Control de calidad de proteínas en el retículo endoplasmático


Por Beatriz Naranjo Martínez, Mª del Carmen Nogales Valenciano y Sara Ortiz Planchuelo. Grado en biología sanitaria – Universidad de Alcalá de Henares
El retículo endoplasmático (RE) es un orgánulo de la célula que regula la síntesis, plegamiento, maduración, estabilización, tráfico y degradación de aproximadamente un tercio de las proteínas totales de la célula. El destino de estas proteínas es ser secretadas por la célula, así como residir en la membrana plasmática, el aparato de Golgi, los lisosomas y el propio RE.
Estas proteínas sufren modificaciones postraduccionales, plegamiento y maduración hasta alcanzar su estado funcional terciario o cuaternario.
El plegamiento ocurre gracias a la intervención de chaperonas y otras enzimas, que reconocen las proteínas desplegadas o mal plegadas y las pliegan para que logren una conformación estable funcionalmente activa. No obstante, este proceso es el punto donde más errores se producen en todo el proceso de síntesis de proteínas. Por ello, el RE posee un sistema de control de calidad del plegamiento, que consiste en exportar únicamente aquellas proteínas plegadas correctamente y dirigir a la degradación las que no lo están, evitando así la acumulación de proteínas aberrantes que puede conducir a la apoptosis.
Un fallo en este control de calidad puede generar múltiples enfermedades relacionadas con el mal plegamiento de proteínas, como diabetes mellitus, hígado graso, neurodegeneración, inflamación y cáncer.
Síntesis de proteínas en el retículo endoplasmático
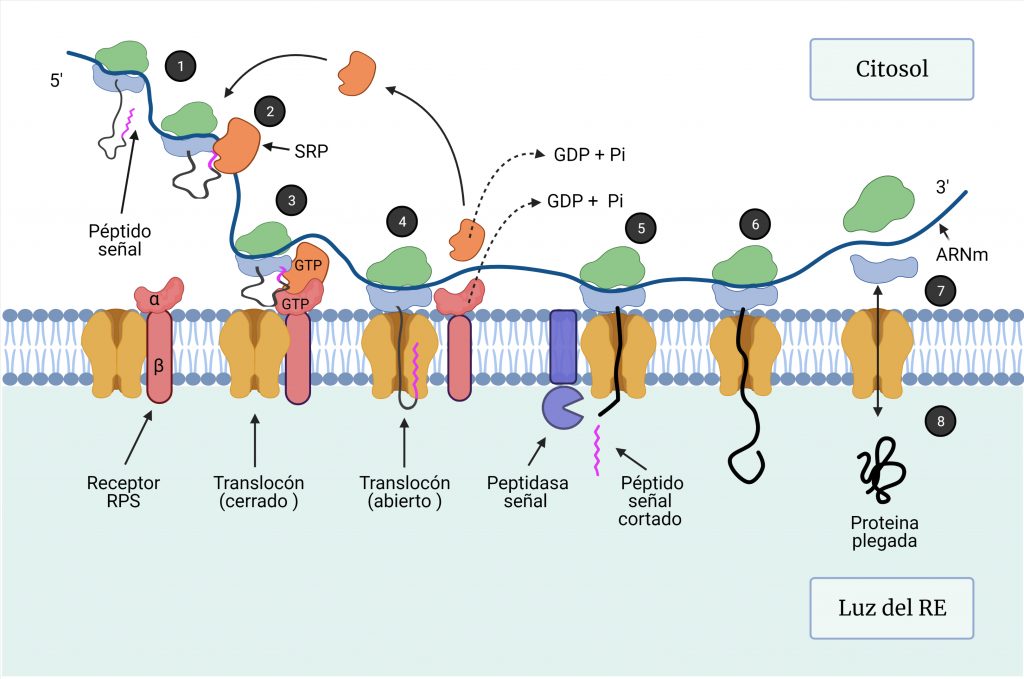
Las proteínas destinadas al RE comienzan su síntesis en los ribosomas libres en el citosol, donde se traduce, en primer lugar, un péptido señal de 16 a 30 aminoácidos, que contiene un núcleo de aminoácidos hidrofóbicos, que es esencial para su función (Figura 1, paso 1).
Una ribonucleoproteína citosólica, la partícula de reconocimiento de señal (SRP), se une a la subunidad 60S y al péptido señal en cuanto éste emerge del ribosoma (Figura 1, paso 2), de manera que detiene la traducción para que el polipéptido naciente pueda ser translocado al RE.
Este complejo se dirige a la membrana del RE, donde el receptor de la SRP reconoce el extremo de SRP no unido al ribosoma (Figura 1, paso 3). La unión del complejo ribosoma-péptido señal-SRP al receptor de SRP conduce a la apertura del complejo Sec61 o translocón en la membrana del RE, que actúa como un poro acuoso. Esta unión al translocón permite la liberación de SRP por la hidrólisis de GTP, la reanudación de la traducción y la entrada del polipéptido al RE a medida que se va sintetizando (Figura 1, paso 4). Todo este proceso requiere ATP. [1,2,3]
Dependiendo del destino de la proteína naciente, la síntesis continua de forma distinta:
- Si la proteína se va a secretar, a medida que la cadena polipeptídica se va alargando, pasa a través del canal del translocón hacia la luz del RE, donde el péptido señal es cortado inmediatamente por la peptidasa señal y posteriormente degradado (Figura 1, paso 5). La cadena continúa sintetizándose (Figura 1, paso 6) hasta la terminación de la traducción del mRNA, de manera que se libera el ribosoma (Figura 1, paso 7) y se cierra el translocón (Figura 1, paso 8).
- Si la proteína va a formar parte de una membrana biológica, poseerá un péptido de señal-anclaje, que posee una región hidrofóbica que le sirve para anclarse a la membrana del RE y no ser escindida. [4]
Plegamiento de proteínas
Una vez el polipéptido emerge del translocón, ha de plegarse para adquirir su conformación funcionalmente activa (estado nativo), la cual es la más estable [5, 6]. Para hacer posible la espontaneidad del proceso, se “ocultan” los residuos hidrófobos de aminoácidos no polares en el núcleo de la proteína y se exponen las cadenas laterales hidrófilas al entorno acuoso.
Primero se produce el plegamiento co-traduccional temprano, en el cual se forman estructuras secundarias. Una vez la proteína se libera del translocón, se produce el plegamiento postraduccional, que predomina sobre el primero y da lugar a las proteínas funcionales.
El proceso de plegamiento ha de llevarse a cabo en un tiempo biológicamente aceptable. Para ello, se limitan el número de conformaciones que pueden adoptar los polipéptidos, mediante la formación de interacciones hidrofílicas como puentes salinos y enlaces disulfuro [5].
