LOS PRIONES


INTRODUCCIÓN
Los priones son proteínas infecciosas capaces de adoptar distintas conformaciones, siendo, por lo tanto, una excepción a la teoría clásica del plegamiento de las proteínas, que implica un único plegamiento para cada secuencia de aminoácidos.
Existen varios tipos de priones, en mamíferos el prión PrP, en el que nos vamos a centrar en este artículo y el responsable de las enfermedades conocidas como encefalopatías espongiformes transmisibles (TSEs).
Esta proteína puede expresarse como PrPc, siendo este el prión con isoforma constitutiva, como PrPsc, que es el prión con isoforma no constitutiva o scrapie, capaz de formar agregados amiloides y provocar las TSEs. Ambas isoformas se tratan de la misma proteínas, están compuestas por la misma secuencia de aminoácidos pero varía su plegamiento, y consecuentemente, su función.

Estas proteínas estás codificadas por el propio genoma del hospedador y presentan las dos conformaciones mencionadas anteriormente. Se denomina prión a la conformación patológica de la proteína, es decir, en mamíferos, la PrPsc es el prión de la PrPc, que considerada la proteína “normal”.
FUNCIÓN BIOLÓGICA
Los priones, o PrP o PrPc (priones con su isoforma constitutiva), son producidas en su forma natural en las neuronas y células de la glía en el cerebro y médula espinal. Además, puede estar presente en otros tejidos, principalmente tejido linfoide, como el bazo y en las células del sistema inmune tales como células dendríticas y linfocitos B
Sin embargo, sus funciones siguen siendo investigadas hoy en día. Numerosos estudios, como el publicado por el National de Recherche Scientifique, han relacionado los priones con los mecanismos de defensa celular contra la tensión oxidativa, centrándose además en determinados ámbitos proteicos, y no solo en las PrP como conjunto.
Este estudio concluyó que las células donde las PrP se expresan más de lo normal, estaban mejor equipadas contra la toxicidad, es decir, oponen mayor resistencia frente al daño del ADN causado por la tensión oxidativa. Este estudio también relaciona las PrP con la regulación en el tejido cerebral de iones de cobre, cuyo equilibrio se ve alterado en condiciones de tensión oxidativa.
También se ha demostrado la función que PrPc cumple en la defensa contra radicales libres. Esto es posible gracias a su capacidad de secuestrar iones cobre del medio celular y su asociación con superóxido dismutasas (SODs), enzimas con función de defensa antioxidante, convirtiendo el radical libre anión superóxido en peróxido de hidrógeno, que obtienen cobre de las PrPc. El cobre cumple un papel importante manteniendo la conformación de PrPc, pues estabiliza las α-hélices de la proteína cuando se encuentra en esta forma cooperativa.
Por otro lado, se han descubierto las funciones fisiológicas que llevan a cabo el prión celular (PrPc) en los circuitos neuronales del hipocampo en un estudio que demostró cómo los ratones con déficit de esta proteína presentaban problemas de memoria y aprendizaje, así como dificultades en la actividad motora y mayores niveles de estrés.
Estos efectos fueron asociados a la disminución del umbral de convulsiones, estimuladas por una serie de agentes proteicos, y al retraso de la maduración de las neuronas, y por lo tanto, de la formación de redes neuronales.
La PrPc también está relacionada con la transducción de señales, pues, estudios han demostrado cómo afecta a los canales de calcio la infección por priones. Esto se debe a que PrPsc tiene la capacidad de formar canales permeables a iones calcio y sodio en las membranas celulares. En otros estudios se observó, además, que la presencia de este péptido aumenta las concentraciones de calcio en las células de de la microglía, provocando así una cascada de señalización.

Además de su función en la transducción de señales, participa en el tráfico subcelular, pues, la PrPc, que se expresa en la membrana plasmáticas, es endocitosis tras su anclaje a la membrana, pero antes, actúa como receptor del ión cobre, provocando también su endocitosis.
De esta manera, se pueden ultimar las funciones múltiples de la PrPc, relacionadas con la salud neuronal, entre la que destacan la importancia de ésta en la función de la interconectividad neuronal, así como en la protección de la sinapsis frente a agentes citotóxicos, producido por el mismo sistema nervioso central.
Sin embargo, cabe destacar la posibilidad de que la forma patogénica de las PrP interfieran en estas funciones, dando lugar a la aparición de la patología de las TSE.
ESTRUCTUTURA Y MECANISMO
Todo el conocimiento que se tiene sobre los priones se debe a las múltiples investigaciones realizadas en la bacteria Escherichia coli. Podemos diferenciar dos estructuras dentro de los priones: los priones con PrPC y con PrPSc.
