INSENSIBILIDAD CONGÉNITA AL DOLOR… ¿PRIVILEGIO O CONDENA?


Andrea Blázquez Arenas, María Chirivì Ramos
Vivir en un mundo sin dolor. ¿Suena bien verdad? No sentir dolor al quemarse, caerse o cortarse al cocinar…podríamos suponer que son todo ventajas. Al fin y al cabo a nadie le gusta sufrir, pero… ¿Cómo sabríamos cuándo ir al medico por una apendicitis o por un hueso roto?
Todo esto lo viven las personas con insensibilidad congénita al dolor (CIP), una enfermedad que afecta al sistema nervioso periférico que, aunque es poco común, sus consecuencias pueden ser mortales, debido a que estas personas no son conscientes de enfermedades que puedan padecer, ya que muchas se manifiestan a través del dolor.
¿CÓMO SABER SI UNA PERSONA PRESENTA CIP?
Las personas con CIP suelen presentar una serie de síntomas generales: (1) (2)
- La principal característica es la incapacidad para sentir dolor. Aunque estas personas pueden distinguir las variaciones en la temperatura, no son conscientes de si se están quemando, lo que hace que sea bastante común que presenten quemaduras.
- Al no ser conscientes del dolor, suelen presentar moretones, heridas (muchas de estas en la boca por morderse sin darse cuenta o en las manos por automutilación de parte de los dedos), e incluso huesos rotos u otras patologías que pueden pasar inadvertidas.
- Muchas personas pueden manifestar anosmia (pérdida de olfato).
¿POR QUÉ SE PRODUCE CIP?
CIP se produce por una alteración de determinados nociceptores. ¿Pero qué son los nociceptores? Estas son las neuronas especializadas en percibir la sensación de dolor cuando se produce un daño tisular.
Hay diferentes mutaciones de genes implicados en el CIP, pudiendo clasificarlos en:
- Genes CIP implicados en el desarrollo de los nociceptores, entre los que destacan:
- Receptor neurotrófico de tirosina quinasa 1 (NTRK1)
- Factor de crecimiento nervioso (NGF)
- Proteína 12 con dedos de zinc de dominio PR (PRDM12)
2. Genes CIP que producen fallos en los nociceptores (no reconocen el daño tisular) entre los que destacan:
- SCN9A
- SCN11A
Las mutaciones que más se suelen dar afectan a los genes SCN9A, NTRK1 y PRDM12.
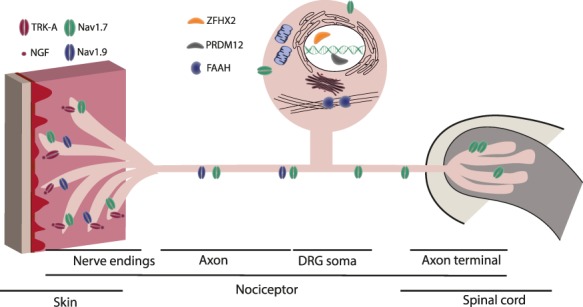
Esquema de un nociceptor en el que se muestra la ubicación subcelular de las proteínas implicadas en CIP. Imagen tomada de Drissi, I., Woods, W. A., & Woods, C. G. (2020). Understanding the genetic basis of congenital insensitivity to pain. British Medical Bulletin.