La formación de estas interacciones co‐ y postraduccionales es catalizada por una maquinaria de modificación y plegamiento de proteínas residente en RE, que comprende una red de chaperonas, enzimas glucosilantes, oxidorreductasas e isomerasas que actúan simultánea o secuencialmente [5, 6].
A menudo las proteínas pequeñas con un solo dominio adoptan su estado nativo de manera espontánea, sin necesidad de la intervención de esta maquinaria, al contrario de lo que ocurre en el caso de proteínas con estructuras más complejas que se plegaran más lentamente y, por tanto, requieren de diferentes enzimas para alcanzar su conformación activa en un tiempo aceptable biológicamente [7].
Chaperonas
Las chaperonas moleculares se definen como «proteínas que interactúan, estabilizan o ayudan a una proteína no nativa a adquirir su conformación nativa, pero no están presentes en la estructura funcional final» [8]. Fueron identificadas por su mayor abundancia después de su exposición al choque térmico, por lo que son conocidas como Hsp, por sus siglas en inglés Heat Shock Proteins [5].
Estos catalizadores moleculares se encuentran en todos los orgánulos y compartimentos en las células encargadas de la síntesis y modificación post-traduccional, dado que se han conservado a lo largo de la evolución [9]. En el RE destacan la Hsp70 BiP (Grp78), la Hsp90 Grp94 (gp96) y las chaperonas de lectina calnexina (CNX) y calreticulina (CRT), que son exclusivas del RE [5]. La mayoría de los factores de plegamiento del RE se unen a Ca2+ y dependen de él para funcionar.
Las chaperonas actúan como enzimas acelerando las etapas limitantes de la reacción de plegamiento, es decir, disminuyen la barrera energética entre el estado nativo y no nativo de la proteína [6]. Para ello, reconocen dominios hidrofóbicos expuestos al medio acuoso, los cuales señalizan que la proteína está mal plegada, es un intermediario de plegamiento o forma parte de un polímero no ensamblado. Una vez reconocidos estos dominios hidrofóbicos, los ocultan y forman interacciones no covalentes con ellos para dar lugar al estado nativo, que es estable contra la agregación multimérica irreversible.
Si la proteína ha alcanzado su conformación nativa, no podrá volver a unirse a las chaperonas, pero si este plegamiento se ha producido de manera incompleta, habrá residuos hidrofóbicos expuestos que permitirán una nueva oportunidad para llevar a cabo el plegamiento correctamente [4].
Así, las chaperonas pueden diferenciar específicamente las etapas de plegamiento para una amplia gama de proteínas y, por tanto, actúan como supervisoras de la calidad del plegamiento proteico [9].
Control de calidad del RE
A pesar de la intervención de las maquinarias de plegamiento proteico, el plegamiento es el punto más propenso a errores desde la transcripción hasta lograr la proteína funcional.
El RE posee una concentración de calcio y un potencial redox mayor que el citosol, lo cual facilita el funcionamiento adecuado de las maquinarias de plegamiento. Esto le otorga la capacidad de realizar un control de calidad del plegamiento, mediante el monitoreo de la cantidad de proteínas mal plegadas en el RE, y un control de cantidad, al eliminar copias excesivas de ciertas proteínas para coordinar su plegamiento con las demandas fisiológicas y patológicas de la célula [9].
Este control consiste en detectar aquellas proteínas que tienen defectos de plegamiento o se encuentran en exceso para dirigirlas a vías de degradación, pasando únicamente a la vía secretora aquellas proteínas correctamente plegadas [10].
En RE de mamíferos existen dos mecanismos distintos de control de calidad: la vía de plegamiento general y la vía específica de glucoproteínas [5].
En ausencia de N-glucanos dentro de los primeros 50 residuos de aminoácidos, la proteína naciente se dirige hacia la vía de plegamiento general, de manera que se une primero a la chaperona BiP (Grp78). En presencia de dichos N-glucanos, la proteína se dirige hacia la vía de plegamiento específica de glucoproteínas, de manera que interacciona con las chaperonas calnexina-calreticulina (CNX / CRT) para sufrir procesos de modificación en el oligosacárido hasta alcanzar el plegamiento adecuado. No obstante, debido a la importancia de BiP en la translocación, ésta puede interaccionar primero de manera transitoria con la glucoproteína [11]. Ambos mecanismos actúan de forma coordinada en la maduración proteica para que únicamente sean exportadas las proteínas funcionales.
Cuando las proteínas alcanzan una conformación estable funcionalmente activa, pueden ser exportadas mediante vesículas al aparato de Golgi.
Las proteínas que permanecen mal plegadas tras varios intentos de plegamiento son finalmente retrotraslocadas al citosol para ser degradadas en el proteasoma mediante la vía de degradación asociada al RE (ERAD). En caso de que se formen cuerpos de inclusión proteicos debido a la agregación, éstos son degradados por autofagia mediante la vía de degradación en lisosomas asociada al RE (ERLAD).
Ante condiciones de estrés reticular, la acumulación de proteínas mal plegadas puede superar un umbral crítico, lo cual induce la activación de la respuesta de proteína desplegada (UPR). Este mecanismo trata de promover la supervivencia celular mediante el restablecimiento de la homeostasis, por un lado, controlando la síntesis y degradación de estas proteínas, y por otro, regulando la transcripción de los factores que intervienen en esta proteostasis.
Cuando el estrés del RE es demasiado alto, la UPR puede no ser suficiente para recuperar la homeostasis, de manera que los niveles de proteínas mal plegadas siguen siendo altos y se activan vías pro-apoptóticas para evitar la supervivencia de células aberrantes [7].
Vía de plegamiento general
Como ya hemos mencionado, en la vía general de plegamiento intervienen una chaperona de la familia Hsp70, BiP, y enzimas de plegamiento entre las que destacan las proteínas disulfuro isomerasas (PDI) (PDIA1), y peptidil prolil cis‐trans isomerasas (PPI) [12].
La proteína de unión a inmunoglobulina (BiP) es la chaperona más abundante y versátil de la célula, considerándose el regulador maestro del plegamiento de proteínas en el RE. Entre sus funciones destacan:
- Interviene en la translocación de cadenas nacientes al lumen reticular.
- Participa en el plegamiento y la oligomerización de proteínas.
- Interviene en la preparación de las proteínas no nativas para ser translocadas al citosol y degradas en el proteasoma.
- Regula la respuesta a proteína desplegada (UPR).
- Ayuda a mantener la homeostasis del Ca2+, lo cual es fundamental para el plegamiento de proteínas, pues la mayoría de los factores son dependientes de Ca2+ [4, 6].