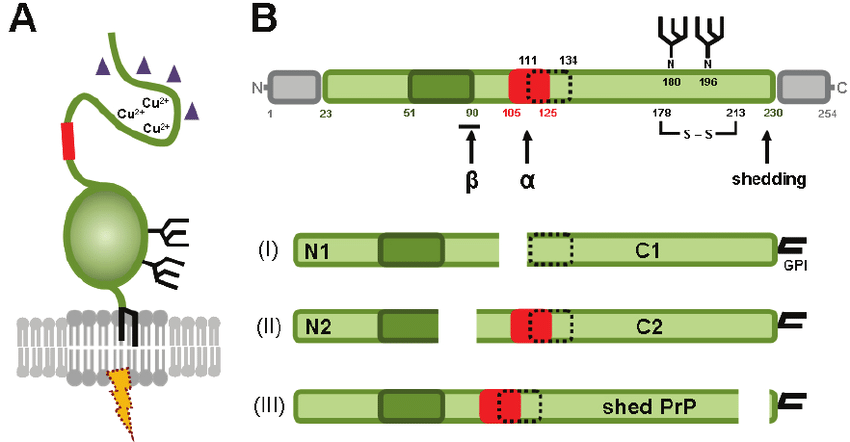
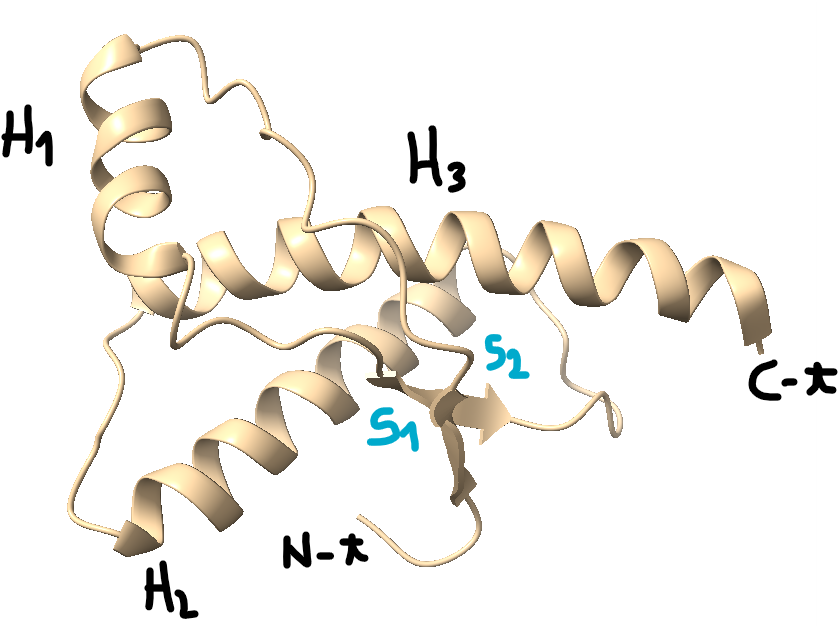
Los PrPc son unas glicoproteínas (aproximadamente 27-30 kD) de la superficie celular que se caracterizan por tener dos hélices alfa (aminoácidos en forma de espiral) y dos cadenas de oligosacáridos. Consisten en priones no infecciosos, que en los seres humanos constan de 209 aminoácidos y contienen un enlace disulfuro. Su estructura es alfa-helicoidal, ya que presenta tanto hélices alfa como láminas beta (hebras planas de aminoácidos). Esta proteína no puede ser separada por centrifugación, ya que no es sedimentable y es fácilmente digerida por la serina proteasa. Esta proteína además es una proteína monomérica formada por monómeros estables, con una resistencia normal y es soluble en detergentes. En la siguiente foto podemos observar esta Prpc, donde las H1,H2 Y H3 son las regiones con alfa hélices y las S1 y S2, las láminas beta antiparalelas
Los PrPsc son unas proteínas infecciones que inducen el cambio de conformación de PrPc a PrPsc con una conformación anormal. Este cambio de conformación en su estructura secundaria se da debido a un plegamiento erróneo espontáneo, una mutación genética en el genoma humano o la exposición de PrPc a Prpsc, lo que conduce a un incorrecto plegamiento de su estructura terciaria. Cuando se produce uno de estos tres casos mencionados y obtenemos PrPsc, a estructura y composición de la proteína cambia radicalmente, y con ello su función. La proteína pasa de ser una proteína alfa-helicoidal soluble con una pequeña concentración de láminas beta a una proteína con un alto porcentaje de láminas beta e insoluble. A diferencia de la forma normal del prion, la forma patógena no es una proteína monomérica, ya que contiene agregados proteicos y monómeros poco estables, así como una resistencia extrema a la radiación y disolventes fuertes y es insoluble en detergentes. Este cambio estructural provoca fallos en el mecanismo de eliminación de proteínas cuyo funcionamiento es malo. Cuando aparecen proteínas que funcionan mal, son degradadas por un proteasoma debido a la acción de la ubiquitina. Sin embargo, debido a la formación de los priones, este complejo se inhibe, lo que permite que los PrPsc no se degraden y se expandan y acumulen en le citosol.