GEN SCN9A
SCN9A es el gen que codifica parte del canal de sodio dependiente de voltaje Nav1.7, el cual está altamente expresado en todo tipo de nociceptores, además de estar presente en las neuronas olfatorias sensitivas. (3)
Las mutaciones en SCN9A causan tanto un dolor excesivo (autosómico dominante) como una ausencia de dolor (autosómico recesivo). En el caso de que se dé una mutación en el gen SCN9A que haga que no se formen los canales de sodio Nav 1.7, se afectaría a la transmisión de las señales de dolor desde la zona de la lesión al cerebro, dando lugar a la insensibilidad al dolor. La pérdida de este canal en las neuronas sensoriales olfativas afectaría la transmisión de señales relacionadas con el olor al cerebro, dando lugar a una pérdida de olfato (anosmia). Todo esto hace que SCN9A sea considerado un elemento esencial en la detección del dolor. (1) (3)
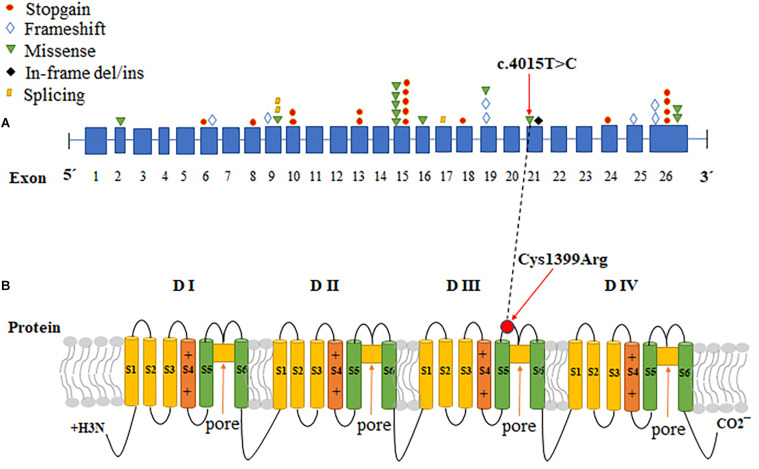
(A) Se representa diversas mutaciones en diferentes exones en SCN9A que dan lugar a CIP. (B) Representación del canal de sodio dependiente de voltaje Nav1.7, codificado por SCN9A y la ubicación de las mutaciones que han sido identificadas. Nav1.7 consta de cuatro dominios similares, cada uno formado por seis segmentos transmembrana a-helicoidales (numerados del 1 al 6). Los segmentos transmembrana 5 y 6 recubren los poros, mientras que en el segmento 4 se encuentra el sensor de voltaje (representado con el +). Las flechas rojas indican la ubicación de una mutación sin sentido identificada. La línea de puntos negra correlaciona el cambio de CDS (región codificante de un gen) con el cambio de proteína en SCN9A. Imagen tomada de Xie, X. H., Tang, J. G., Liu, Z. H., Peng, S. J., Yuan, Z. Z., Gu, H., … & Tan, Z. P. (2021). Case Report: Mutant SCN9A Susceptible to Charcot Neuroarthropathy in a Patient With Congenital Insensitivity to Pain. Frontiers in neuroscience, 15.
¿Cómo funciona el canal Nav1.7?
El dolor se produce por daño tisular, el cual puede ser causado, por ejemplo, por presión, acidez, alta temperatura, y los nociceptores se encargan de detectarlos (salvo el daño por radiación). Estos nociceptores van a permitir la entrada de una pequeña cantidad de sodio en su interior. Se supone que Nav1.7 está ubicado en los extremos del nociceptor, integrando señales de daño tisular locales. Si estas señales son suficientes, se produce la apertura de los canales y los iones de sodio fluyen hacia la célula, produciendo una lenta despolarización de la membrana de alrededor de estos canales. Esto puede llevar a que se genere un potencial de acción por la activación de Nav1.8, y desde allí se transmita a la médula espinal. (4)
Síntomas y características
Las personas con SCN9A -CIP no experimentan dolor inflamatorio, agudo y, de forma significativa, dolor neuropático. Sin embargo, la quimioterapia y el dolor óseo por cáncer sí que se da ya que este no depende del SCN9A. Por otro lado, uno de los síntomas más característico comentado anteriormente es la anosmia o pérdida de olfato, debido a que esta mutación afecta a los receptores sensoriales olfativos. (1) (4)
Aunque SCN9A se expresa en los islotes de Langerhans, en las células β (productoras de insulina), la diabetes no se desarrolla ni en SCN9A -CIP humano ni en ratón, por lo que se cree que en estas células Nav1.7 es redundante. (4)
Por otro lado, se ha demostrado en otros estudios, mediante el uso de imágenes de calcio in vivo y registro extracelular, que los ratones knockout para el canal de sodio Nav 1.7 tienen una actividad nociceptiva normal, pero la transmisión de la sinapsis de los nociceptores en la medula espinal se reduce en gran parte por un mecanismo dependiente de opioides (regulación positiva de un sistema opioide endógeno), lo que contribuye a la ausencia de dolor en individuos con CIP. (5) (6)