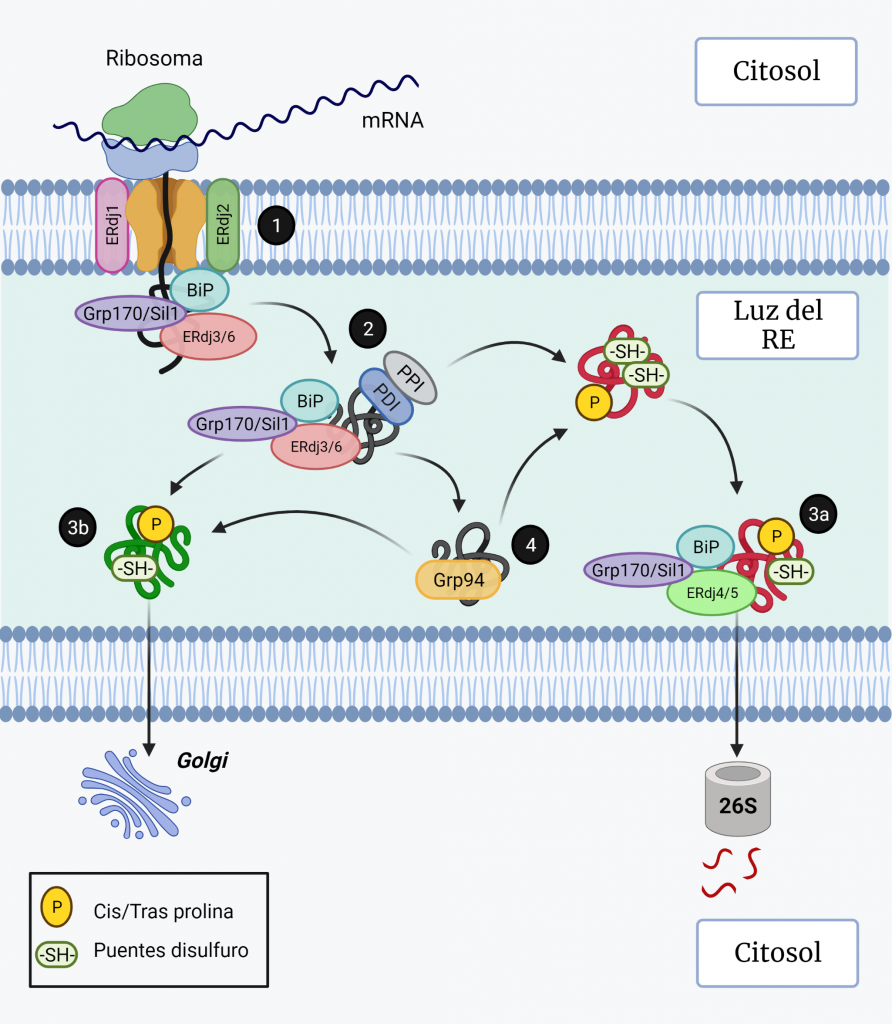
La actividad ATPasa de BiP está regulada por las proteínas J (ERdj1–7) y factores de intercambio de nucleótidos (Grp170 y Sil1). Los ERdjs actúan reclutando a BiP para realizar diferentes procesos como la traslocación (ERdj1 y ERdj2) (Figura 3, paso 1), el plegamiento (con ERdj3 y ERdj6) (Figura 3, paso 2) y la degradación (ERdj4 y ERdj5) (Figura 3, paso 3a) [8].
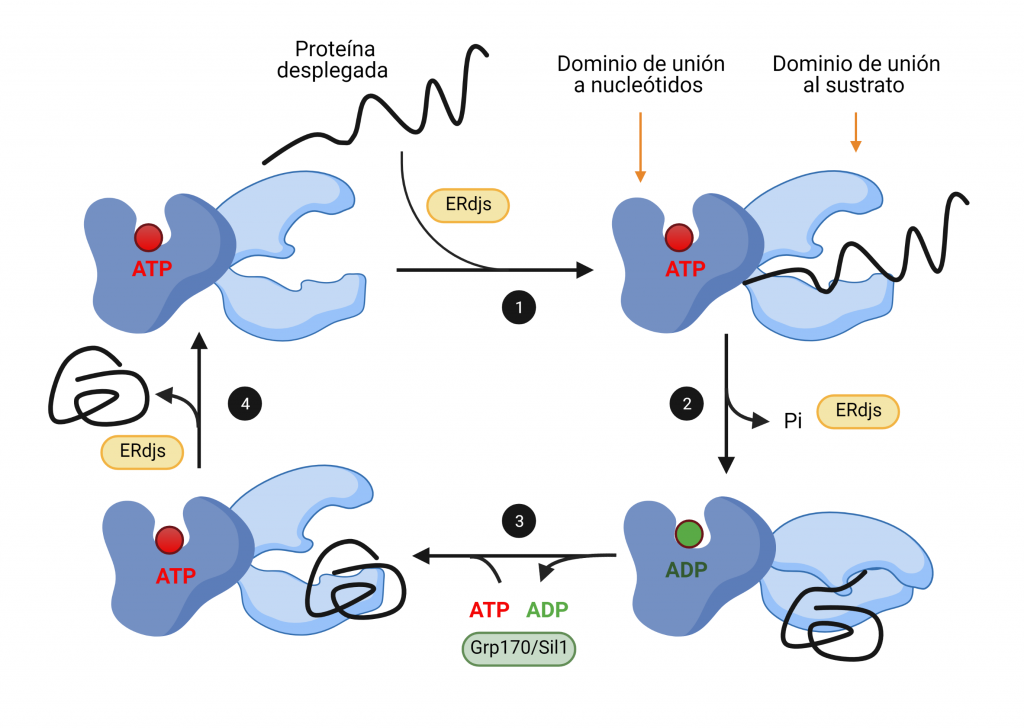
Cuando se une ATP al dominio de unión de nucleótidos de BiP, el dominio de unión a sustrato alcanza su conformación abierta, en la cual se une de manera transitoria a las regiones hidrofóbicas que están expuestas en la proteína plegada incompletamente (Figura 2, paso 1). Al hidrolizarse este ATP, se desacoplan los dominios, y este último adquiere su conformación cerrada, uniéndose de forma más estrecha a su proteína sustrato, de manera que facilita su plegamiento (Figura 2, paso 2). Las proteínas ERdj intervienen en ambos pasos, en primer lugar, transfiriendo las proteínas desplegadas y, en segundo lugar, produciendo la hidrólisis de ATP.
El ADP resultante de la hidrólisis es sustituido por ATP gracias a la intervención de factores de intercambio de nucleótidos (Figura 2, paso 3). Esto produce un cambio conformacional que libera la proteína diana, permitiendo que la chaperona sea reutilizada tras la intervención de las proteínas ERdj (Figura 2, paso 4).
Grp94 es otra chaperona de la familia Hsp90, sin actividad ATPasa, que se une a los sustratos después de BiP (Figura 3, paso 4) y es esencial en muchos procesos en el RE, incluido el plegamiento de proteínas, el control de calidad del RE, la respuesta al estrés y la amortiguación de Ca2+ [5, 6].
