unida a la estructura generada en el paso 9. No obstante, también podría darse el caso de que una polimerasa desplazase esa región y se uniera el cebador por esa región, y de nuevo, según sintetice, desplazará a la otra hebra. Esta hebra desplazada tendrá la misma capacidad que las otras ya dichas, y una de sus regiones podrá autohibridar (B1c complementa a B1), pudiéndose unir otra polimerasa.
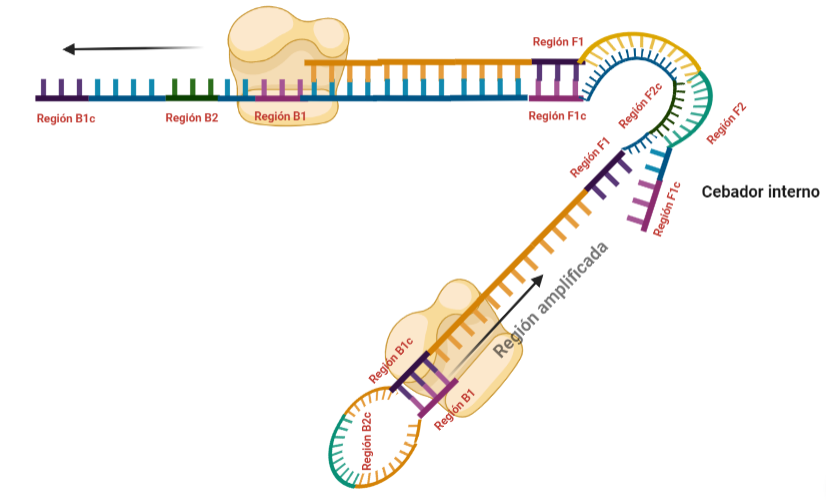
PASO 11: La polimerasa que se había unido en el bucle nuevo formado, por su misma actividad, desplazará a la hebra que se encuentre, y con esto, se formará dos estructuras: una nueva mancuerna (derivada de la hebra que desplaza la polimerasa y que de nuevo consigue autohibridar) y una cadena con un bucle y que consigue tener dos regiones de interés iniciales. De la estructura en mancuerna, pasará lo mismo que en el paso 8, pero ahora, el extremo 3′ lo cede la región B1, por lo que a partir de aquí, los pasos se repiten, pero de manera opuesta. A la otra estructura, se le podrá unir el cebador BIP (debido a que su bucle presenta la región B2c, complementaria a la región B2 del cebador)
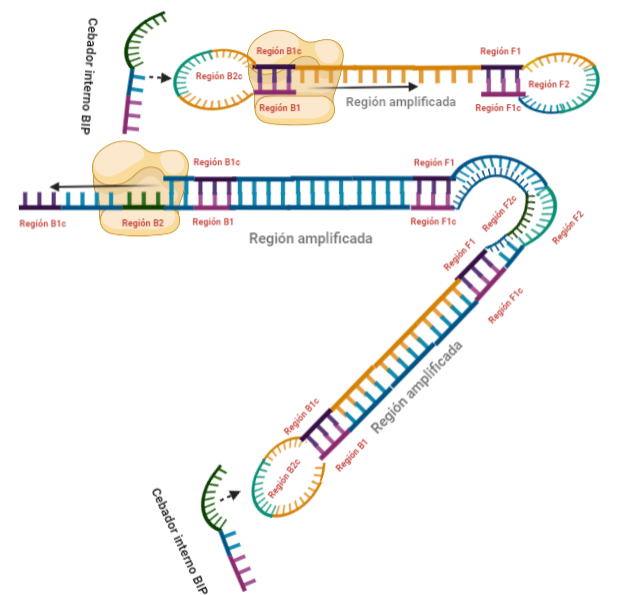
PASOS SIGUIENTES: A partir de aquí, las estructuras que se forman son cada vez más complejas, y creemos que no es necesario explicarlas, pues con entender lo que se ha explicado en pasos anteriores, es suficiente para lograr entender el fundamento de la técnica: diseño de cebadores que permitan autohibridamientos posteriores y actividad desplazadora de cadena de la polimerasa. Estas dos cosas resumen el fundamento de la técnica, y sin ellas, no puede desarrollarse una LAMP. Además, otro aspecto importante en esta técnica, es que la amplificación que se produce es exponencial, generando muchas estructuras secundarias y muy variadas que comparten una cosa entre sí, y es que presentan al menos una región que es la que inicialmente era la de interés.