Se ha comprobado también que la analgesia producida se revierte con la aplicación central (no periférica) de antagonistas opioides. Esto no ocurre en la neuronas sensoriales olfativas, ya que son independientes de los opioides. En humanos con mutación nula de estos canales de sodio se da una analgesia reversible con naloxona (antagonista de opioides). La inhibición de la liberación de neurotransmisores es, por tanto, el principal mecanismo por el que se produce anosmia y pérdida de sensación de dolor en mutantes nulos de Nav 1.7 en humanos y ratones. (6)
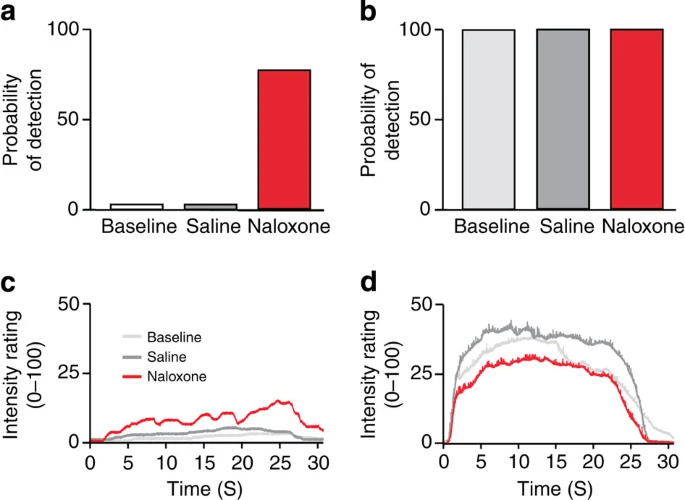
En las diferentes graficas se representa la percepción del dolor fásico. Se evaluó la probabilidades de detectar pulsos de calor (percepción del dolor fásico) en un paciente con Nav 1.7 nulo (caso a), además de en tres controles sanos de la misma edad (caso b). En el paciente Nav 1.7 nulo no se detectó ningún estímulo en las condiciones de referencia y salinas. La probabilidad de detectar el estímulo aumentó drásticamente al 80 % de los estímulos detectados durante la infusión de naloxona, alcanzando casi unos niveles de detección similares a los de los controles sanos emparejados. El dolor tónico fue provocado por estímulos láser de 25 s, a la vez que los que los sujetos fueron calificando su intensidad (0 = ninguna sensación, 100 = el peor dolor imaginable). La naloxona mejoró fuertemente las sensaciones de dolor tónico en el paciente sin Nav 1.7 (caso c) a lo largo del tiempo de la estimulación, sin efecto en los sujetos de control (caso d). Imagen tomada de Minett, M. S., Pereira, V., Sikandar, S., Matsuyama, A., Lolignier, S., Kanellopoulos, A. H., … & Wood, J. N. (2015). Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1. 7. Nature communications, 6(1), 1-8.
GEN PRDM12
Las enfermedades provocadas por el grupo de genes que causan CIP por falta de desarrollo de los nociceptores se conocen como neuropatías autonómicas y sensoriales hereditarias (HSAN). En este tipo de neuropatías hay una disminución de fibras A-delta y/o C. Estas últimas son fibras sensoriales aferentes que forman parte del sistema nervioso periférico cuya función es detectar estímulos nociceptivos (dolorosos). En el caso de PRDM12 hay una disminución de fibras A-delta pero no de fibras C. (3)
Esta enfermedad por mutación del gen PRDM12 se da en individuos que presentan la mutación bialélica (es una enfermedad autosómica recesiva).
El gen PRDM12 es el gen que codifica para la proteína 12 con dedos de zinc de dominio PR. Este es un regulador transcripcional, es decir, regulan la expresión de otros genes que actúan en la diferenciación de las células indiferenciadas de la cresta neural durante el desarrollo del individuo a una célula precursora de neuronas sensitivas. Regulan la expresión de los genes gracias a su capacidad de modificar la cromatina. (7) PRDM12 presenta un dominio PR (relacionado con el dominio SET, que codifica para la actividad metiltransferasa), 3 dominios de tipo dedo de zinc y una región polialanina en el extremo C-terminal de la proteína. (8)
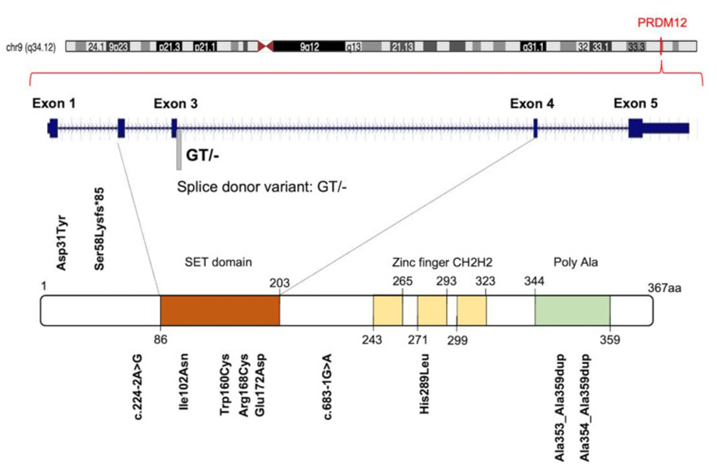
Ilustración gráfica de las diferentes estructuras que conforman la proteína PRDM12, con la localización de las mutaciones descubiertas que causan CIP. Imagen tomada de Rienzo, M., Di Zazzo, E., Casamassimi, A., Gazzerro, P., Perini, G., Bifulco, M., & Abbondanza, C. (2021). PRDM12 in Health and Diseases. International journal of molecular sciences, 22(21), 12030.