Además de estas chaperonas, en el plegamiento intervienen enzimas, destacando las peptidil prolil cis‐trans isomerasas (PPI) y las disulfuro isomerasas (PDI).
Las peptidil prolil cis‐trans isomerasas (PPI) catalizan la isomerización del enlace peptídico que precede a los residuos de prolina desde la conformación trans a la cis y viceversa. Esto es importante porque, durante el plegamiento, pueden ser necesarias múltiples isomerizaciones cis-trans hasta alcanzar la estructura proteica correcta. Este cambio conformacional es extremadamente lento, por lo que se considera un paso limitante en el plegamiento de proteínas [5].
Las disulfuro isomerasas (PDI) catalizan la formación, reducción e isomerización de puentes disulfuro, que estabilizan la estructura de la proteína nativa y los complejos oligoméricos. Esta reacción también es un paso limitante de la velocidad en el plegado [12].
Cuando las proteínas están bien plegadas, salen del RE y viajan al Golgi (Figura 3, paso 3b), mientras que las proteínas mal plegadas se dislocan al citosol para su degradación [5].
Vía de plegamiento específica de glucoproteínas
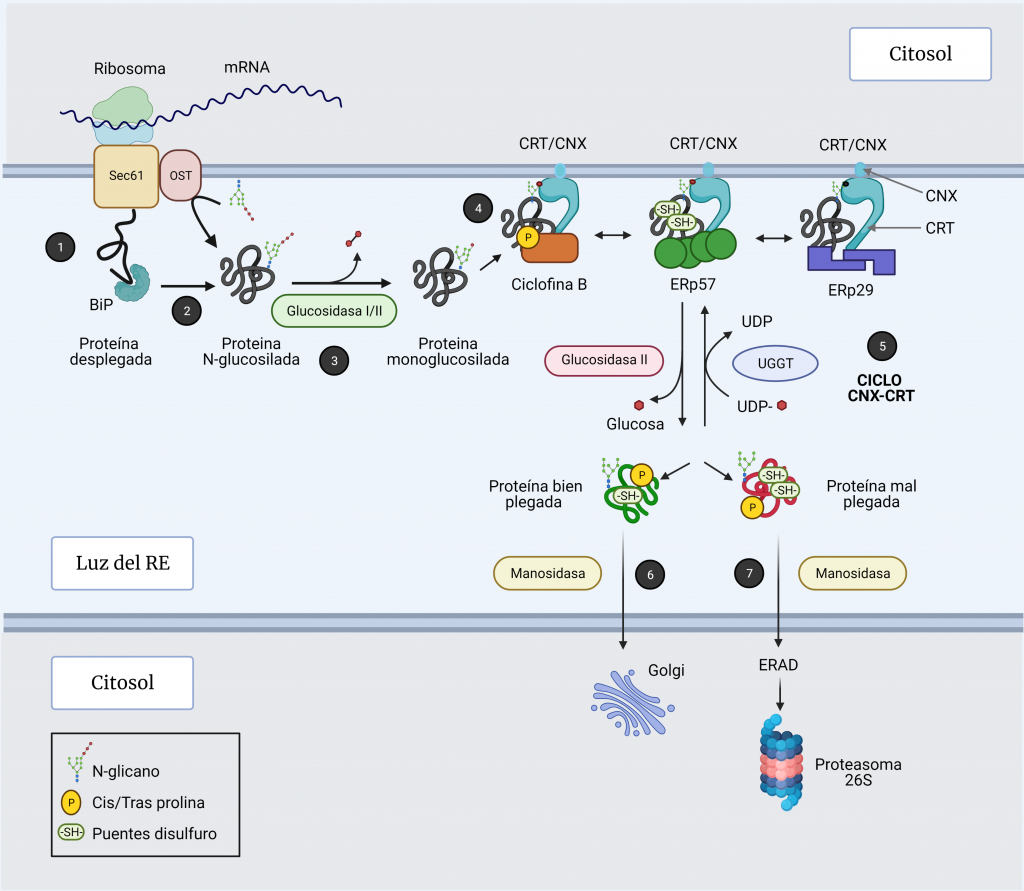
La mayoría de las proteínas solubles y de membrana que se dirigen a la vía secretora reciben glucanos ligados a N a medida que se traducen y translocan al RE (Figura 4, paso 1). Esto juega un papel crucial en el plegamiento de proteínas en el RE, principalmente a través de su interacción con calnexina (CNX) y calreticulina (CRT) [12, 13].
La N-glucosilación comienza cuando la oligosacariltransferasa (OST), una enzima anclada al translocón, transfiere un complejo de 14 azúcares (2 moléculas de N-acetil-glucosamina, 9 moléculas de manosa y 3 moléculas de glucosa) de un dolicol pirofosfato de la membrana del RE al residuo N de asparagina (Asn) de una secuencia aceptora (N-glicosilación) (Figura 4, paso 2) [5, 7, 12].
Las chaperonas calnexina y calreticulina (CNX / CRT) únicamente se unen a intermediarios proteicos monoglucosilados. Estos intermediarios se logran gracias a la previa escisión secuencial de las dos primeras glucosas de las glucoproteínas por las glucosidasas I y II (Figura 4, paso 3).
La calnexina es una proteína integral de la membrana del RE y la calreticulina es su parálogo soluble en el lumen de éste. Ambas proteínas poseen un dominio de unión a glucanos similar a lectina y un brazo flexible, el dominio P, que recluta a otras chaperonas. Además, se estabilizan cuando se unen a calcio [4].
A través de sus dominios P, se unen a chaperonas de función específica como ERp57, que es una disulfuro isomerasa (PDI) [14]; ciclofilina B, que es una peptidil prolil cis‐trans isomerasa (PPIasas); y ERp29, que tiene función de chaperona general (Figura 4, paso 4) [7, 12]
Una vez realizada la función de plegamiento, la glucosidasa II recorta la glucosa restante, liberando la proteína del complejo de chaperonas (Figura 4, paso 5).
La glucoproteína glucosiltransferasa (UGGT1) reconoce imperfecciones estructurales en las glucoproteínas que no han conseguido alcanzar su estado nativo tras el plegamiento y monoglucosila de nuevo sus cadenas laterales a partir del donador UDP-glucosa, de manera que pueden unirse otra vez a la calnexina-calreticulina para intentos de plegamiento adicionales (Figura 4, paso 5) [12].
Además, esta proteína monoglucosilada también actúa como sustrato de la glucosidasa II, que compite con UGGT1 con efectos opuestos. La situación en el RE va a condicionar la disponibilidad de estas dos enzimas y, por tanto, el desplazamiento de este equilibrio; por ejemplo, cuando se produce estrés en el retículo aumenta la expresión de UGGT1, por lo que este equilibrio se desplaza hacia la monoglucosilación para que se incremente la tasa de plegamiento.
