TYRION LANNISTER Y EL ENANISMO EN JUEGO DE TRONOS


Blog escrito por Daniel Lozano Mínguez y Patricia Lomba Hernández, alumnos de Biología Molecular de 3º de Biología Sanitaria, Universidad de Alcalá de Henares.

INTRODUCCIÓN: ACONDROPLASIA
¿Alguna vez te has preguntado a qué se debe la apariencia de Tyrion Lannister en Juego de Tronos, lo que le brinda su mote de “medio-hombre”? La realidad es que tanto el personaje como el actor que lo interpreta (Peter Dinklage) padecen una enfermedad conocida como acondroplasia, una forma del coloquialmente llamado “enanismo”.
La acondroplasia es un trastorno genético que afecta al crecimiento óseo, es de herencia autosómica dominante. Este trastorno es causado por una mutación en un gen relacionado con el factor de crecimiento del fibroblasto, afectando la osificación endocondral. Clínicamente, se aprecia una talla baja con desproporción anatómica, macrocefalia, acortamiento de extremidades y deformidades esqueléticas [1]. Además, la acondroplasia es la causa más frecuente de enanismo, siendo responsable de un 70% de los casos [2].
QUÉ OCURRE EN LOS GENES
La acondroplasia es debida a la mutación G380R, que afecta al gen FGFR3 (factor de crecimiento fibroblástico)[5] con locus 4p16.3, que codifica una proteína llamada receptor 3 del factor de crecimiento de los fibroblastos. Sobre esta proteína actúa un factor de crecimiento responsable del alargamiento de los huesos. Cuando el factor de crecimiento no puede actuar correctamente por la ausencia de su receptor, el crecimiento de los huesos en el cartílago de la placa de crecimiento se hace más lento [3].
Los cuatro receptores de FGFs (FGFRs1-4) pertenecen a la familia de receptores de tirosina quinasa. Tienen una afinidad variable por los factores de crecimiento de los fibroblastos (FGF). Los factores se localizan en el exterior de la membrana y c. El transcrito de este gen contiene un marco de lectura abierto (ORF) de 2520 nucleótidos con 19 exones y 18 intrones [7]. La proteína codificada tiene 3 dominios:
- Dominio extracelular de unión a los FGFs, con tres subdominios glicosilados de Ig
- Dominio transmembrana hidrofóbico
- Dominio catalítico intracelular con actividad tirosina quinasa
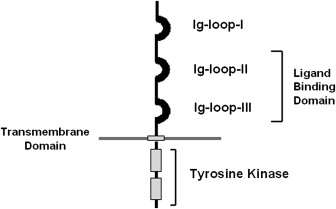
Cuando se une el ligando, se activa el receptor y se dimeriza, produciendo un cambio de conformación que activa el dominio catalítico. Así, se autofosforilan los residuos de tirosina presentes. Estos residuos fosforilados activan la cascada de proteínas quinasas asociadas a microtúbulos (MAP), lo que da lugar a la activación de diferentes factores de transcripción intranucleares. Una mutación en este dominio catalítico es, en la mayoría de los casos, la causa de la acondroplasia. [7]
En un estudio que contaba con 20 pacientes con acondroplasia, se vio que 19 de ellos habían sido casos de mutaciones nuevas y 1 era hereditario. En el 95% de los casos, la mutación era por un cambio de guanina por adenina en el nucleótido 1138 (codón 380)[5], mientras que el 5% restante se correspondía con una transgresión de guanina por citosina en ese mismo nucleótido. Las dos provocan un cambio en el primer nucleótido, haciendo que el codón GGG, que normalmente codifica para glicina, codifique para otro aminoácido, arginina en ambos casos [4].
Este nucleótido se considera el más sensible para la mutación en todo el genoma humano. Esto se da sobre todo en casos en los que el padre es mayor, lo que sugiere que el alelo mutado es de origen paterno. Otras mutaciones en este nucleótido se relacionan con varias enfermedades esqueléticas, como queratosis seborreica, nevus epidémicos y carcinomas uroteliales [7].
CLÍNICA
Los efectos clínicos que esto supone sobre el paciente consisten en una estatura por debajo de lo común, siendo la media en adultos de 131 ± 5,6 cm para los hombres y de 124 ± 5,9 cm para las mujeres. Incluyen además rasgos faciales característicos con una cabeza grande, frente prominente, cara pequeña, puente nasal plano, paso nasal estrecho y mandíbulas prominentes. Sin embargo, su actividad neurológica es normal y pueden llevar a cabo vidas corrientes y productivas. [7]