De una manera más visual, las estructuras que se forman, pueden ser observadas en este vídeo, tomado de New England Biolabs:
VARIACIONES DE LA TÉCNICA Y MÉTODOS DE DETECCIÓN
Como variante de LAMP tenemos la RT-LAMP que mediante la incorporación de una polimerasa con actividad retrotranscriptasa o trancriptasa inversa (siendo la polimerasa de elección la Bts 3.0) a la mezcla de reacción, nos permitirá analizar muestras de RNA. Es esta técnica la que también se está empezando a emplear en la detección del virus SARS-CoV-2.
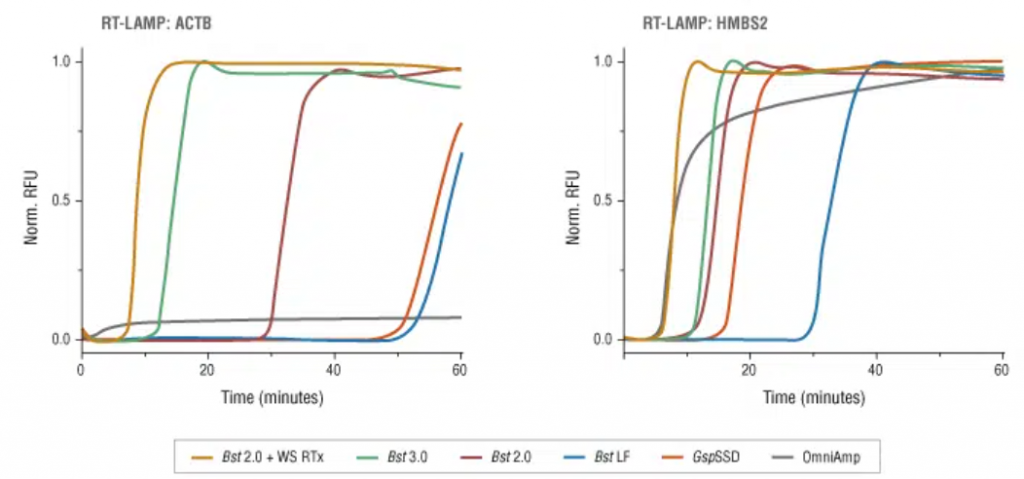
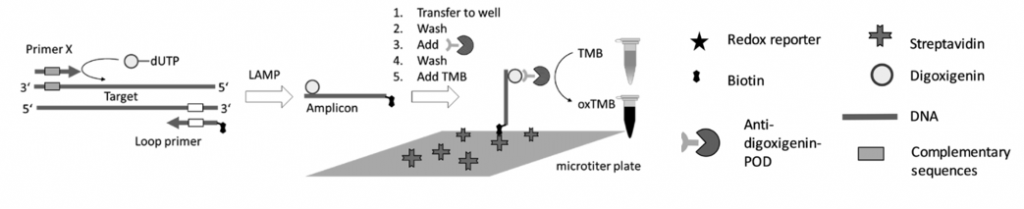
Tras realizar la LAMP, deberemos demostrar si se ha producido la amplificación, y para ello existen varias formas de poder hacerlo:
- Una de ellas es acoplar una LAMP (o RT-LAMP) a un método de bioluminiscencia en tiempo real (BART), lo que da lugar a LAMP-BART y a RT-LAMP-BART. Mediante este método se va detectando la amplificación a la vez que esta va ocurriendo.