Por tanto, UGGT1 actúa como un sensor de reconocimiento de proteínas mal plegadas, pues determina si una proteína sale del RE o se retiene para una mayor intervención. [7, 15]
Una proteína puede recorrer el ciclo CNX/CRT numerosas veces hasta que alcanza su conformación nativa. Para salir de este ciclo, el residuo de manosa más externo del brazo que contiene glucosa es eliminado por una manosidasa (Figura 4, paso 6 y 7), evitando que la UGGT1 vuelva a glucosilar.
Si las proteínas consiguen un plegamiento correcto, tras esta escisión, pueden ser exportadas fuera del RE [7]. Sin embargo, si las proteínas no consiguen plegarse adecuadamente tras varios ciclos, se dirigen a la vía de degradación de proteínas asociada a RE (ERAD).
ERAD
Cuando las proteínas no adquieren su forma nativa tras varios intentos de plegamiento, las células activan la vía de degradación asociada al RE (ERAD) para evitar que las proteínas se acumulen dando lugar al estrés del RE, el cual pondría en peligro la supervivencia celular. En este mecanismo las proteínas mal plegadas son retrotranslocadas al citosol para su posterior ubiquitinación y degradación por el proteosoma 26S, pues el RE no posee mecanismos de degradación [1].
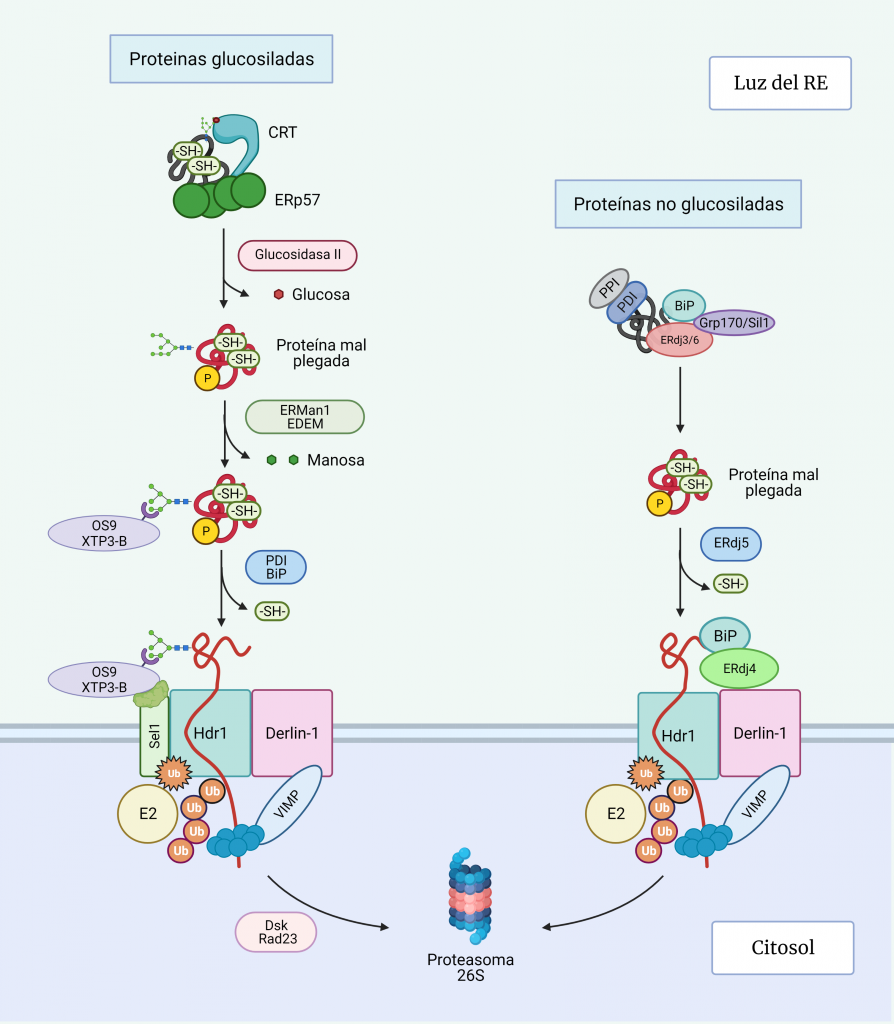
Vía ERAD de proteínas glucosiladas
Salida del ciclo de calnexina/calreticulina
Se cree que la salida de las glucoproteínas mal plegadas del ciclo de CXN/CRT ocurre cuando la α1,2-manosidasa I del RE (ERManI) corta el residuo de manosa más externo del brazo que contiene glucosa, dando lugar a un residuo de 8 manosas en vez de 9 como el glucopéptido original, de manera que la UGGT1 no puede volver a monoglucosilar. [17, 18, 19].
No obstante, esta desmanosidación también ocurre en las proteínas bien plegadas, por lo que una proteína cuyo N-glicano se recorta de 9 a 8 manosas puede ser sustrato tanto para el transporte a Golgi como para ERAD [20].
Recientemente se ha demostrado que, si la proteína se pliega adecuadamente, se incorpora a vesículas COPII que se dirigen al Golgi. Sin embargo, si no se pliega, las proteínas similares a la α-manosidasa I (EDEM1-3) reconocen el glucano recortado y producen un recorte adicional de manosa que conducirá a los siguientes pasos de la vía de degradación (Figura 5) [18, 20].
A partir de aquí, ERAD ocurre en un proceso de múltiples pasos que comprende el reconocimiento, translocación y ubiquitinación de proteínas ER para la degradación proteasomal citosólica.
Reconocimiento
La acción secuencial de ERManI y EDEM1 en glicoproteínas mal plegadas da como resultado la exposición de un resto de manosa unido a α1,6, que es reconocido por el dominio receptor de manosa-6-fosfato de las lectinas: osteosarcoma amplificado 9 (OS-9) y proteína transactivada XTP3 (XTP3-B). Estas lectinas junto con los sustratos ERAD son reclutados Sel1L, una proteína asociada con el complejo de retrotranslocación [17, 18].
Translocación y ubiquitinación
Antes de la translocación, las proteínas deben abrir su estructura parcialmente plegada, mediante la ruptura de los puentes disulfuro por disulfuro isomerasas (PDI) y BiP.
En el complejo de retrotranslocación de la membrana del RE destaca la proteína Derlin-1, que forma canales de retrotranslocación en la membrana del RE [20].
Como se ha mencionado, OS-9 y XTP3-B unidos a la proteína desplegada son reclutados por la SEL1L, que se encuentra asociada con Hrd1, una ubiquitina ligasa transmembrana que a su vez está unida al complejo de retrotranslocación.
La ubiquitinación de Hdr1 produce un cambio conformacional en éste, que permite la inserción del polipéptido por el canal Hdr1. A medida que la proteína emerge en el citosol, será poli-ubiquitinado por Hrd1 y las enzimas E2 de conjugación de ubiquitina asociadas. La AAA-ATPasa p97, se recluta en la membrana del RE a través de su asociación con VIMP y proporciona la energía necesaria para extraer las proteínas de la membrana del RE.