En la infancia suele darse hipotonía, lo que da lugar a un retraso en el desarrollo de algunas funciones básicas como caminar o el habla. Otra particularidad es que sus extremidades, especialmente las superiores, están acortadas con respecto al tamaño medio. Por otro lado, los dedos de las manos se encuentran engrosados en la base y tienen la misma longitud, adoptando una forma denominada mano en tridente. [7]
HERENCIA
La acondroplasia se puede heredar como un rasgo autosómico dominante. Esto significa que, si un niño recibe el gen defectuoso de uno de los padres, presentará el trastorno. Si uno de los padres padece acondroplasia, el bebé tiene un 50% de probabilidad de heredar el trastorno. Si ambos padres tienen la enfermedad, las probabilidades de que el bebé resulte afectado aumentan al 75%. Sin embargo, la mayoría de los casos (aproximadamente 80%) aparecen como mutaciones espontáneas. Esto quiere decir que dos progenitores que no tengan acondroplasia pueden engendrar un bebé con la afección, como es el caso de Tyrion Lannister. [6]
También puede darse el caso de que dos padres que padecen acondroplasia tengan un hijo que no presente la enfermedad. Esto ocurre tan solo en un 25% de los casos al recibir el alelo no mutado por parte de la madre (50%) y el alelo no mutado por parte del padre (50%) → 0,5 x 0,5 = 0,25. Un ejemplo de esto es el famoso influencer estadounidense Peet Montzingo, cuyos padres y hermanos presentan la enfermedad, pero él no. Este, en sus redes sociales, sube contenido sobre su curiosa vida diaria junto a su familia. Además, ha publicado un libro titulado “Little imperfections” en el que cuenta su experiencia personal creciendo como la única persona alta en una familia de personas pequeñas.
DATO CURIOSO

Un claro ejemplo de acondroplasia lo podemos ver en el famoso cuadro de Las Meninas, de Velázquez. En este, se aprecia claramente que María Bárbola (o Maribárbola, la chica de la derecha) padecía de acondroplasia. Esta era una de los 40 enanos y bufones de la corte, y parece que Velázquez tenía una relación especial de amistad con ella. Justo delante de Maribárbola aparece Nicolasito Pertusato, un italiano que padecía esta misma enfermedad. Cuando se pintó el cuadro, este tenía unos 15 o 16 años, sin embargo, aparenta tener 8 o 9. [8]
BIBLIOGRAFÍA
- Hernández-Motiño, L. C., Sujey Brizuela, Y., Vizcarra, V., Cruz Revilla, R., Jamaica Balderas, L., & Karam Bechara, J. (2012). Acondroplasia-estenosis del canal medular-una complicación neurológica. Boletín médico del Hospital Infantil de México, 69(1), 46-49.
- Arregui, S. F. (2009). El estigma social del enanismo óseo consecuencias y estrategias de afrontamiento (Doctoral dissertation, Tese (Doctorado em Psicologia Social), Departamento de Psicología Social y de las Organizaciones, Facultad de Psicología, Madrid, ESP).
- Guas, H. R., Labore, M. D., & Arguello, M. J. (2011). El mosaicismo germinal como posible mecanismo etiológico de la Acondroplasia y el síndrome Saethre-Chotzen. Presentación de dos familias. Revista Cubana de Genética Comunitaria, 5(3), 139-144.
- Castro, Á., Gutiérrez, A., Rodríguez, L. F., Tatiana, P., Velasco, H., Arteaga, C., … & Giraldo, A. (2010). Análisis mutacional de la acondroplasia en 20 pacientes colombianos. Revista de la Facultad de Medicina, 58(3), 185-190.
- Román, A. P. (2009). Displasias óseas. El residente, 4(1), 5-9.
- Legare, J. M. (1998). Achondroplasia. In M. P. Adam (Eds.) et. al., GeneReviews®. University of Washington, Seattle.
- Richette, P., Bardin, T., & Stheneur, C. (2008). Achondroplasia: from genotype to phenotype. Joint bone spine, 75(2), 125–130. https://doi.org/10.1016/j.jbspin.2007.06.007
- Buroni, J., & Buroni, M. (2016). Las patologías que plasmó Velázquez en “Las Meninas”. Alm Med Cult.[Internet], 42-53.

Muy interesante y claro, y muy chulos los ejemplos