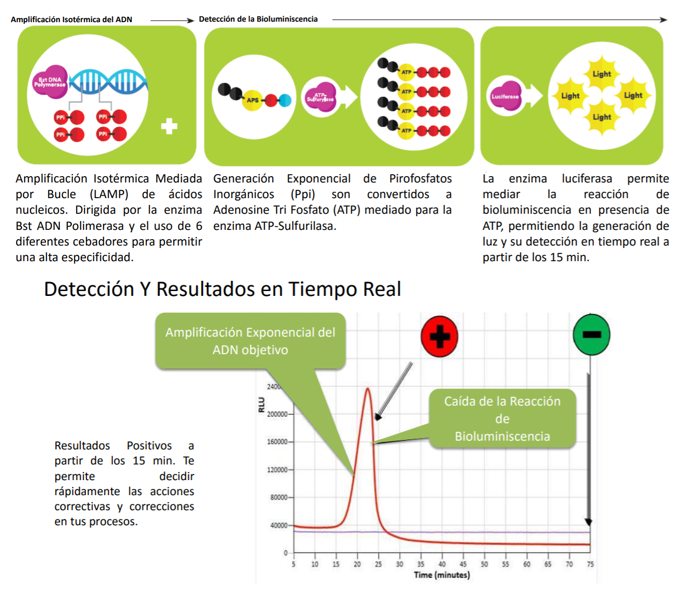
- Una detección más clásica es mediante turbidimetría y visualización directa. En la reacción de la polimerasa se libera pirofosfato inorgánico (PPi) y la LAMP genera mucho producto de amplificación (la reacción es exponencial), por lo que la cantidad de PPi será inmensa. Este pirofosfato reacciona con el magnesio que formaba parte de mezcla de reacción y forma pirofosfato de magnesio, que enturbia el medio en el que se encuentra. El pirofosfato de magnesio, y por tanto, la turbidez, es proporcional a la cantidad de producto amplificado. Existen métodos que pueden pueden medir de manera cuantitativa la turbidez de un medio.

- La electroforesis en gel de agarosa, es una técnica empleada para visualizar la amplificación de una LAMP. A nivel de diagnóstico, esta técnica no se suele emplear debido al tiempo que tarda en correr un gel. El resultado en una electroforesis de una LAMP se asemeja a una escalera (no una banda continua como una PCR), debido a la cantidad de estructuras secundarias que se forman durante la amplificación.
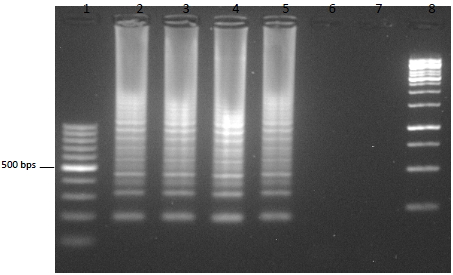
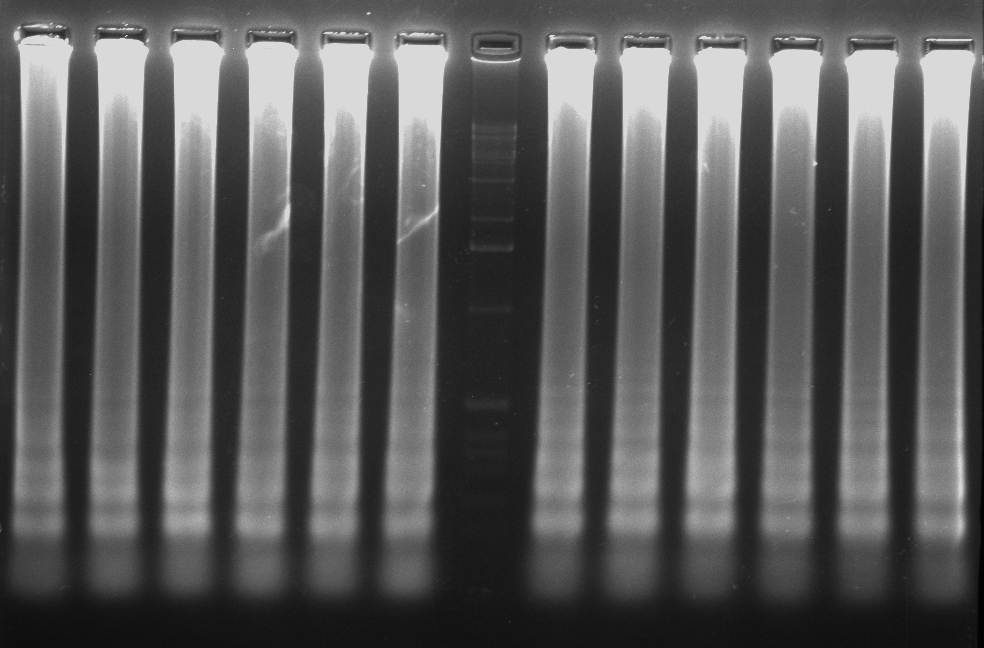
- Otras formas de detección son mediante el uso de fluorocromos, la técnica del quencher-fluróforo, la detección colorimétrica al final de la reacción, mediante sondas específicas…

El siguiente artículo se muestra una revisión de todos los métodos empleados para la detección de los productos amplificados por la técnica LAMP: Loop-mediated isothermal amplification (LAMP) – review and classification of methods for sequence-specific detection
¿POR QUÉ ES NECESARIO CONOCER EL FUNDAMENTO DE UNA TÉCNICA?