Finalmente, la cadena polipeptídica es dirigida hacia este proteasoma por Dsk2 y Rad23, una vez la N-glicanasa ha recortado el glicano restante de la cadena polipétidica para que esta última pueda entrar por el poro del proteasoma dónde finalmente serán degradadas. [20, 21].
Vía ERAD de proteínas no glucosiladas
La vía ERAD de las proteínas no glicosiladas está menos estudiada, pero fundamentalmente difiere del proceso de las glucosiladas en el reconocimiento de los sustratos desplegados (Figura 5). La decisión de dirigir las proteínas a la vía de degradación implica la unión de la proteína a las co-chaperonas pro-degradación: ERdj4, que está asociado con Derlin1, y ERdj5, que actúa como disulfuro isomerasa, induciendo un mayor despliegue de las proteínas para dirigirlas a ERAD, mientras que ERdj4 está asociado con Derlin1 [20].
ERLAD
Las proteínas mal plegadas que no pueden ser reconocidas y degradadas por ERAD, son autofagocitadas para su degradación en lisosomas asociada al ER (ERLAD) [16, 22].
Estrés del RE
La eficiencia del plegamiento de proteínas en el RE se puede ver alterada por un amplio grupo de alteraciones celulares que conducirán a la acumulación de proteínas mal plegadas dentro del orgánulo que, si supera un umbral crítico, provocará estrés en el RE. Las condiciones que desencadenan este estrés incluyen: falta de nutrientes, hipoxia, mutaciones puntuales en proteínas secretadas que intervienen en el plegamiento o causan agregación y pérdida de la homeostasis del calcio. Así, las células han desarrollado un sofisticado sistema para detectar y responder frente al estrés antes de que peligre su supervivencia [23].
UPR determina el destino celular bajo estrés en el RE
Ante el estrés del RE, las células activan una compleja red de vías de señalización intracelular interconectadas, la respuesta de proteína desplegada (UPR). Esta vía es iniciada por tres transductores ubicados en la membrana del RE, PERK, IRE1 (α y β) y ATF6, que contienen un dominio luminal RE capaz de detectar directa o indirectamente la acumulación crítica de proteínas mal plegadas, de manera que transmiten esta información al citoplasma y al núcleo [23, 24].
La UPR se activa para restaurar la homeostasis del plegamiento de proteínas del RE, pero si el estrés ER no se mitiga y amenaza la supervivencia celular, la UPR desencadena la apoptosis [25]. Esta vía se activa para restaurar la homeostasis del plegamiento de proteínas del RE.
UPR adaptativa: hacia la supervivencia celular
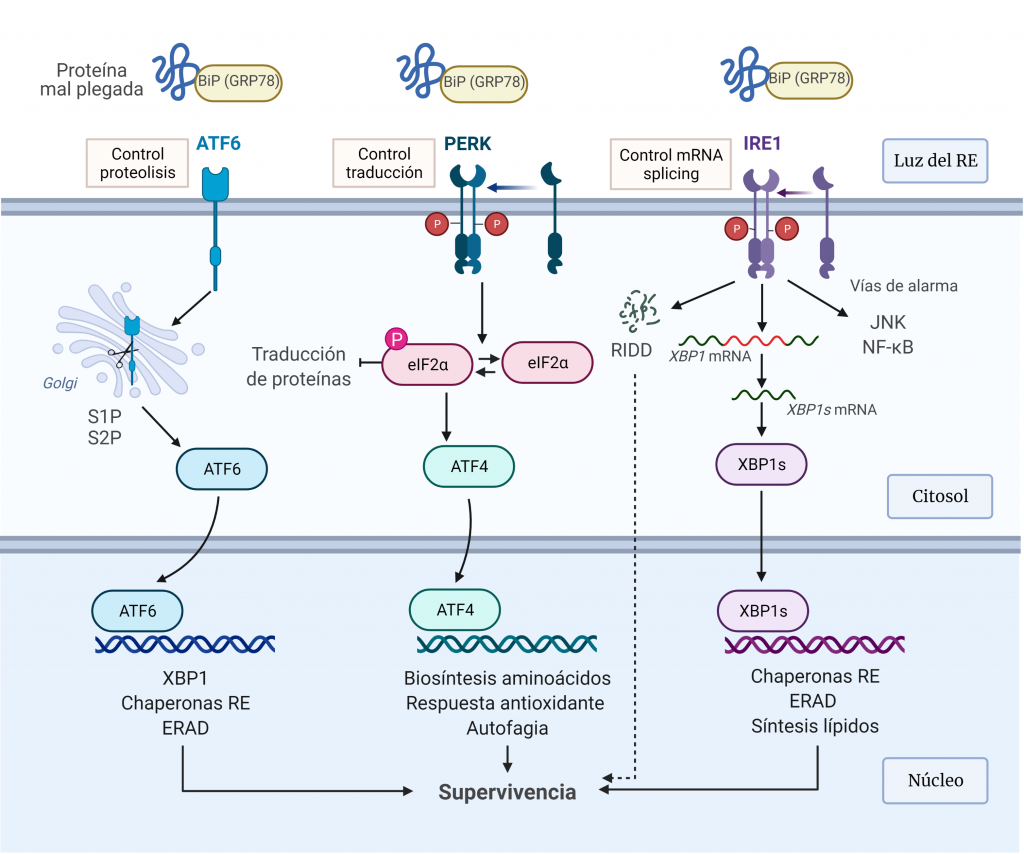
Ante el estrés agudo del RE, la UPR se activa para restaurar la homeostasis del plegamiento de proteínas del RE. Esto implica, en primer lugar, la expansión de la membrana del RE y un aumento en el plegamiento y transporte de proteínas en el RE, y, posteriormente, la atenuación transitoria de la síntesis de proteínas y un aumento en la degradación de proteínas asociadas al RE (Figura 6).
IRE1α
IRE1α (proteína 1α que requiere inositol) es una glicoproteína transmembrana del RE, que posee un dominio citoplasmático con actividad quinasa y RNasa, y un dominio luminal que detecta a las proteínas mal plegadas [4].
Esta proteína mantiene en un estado reprimido en condiciones sin estrés a través de una asociación con BIP (26). Durante el estrés del retículo endoplásmico, BIP se disocia para unirse a las proteínas mal plegadas. Esto conduce a la fosforilación y dimerización parcial de IRE1α, que estimula su actividad RNasa que realizar el corte y empalme de la proteína de unión a caja X (XBP1).
XBP1s controla la transcripción de genes que codifican proteínas implicadas en el plegamiento de proteínas, la degradación asociada a RE (ERAD), el control de calidad de proteínas y la síntesis de fosfolípidos, esta última para dar como resultado una expansión del RE.