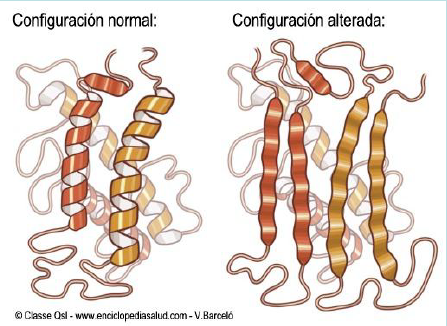
Una hipótesis de porque se produce este cambio radical es la sustitución del aminoácido leucina por prolina, ya que la presencia de prolina impide la formación de la estructura secundaria, produciendo la desestabilización de la hélice alfa y aumentando la lámina beta, pero aún no se sabe con exactitud.
Hay que destacar que tanto la forma normal de los priones como la patógena tienen la misma secuencia de aminoácidos, ya que derivan del mismo gen, el cual se localiza en el brazo corto del cromosoma 20, sin intrones y es un gen autosómico dominante.
IMPLICACIONES BIOMÉDICAS, AMBIENTALES Y TERMOLÓGICAS: ENFERMEDADES PRODUCIDAS POR PRIONES
Como se ha explicado anteriormente, es la conformación de PrPsc la que le aporta su capacidad de ser infecciosa, al ser una versión modificada de PrP que ya no tiene su función fisiológica original, pero que adquirió la capacidad de autorreplicarse mediante la conversión de proteínas PrPc (que usa como molde) en más copias de sí misma. Otra diferencia entre estas dos isoformas, es la resistencia que PrPsc opone a los tratamientos con proteasas, de la cual, PrPc carece, pues es totalmente degradada por estas enzimas.
A su vez, la conversión de PrPc a PrPsc es dependiente de chaperonas, pues se ha demostrado que esta conversión, en ausencia de chaperonas es muy lenta. En estudios in vitro se ha observado cómo las chaperonas reconocen y unen las PrPc con PrPsc, al producirse esta asociación , la reacción de conversión se acelera. De esta misma manera, las chaperonas son también capaces de interferir en la reacción de conversión uniéndose a la PrPc, estabilizando la conformación de PrPc. Este último tipo de actuación se ha detectado en en las chaperonas Bip, mientras que la anterior, en las chaperonas Hsp60 y Hsp104, cuyas estructuras podemos encotrar debajo, respctivamente.

Las enfermedades provocadas por este tipo de proteínas son estudiadas a conciencia desde 1985, después de la epidemia que se vivió en Reino Unido de encefalopatía bovina espongiforme, una enfermedad provocada por priones.
Se trata de las encefalopatías espongiformes transmisibles (TSEs), las enfermedades producidas por priones. Estas son enfermedades neurodegenerativas letales y que carecen de tratamiento en la actualidad, con periodo de incubación lento, pudiendo tardar incluso décadas en manifestar síntomas, pues se tratan de proteínas del mismo organismo, tienen la misma secuencia de aminoácidos que PrPc, de manera que no produce respuesta inflamatoria o inmune.
Se producen por la acumulación de priones con la conformación PrPsc en el sistema nervioso, que provoca la apoptosis neuronal.
Tras años de estudio, se ha concluido que, en humanos, las enfermedades por priones pueden tener un origen infeccioso, como ocurre en el caso del Kuru o de la enfermedad de Creutzfeldt-Jakob iatrogénica; así como también pueden tener un origen hereditario o condicionado por mutaciones, tal y como sucede con las formas familiares de enfermedad de Creutzfeldt-Jakob, Gertssman-Stràussler-Scheinker y el insomnio familiar fatal. Estas son las únicas enfermedades conicidad que pueden ser genéticas e infecciosas a la vez.
En cualquier caso las PrPsc, con isoforma no constitutiva, inducen su conformación a las PrPc, de manera que adoptan la confirmación patogénica. Esta es una reacción autocatalítica que contradice el dogma central de la biología: se produce una transmisión de información a través de los cambios conformacionales de una proteína y no a través de un ácido nucleico. Los priones contienen información en su conformación PrPsc de manera análoga a los genes, comportándose como genes, además de como agentes infecciosos.