A parte de la curiosidad que puede despertar, al menos en nosotros, conocer lo que está pasando a nivel molecular en cualquier proceso que podamos estudiar (¿no os ha pasado que os habéis imaginado a las partículas, átomos y moléculas como si tuvieran vida propia? Cuando realmente somos fruto de un conjunto de interacciones aleatorias), consideramos que saber qué es lo que ocurre en una técnica es esencial por dos cosas:
- Primero, porque si sabemos que ocurre en cada momento, es más fácil una mejor comprensión de los resultados, pudiendo detectar antes tus fallos experimentales.
- Segundo, porque la ciencia está en constante evolución, y sabiendo qué ocurre en cada momento, podemos conseguir modificarla para adaptarla a nuestro experimento, y por ejemplo, surgir variantes de la técnica original (como la RT-LAMP), o incluso, generar nuevas técnicas. Por ejemplo, ¿Cuál es uno de los inconvenientes de una técnica ELISA? Su sensibilidad. Si hibridamos una LAMP con una ELISA (técnica LAMP-ELISA), podremos conseguir que su sensibilidad aumente.
APLICACIONES
Esta técnica se está usando para detectar la presencia de DNA y de RNA en diversas muestras y para el diagnóstico clínico.
Algunos ejemplos son:
- Detectar RNA de SARS-CoV-2. En varios países se acepta el uso de esta técnica a la hora de viajar al extranjero como prueba para detectar el SARS-CoV-2.
- Detectar otros virus de RNA como el virus del Zika, virus de la gripe A (Influenzavirus A) y B (Influenzavirus B).
- Detectar parásitos como Schistosoma, Loa loa y Mansonella perstans.
- Analizar la calidad de los alimentos y del agua.
- Detectar, en enfermos con cáncer de mama, células tumorales en el ganglio centinela durante el intraoperatorio.
CONCLUSIÓN
Con esta entrada al blog hemos querido hacer un repaso básico pero detallado de la técnica LAMP, para que podáis conocer una técnica que no se cuenta en clase pero que creemos que en un futuro os podrá servir de ayuda en vuestros trabajos o labores de científicos.
Pese a que es poco conocida, creemos que dentro de unos años se explotará lo suficiente para que se empiece a enseñar en las universidades, pues como ya indicamos, tiene múltiples ventajas respecto a otras técnicas (como la PCR), permitiendo amplificar de una forma más rápida, específica, sensible y fiable sin la necesidad de ningún aparato salvo uno que mantenga unas temperaturas contantes.
Desde esta entrada fomentamos también para futuros seminarios de la asignatura de Biología Molecular a que habléis de técnicas, ya que al final, sin técnicas no hay experimentación.