IRE1α también media la descomposición del ARNm para reducir la carga de plegamiento de proteínas en el RE, lo que se denomina descomposición dependiente de IRE1 regulada (RIDD). Por otro lado, induce ‘vías de estrés de alarma’, incluidas las impulsadas por la quinasa N-terminal JUN (JNK) y el factor nuclear κB (NF-κB), mediante la unión a proteínas adaptadoras [25].
PERK
PERK (quinasa del RE pancreático)es una proteína transmembrana, que reprime la traducción de proteínas en éste. Esta proteína fosforila la serina 51 de eIF2α (subunidad α del factor de inicio de la traducción 2 en eucariotas) en respuesta al estrés.
La fosforilación de eIF2α, por un lado, reduce la formación de complejos de iniciación de la traducción y, por tanto, lleva a una disminución de la traducción general. Este control permite reducir el estrés del RE mediante la reducción del número de proteínas mal plegadas [9]. Por otro lado, la fosforilación de eIF2α permite la transcripción de ATF4, un factor de transcripción que actúa sobre genes implicados en el metabolismo de aminoácidos, las respuestas frente a estrés oxidativo, la autofagia y la apoptosis [25].
ATF6
ATF6 (factor de transcripción activador 6) es una proteína transmembrana del RE que se localiza en células no estresadas. En las células sometidas a estrés RE, ATF6 es transportado al aparato de Golgi donde es escindido por las proteasas S1P y S2P para producir un fragmento citosólico soluble (ATF6f), que ingresa al núcleo para inducir la expresión de genes diana relacionados con ERAD y el plegamiento de proteínas como los genes que codifican para las chaperonas BiP. (9) [25].
Estas ramas de señalización de la UPR no se activan de forma simultánea, sino que la activación de ATF6α e IRE1α ocurre de inmediato y disminuye con el tiempo, mientras que la activación de PERK sigue a la de ATF6α e IRE1α y permanece durante el estrés crónico del RE.
En condiciones normales, BiP interactúa con los dominios luminales de estos transductores, actuando como regulador negativo de su activación. Ante el estrés del RE, BiP se une a las proteínas mal plegadas, lo que permite su liberación de los transductores. La liberación de BiP de IRE1 y PERK permite su homodimerización y activación, mientras que su liberación de ATF6 permite el transporte de éste al compartimento de Golgi para la proteólisis intramembrana regulada. Esta activación regulada por BiP proporciona un mecanismo directo para detectar la capacidad de plegamiento del RE. Además, las proteínas mal plegadas pueden actuar como ligandos activadores para estos sensores de estrés a través de la unión directa al dominio luminal de IRE1α y PERK.
Estos vías de transducción actúan como bucles de retroalimentación homeostática para atenuar el estrés del ER, de manera que, si tiene éxito en reducir la cantidad de proteína mal plegada, la señalización de la UPR se atenúa y la célula sobrevive.
Algunos factores ambientales, el envejecimiento y mutaciones genéticas puede dar lugar a un mal funcionamiento de la UPR, lo cual se ha demostrado que induce enfermedades relacionadas con el plegamiento anómalo de proteínas como diabetes, ateroesclerosis, neurodegeneración, inflamación y cáncer.
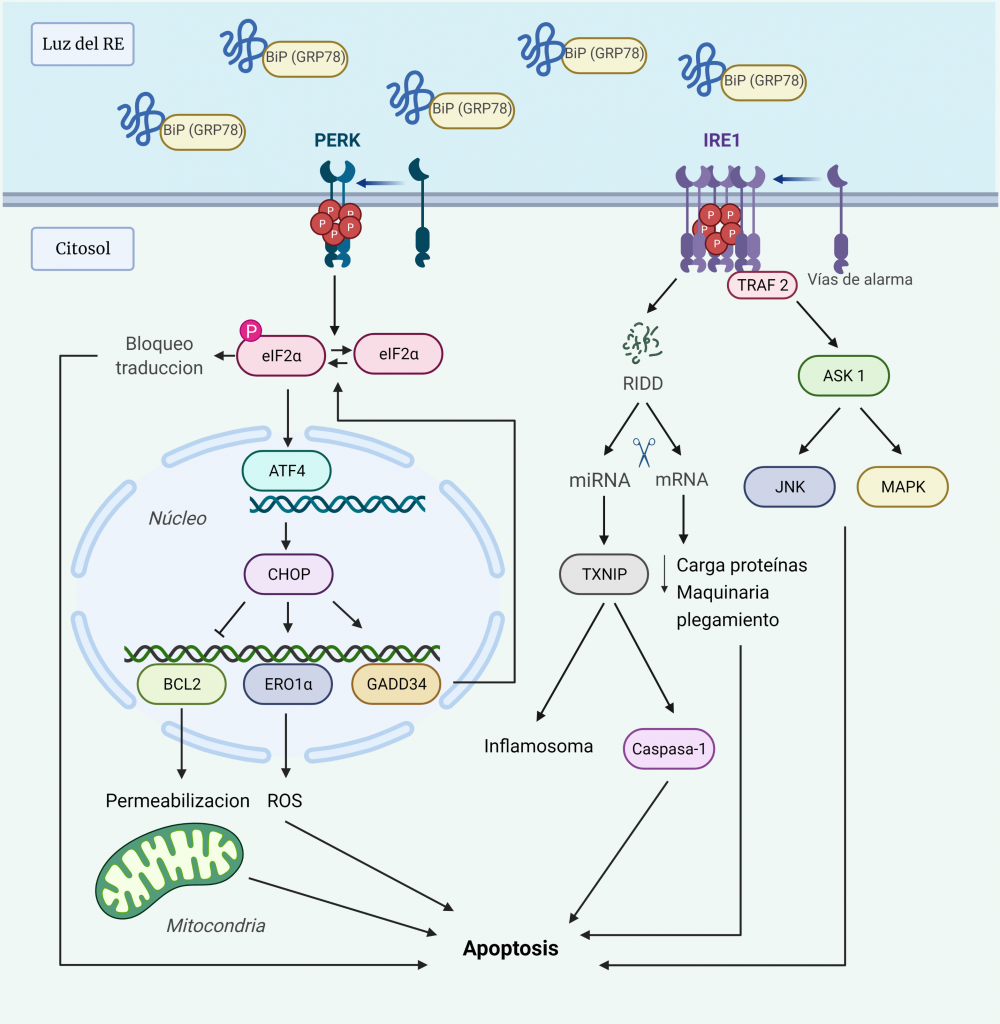
UPR terminal: hacia la muerte celular
Ante el estrés del RE prolongado, las respuestas adaptativas pueden resultar insuficientes para restaurar la homeostasis del plegamiento de proteínas, de manera que la UPR activa una vía de señalización alternativa proapoptótica, que induce la muerte celular para evitar la supervivencia de células aberrantes.