La patogénesis de los priones es un proceso complejo que puede dividirse en las siguientes fases:
· Infección y replicación en los tejidos periféricos. Los tejidos que actuarán como vectores de transmisión se contaminan y el prión se replica.
· Entrada por vía oral y transporte al sistema nervioso central. La entrada de prpsc por via oral es capaz de iniciar la reacción de cambio conformacional, convirtiendo las moléculas de prpc intestinales en mas prpsc, que invade entonces los nervios periféricos cercanos, que también expresan prpc, con lo cual se va propagando hasta que llega al sistema nervioso central.
· Neurodegeneración. Muchas enfermedades neurodegenerativas involucran el procesamiento anormal de proteínas neuronales y la acumulación de proteínas mal plegadas. La neurodegeneración producida por los priones 2es un proceso multifactorial que involucra acumulación de proteínas anormales en amiloides y agresomas, estrés oxidativo y apoptosis.
INSOMNIO FAMILIAR FATAL
Los síntomas que presentan, en general, estas enfermedades son disautonomía, disfunción motora, ataxia cerebral, demencia e insomnio progresivo, el cual aparece únicamente en el FFI. El FFI (Insomnio familiar letal) es una enfermedad muy rara producida por priones la cual es de herencia dominante, la cual provoca un insomnio que empeora con el paso del tiempo, acompañado de demencia y alucinaciones tanto visuales como auditivas. Esta enfermedad tiene una incidencia de 100 casos en todo el mundo, y 30 de ellos se encuentran en España, en concreto en una pequeña población de Euskadi.
Otro de los síntomas es atrofiamiento del tejido nervioso por la aparición de espongiosis y deposiciones de placas de tipo amiloide, similares a la amiloidosis que se da en la enfermedad del Alzheimer, provocada también por el plegamiento anómalo de una proteína.
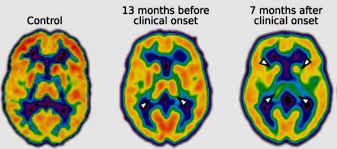
glucosa en el tálamo es menor que en el control (flecha)
Como se ha mencionado anteriormente, estos síntomas aparecen tras un largo período de tiempo.
Se cree que el 10% de los casos de TSEs son producidos por mutaciones, que apoyan el cambio espontáneo de conformación PrPc a patogénica PrPsc sin la existencia previa de PrPsc, en la línea germinal en el gen PRP (que codifica para la proteína PrPc, y por tanto, para todas sus conformaciones), expresado en el cromosoma 20.
Por otro lado, las variantes esporádicas de las TSEs son producidas por una conversión espontánea de PrPc a PrPsc sin necesidad de ninguna mutación en el gen PRP o la presencia de PrPsc del exterior. Este cambio se puede deber a una respuesta a algún cambio en el ambiente, aunque también es posible que sea producido por alguna mutación no detectada.
LESIONES EN EL SISTEMA NERVIOSO PRODUCIDAS POR PRIONES
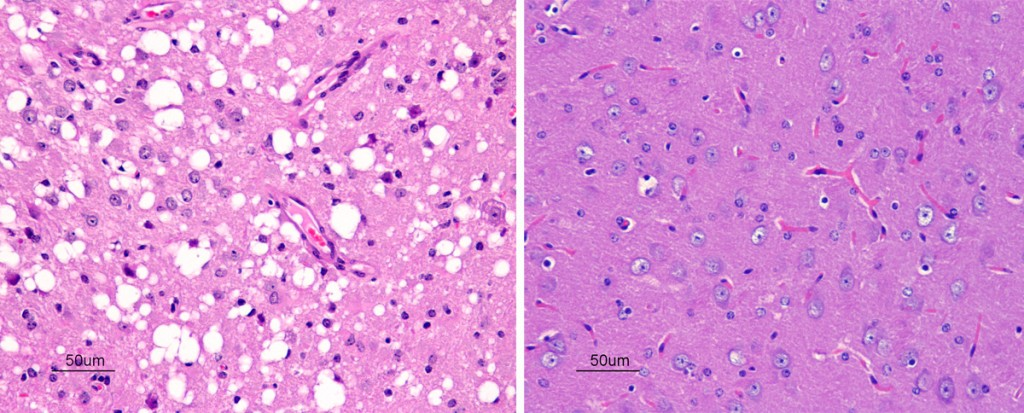
En las encefalopatías espongiformes, aparece una segregación de amiloides insolubles y resistentes a proteasas, compuestos por PrPsc. Debido a la apoptosis de la célula como respuesta, dichos complejos son acumulados en el exterior celular.