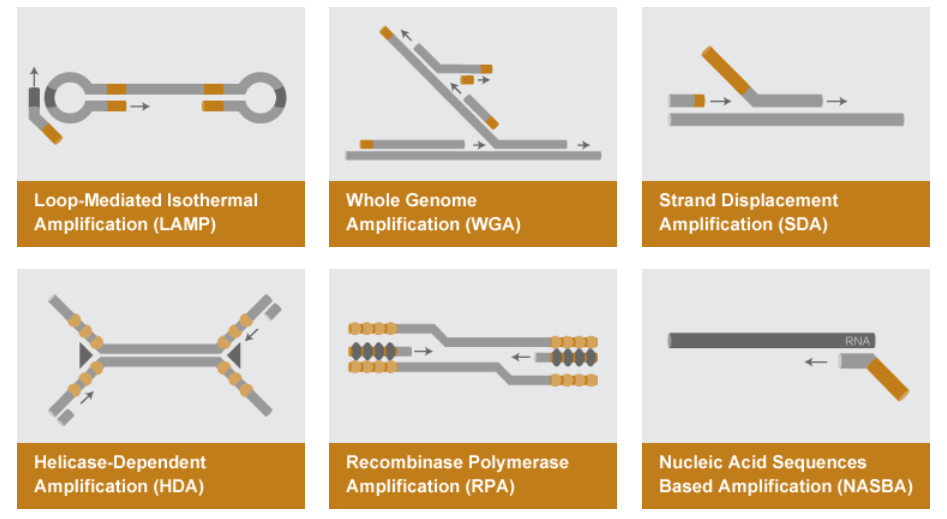
Para finalizar, te dejamos estas preguntas para que te las respondas tú mismo: ¿Qué te ha parecido esta técnica de amplificación? ¿Sabías que existen otras técnicas de amplificación de ácidos nucleicos como la NASBA, SAMART, SDA, RCA, HDA o la SPIA? Desde aquí proponemos temas para futuras entradas al blog.
BIBLIOGRAFÍA
- Brazas, R., 2017. LavaLAMP™ DNA Component Kit. [online] Lucigen.com. https://www.lucigen.com/docs/manuals/MA171_LavaLAMP_DNA_Component_Kit_User_Manual.pdf
- Ito, M., Watanabe, M., Nakagawa, N., Ihara, T., & Okuno, Y. (2006). Rapid detection and typing of influenza A and B by loop-mediated isothermal amplification: comparison with immunochromatography and virus isolation. Journal of virological methods, 135(2), 272–275. https://doi.org/10.1016/j.jviromet.2006.03.003
- Kiefer, J. R., Mao, C., Hansen, C. J., Basehore, S. L., Hogrefe, H. H., Braman, J. C., & Beese, L. S. (1997). Crystal structure of a thermostable Bacillus DNA polymerase I large fragment at 2.1 A resolution. Structure (London, England : 1993), 5(1), 95–108. https://doi.org/10.1016/s0969-2126(97)00169-x
- Loreto, M., 2016. Diseño y puesta a punto de un método LAMP (Loopmediated isothermal amplification) para el diagnóstico de enfermedades producidas por cestodos. [online] Gredos.usal.es. https://gredos.usal.es/bitstream/handle/10366/131526/TG_MegidoDom%C3%ADnguezL.pdf
- New England Biolabs, 2022. Loop-Mediated Isothermal Amplification. [online] International.neb.com. https://international.neb.com/applications/dna-amplification-pcr-and-qpcr/isothermal-amplification/loop-mediated-isothermal-amplification-lamp%20
- Notomi, T., Okayama, H., Masubuchi, H., Yonekawa, T., Watanabe, K., Amino, N., & Hase, T. (2000). Loop-mediated isothermal amplification of DNA. Nucleic acids research, 28(12), E63. https://doi.org/10.1093/nar/28.12.e63
- Parada, D., & Hernández, P. (2014). Método de amplificación de ácido nucleico en un paso en el ganglio centinela del cáncer de mama. Revista Venezolana de Oncología, 26(2), 62-69.
- Park, G. S., Ku, K., Baek, S. H., Kim, S. J., Kim, S. I., Kim, B. T., & Maeng, J. S. (2020). Development of Reverse Transcription Loop-Mediated Isothermal Amplification Assays Targeting Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). The Journal of molecular diagnostics : JMD, 22(6), 729–735. https://doi.org/10.1016/j.jmoldx.2020.03.006
- Sánchez, E., Nina, M., Aguirre, P., Arce, M., Toro, N., & Vilela, R. (2014). Amplificación isotérmica mediada por LOOP (LAMP) de ácidos nuclecios en el diagnostico clínico. Revista CON-CIENCIA, 2(1), 127-140. http://www.scielo.org.bo/scielo.php?script=sci_arttext&pid=S2310-02652014000100014&lng=es&tlng=es.
- Wang, X., Seo, D. J., Lee, M. H., & Choi, C. (2014). Comparison of conventional PCR, multiplex PCR, and loop-mediated isothermal amplification assays for rapid detection of Arcobacter species. Journal of clinical microbiology, 52(2), 557–563. https://doi.org/10.1128/JCM.02883-13

 Entrada siguiente
Entrada siguiente