Aun no se conocen los detalles moleculares de esta vía, pero hay evidencias de que las dos quinasas UPR, PERK e IRE1α, involucran un conjunto de salidas pro-apoptóticas que conducen a la degeneración celular si el estrés del RE no se puede resolver (Figura 7) [10, 27].
La hiperactivación de PERK conduce a la pausa prolongada de la traducción de proteínas, debido a la fosforilación de eIF2α, lo cual es incompatible con la supervivencia. Además, PERK induce la transcripción de ATF4, que activa a su vez la transcripción de CHOP (proteína homóloga C/EBP), un factor de transcripción que media la muerte celular mediante la regulación de la expresión de varias proteínas:
- Inhibe la expresión de las proteínas anti-apoptóticas de la familia BCL-2, lo que conduce a la activación de proteínas BH3. Éstas desactivan las proteínas protectoras mitocondriales y las proteínas pro-apoptóticas BAX y BAK para que permeabilicen la membrana mitocondrial externa.
- Activa la expresión de ERO1α (ER oxidorreductina 1), que crea un entorno hiperoxidante en el RE, perjudicando el plegamiento de proteínas y, por tanto, aumenta la señal a favor de la muerte.
- Activa la expresión de GADD34 (proteína inducible por daño del ADN), que media la desfosforilación de eIF2α, de manera que se reanuda la síntesis de proteínas, aumentando la carga de proteínas en el RE. Por tanto, se forma un bucle de retroalimentación al amplificar la señal tóxica a favor de la muerte [28].
La hiperactivación de IRE1α fosforilada conduce a la formación de oligómeros en vez de homodímeros, lo cual tiene los siguientes efectos:
- Su dominio RNasa tiene afinidad por los sustratos RIDD (desintegración de ARNm dependiente de IRE1), actuando como una endonucleasa de cientos de ARNm localizados en ER, de manera que agota la carga de proteínas en el RE y los componentes de la maquinaria de plegamiento, empeorando aún más el estrés.
- La actividad RNasa reduce los niveles de microARN que normalmente reprimen dianas pro-apoptóticas, como la proteína pro-oxidante TXNIP (proteína que interactúa con tiorredoxina), cuyo aumento activa el inflamasoma y una vía pro-apoptótica dependiente de Caspasa-1.
- Recluta TRAF2 (receptor 2 asociado al receptor del factor de necrosis tumoral), que activa ASK1 (quinasa 1 reguladora de la señal de apoptosis). Ésta activa a su vez JNK (quinasa terminal c-Jun NH2) y MAPK (proteína quinasa activada por mitógenos p38) que, mediante fosforilación, activan la BH3 pro-apoptótica e inhibe la BCL-2 anti-apoptótica [10, 27].
Bibliografía
(1) Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci 2016 -01;73(1):79-94.
(2) Brodsky JL. Translocation of proteins across the endoplasmic reticulum membrane. Int Rev Cytol 1998;178:277-328.
(3) Zapun A, Jakob CA, Thomas DY, Bergeron JJ. Protein folding in a specialized compartment: the endoplasmic reticulum. Structure 1999 -08-15;7(8):173.
(4) Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell 2006 -05-05;125(3):443-451.
(5) Ellgaard L, McCaul N, Chatsisvili A, Braakman I. Co- and Post-Translational Protein Folding in the ER. Traffic 2016 -06;17(6):615-638.
(6) Redondo Juárez P. Fallos en el control de calidad en la síntesis de proteínas: origen de enfermedades raras. Mistakes in quality control of protein synthesis: the origin of rare diseases 2016.
(7) Lamriben L, Graham JB, Adams BM, Hebert DN. N-Glycan-based ER Molecular Chaperone and Protein Quality Control System: The Calnexin Binding Cycle. Traffic 2016 -04;17(4):308-326.
(8) Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol 2009 -06;16(6):574-581.
(9) Fu XL, Gao DS. Endoplasmic reticulum proteins quality control and the unfolded protein response: the regulative mechanism of organisms against stress injuries. Biofactors 2014 Nov-Dec;40(6):569-585.
(10) Hetz C, Papa FR. The Unfolded Protein Response and Cell Fate Control. Molecular cell 2018 Jan 18,;69(2):169-181.
(11) Molinari M, Helenius A. Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science 2000 -04-14;288(5464):331-333.
(12) Kozlov G, Gehring K. Calnexin cycle – structural features of the ER chaperone system. FEBS J 2020 -10;287(20):4322-4340.
(13) Price JL, Culyba EK, Chen W, Murray AN, Hanson SR, Wong C, et al. N-glycosylation of enhanced aromatic sequons to increase glycoprotein stability. Biopolymers 2012;98(3):195-211.
(14) Oliver JD, Roderick HL, Llewellyn DH, High S. ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol Biol Cell 1999 -08;10(8):2573-2582.
(15) Soldà T, Galli C, Kaufman RJ, Molinari M. Substrate-specific requirements for UGT1-dependent release from calnexin. Mol Cell 2007 -07-20;27(2):238-249.
(16) Fregno I, Molinari M. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Critical Reviews in Biochemistry and Molecular Biology 2019 March 4,;54(2):153-163.
(17) Wang Q, Groenendyk J, Michalak M. Glycoprotein Quality Control and Endoplasmic Reticulum Stress. Molecules 2015 -07-28;20(8):13689-13704.
(18) Roth J, Zuber C. Quality control of glycoprotein folding and ERAD: the role of N-glycan handling, EDEM1 and OS-9. Histochem Cell Biol 2017 -02;147(2):269-284.
(19) Caramelo JJ, Parodi AJ. Getting in and out from calnexin/calreticulin cycles. J Biol Chem 2008 -04-18;283(16):10221-10225.
(20) Oikonomou C, Hendershot LM. Disposing of misfolded ER proteins: A troubled substrate’s way out of the ER. Mol Cell Endocrinol 2020 -01-15;500:110630.
(21) Hwang J, Qi L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem Sci 2018 -08;43(8):593-605.
(22) Grumati P, Dikic I, Stolz A. ER-phagy at a glance. J Cell Sci 2018 -09-03;131(17).
(23) Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011 -11-25;334(6059):1081-1086.
(24) Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016 -01-21;529(7586):326-335.
(25) Liu CY, Kaufman RJ. The unfolded protein response. J Cell Sci 2003 -05-15;116(Pt 10):1861-1862.
(26) Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012 -01-18;13(2):89-102.
(27) Oakes SA, Papa FR. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu Rev Pathol 2015;10:173-194.
(28) Nam SM, Jeon YJ. Proteostasis in the Endoplasmic Reticulum: Road to Cure. Cancers (Basel) 2019 -11-14;11(11).

Gracias a estas ilustraciones y la sencillez en la redacción, he podido comprender al fin en detalle el plegamiento de las proteínas y su importancia!
Muchas gracias me habéis salvado el cuatrimestre.
Buena redacción y buen trabajo, se nota el esfuerzo que hay detrás de este artículo.