Estas deposiciones son típicas de enfermedades neurodegenerativas como el Parkinson o el Alzheimer, y debido a alguna mutación, también existen amiloidosis hereditarias.
Por otro lado, también se detecta espongiosis en la sustancia gris y en la sustancia blanca del sistema nervioso central.
La espongiosis observada en las TSEs produce una vacuolización del citoplasma de las neuronas de manera que el núcleo queda en la periferia celular provocada por una sobreexpresión de las acuoporinas 1 y 4.
REFERENCIAS
Du moratoire, C. de S. (s/f). dans les laboratoires de recherche sur les prions infectieux. Gouv.fr. Recuperado el 5 de febrero de 2023, de https://www.enseignementsup-recherche.gouv.fr/sites/default/files/2022-01/rapport-ig-sr-2022-011-16322.pdf
Herms, J. W., Madlung, A., Brown, D. R., & Kretzschmar, H. A. (1997). Increase of intracellular free Ca2+ in microglia activated by prion protein fragment. Glia, 21(2), 253–257. https://doi.org/10.1002/(sici)1098-1136(199710)21:2<253::aid-glia8>3.0.co;2-7
JAWETZ. (1987). Review Medical Microbiology (17a ed.). Appleton and Lange.
Jin, T., Gu, Y., Zanusso, G., Sy, M., Kumar, A., Cohen, M., Gambetti, P., & Singh, N. (2000). The chaperone protein BiP binds to a mutant prion protein and mediates its degradation by the proteasome. The Journal of Biological Chemistry, 275(49), 38699–38704. https://doi.org/10.1074/jbc.M005543200
Kaytor, M. D., & Warren, S. T. (1999). Aberrant protein deposition and neurological disease. The Journal of Biological Chemistry, 274(53), 37507–37510. https://doi.org/10.1074/jbc.274.53.37507
La falta de proteína priónica celular puede provocar síntomas de epilepsia y déficits de aprendizaje – Universitat de Barcelona. (s/f). Recuperado el 5 de febrero de 2023, de https://www.ub.edu/web/ub/es/menu_eines/noticies/2022/03/018.html
Linden, R., Martins, V. R., Prado, M. A. M., Cammarota, M., Izquierdo, I., & Brentani, R. R. (2008). Physiology of the prion protein. Physiological Reviews, 88(2), 673–728. https://doi.org/10.1152/physrev.00007.2007
Mabbott, N. A., & Bruce, M. E. (2001). The immunobiology of TSE diseases. The Journal of General Virology, 82(Pt 10), 2307–2318. https://doi.org/10.1099/0022-1317-82-10-2307
Matamoros-Angles, A., Hervera, A., Soriano, J., Martí, E., Carulla, P., Llorens, F., Nuvolone, M., Aguzzi, A., Ferrer, I., Gruart, A., Delgado-García, J. M., & Del Río, J. A. (2022). Analysis of co-isogenic prion protein deficient mice reveals behavioral deficits, learning impairment, and enhanced hippocampal excitability. BMC Biology, 20(1), 17. https://doi.org/10.1186/s12915-021-01203-0
National center for biotechnology information. (s/f). Nih.gov. Recuperado el 5 de febrero de 2023, de http://www.ncbi.nlm.nih.gov
Prusiner, S. B. (2017). Madness and memory: The discovery of prions–A new biological principle of disease. Yale University Press. https://doi.org/10.12987/9780300199260
RCSB Protein Data Bank. (s/f). 1QLX. Rcsb.org. Recuperado el 5 de febrero de 2023, de https://www.rcsb.org/structure/1QLX
Sakudo, A., Ano, Y., Onodera, T., Nitta, K., Shintani, H., Ikuta, K., & Tanaka, Y. (2011). Fundamentals of prions and their inactivation (review). International Journal of Molecular Medicine, 27(4), 483–489. https://doi.org/10.3892/ijmm.2011.605
Velayos, J. L., Irujo, A., Cuadrado-Tejedor, M., Paternain, B., Moleres, F. J., & Ferrer, V. (2010). La proteína priónica celular en el sistema nervioso central de mamíferos. Correlatos anatomoclínicos. Neurologia (Barcelona, Spain), 25(4), 228–233. https://doi.org/10.1016/j.nrl.2009.12.004
Cortelli 2006. Estudio longitudinal con 18FDG-PET
Trabajo realizado por los estudiantes de Biología Sanitaria (UAH): Mar Melián Rodríguez y Álvaro Mínguez Romero.
 Entrada anterior
Entrada anterior