Los dedos de zinc que presenta esta proteína interactúan con una metiltransferasa (enzima que tiene la capacidad de metilar) EHMT2 conocida como G9a. Esta última se encarga de metilar la lisina en posición 9 de la histona H3 durante el periodo de formación de nuevas neuronas (neurogénesis). Se cree que esta metiltransferasa inhibe la proliferación de las células. (7)
La proteína PRDM12 funcional, junto a otros factores proneuronales de neurogenina, es esencial para el desarrollo de nociceptores en los ganglios sensoriales ya que regulan la iniciación y mantenimiento de NTRK1. Este último es el gen que codifica para el receptor de tirosina quinasa neurotrófica (TRKA), el cual interactúa con su ligando, el factor de crecimiento nervioso (NGF), factor necesario para la diferenciación y la supervivencia de los nociceptores. (9)
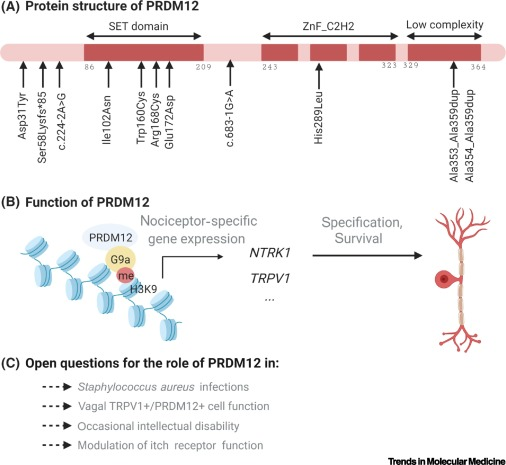
(A) Ilustración gráfica de las diferentes estructuras que conforman la proteína PRDM12. (B) Mecanismo de acción de cómo PRDM12 se une a la metiltransferasa G9a para metilar la lisina 9 de la histona 3 (H3K9) para controlar la transcripción de genes esenciales para el desarrollo y supervivencia de la neurona nociceptora (NTRK1 y TRPV1 entre otros). (C) Implicación de PRDM12 en diferentes funciones, según los síntomas observados por mutación de este. Imagen tomada de Imhof, S., Kokotović, T., & Nagy, V. (2020). PRDM12: new opportunity in pain research. Trends in Molecular Medicine, 26(10), 895-897.
Aquellos individuos que presentan una mutación en PRDM12 (estas se pueden dar en diferentes partes de la proteína) pueden provocar la pérdida de la función de la proteína por un mal plegamiento de esta, así como la agregación de estas entre sí impidiendo que lleven a cabo su función epigenética. De esta forma no pueden interaccionar con otras proteínas e inhiben la desmetilación de la H3. Es así como no se puede dar la señalización aguas abajo controlada por PRDM12. (10) Las células que en condiciones normales se diferenciarían a nociceptores, llevan a cabo la apoptosis. (9)
Se cree que PRDM12 no se une directamente al ADN, sino que se une mediante otros factores de transcripción que a día de hoy se desconocen. El mecanismo de acción de esta proteína aún no está claro y se debe investigar más para sacar conclusiones.
Síntomas y características
Las personas que padecen esta enfermedad suelen presentar una insensibilidad al dolor no global, infecciones por Staphylococcus aureus (se ha visto que algunos individuos presenta una inmunidad reducida frente a este microrganismo, lo que provoca a la aparición de infecciones recurrentes), no suelen presentar reflejo corneal y una producción de lagrimas alterada y dificultades con la regulación de la temperatura. En este caso los individuos suelen presentar un intelecto normal. (2)
PRDM12 es esencial sobre todo en la etapa de desarrollo temprano del individuo pero se sigue expresando en los nociceptores de individuos adultos. En caso de que su expresión en adultos sea importante para el mantenimiento de la función de los nociceptores y siga actuando como factor de transcripción, podría ser una interesante diana analgésica para tratar el dolor crónico. (3)
GEN NTRK1 (TRKA)
La patología producida por la mutación del gen NTRK1 fue la primera identificada de todas las CIP, siendo además la más común. (11)
Las mutaciones (de tipo bialélica – enfermedad autosómica recesiva) en el gen NTRK1 producen una CIPA caracterizada por insensibilidad al dolor, discapacidad intelectual, una mayor predisposición para contraer infecciones por Staphylococcus aureus y anhidrosis (incapacidad de sudar). A causa de la anhidrosis, se da una mayor predisposición de adquirir fiebre recurrente, siendo este un signo inicial de NTRK-CIPA. Sin embargo, la vibración y posición, además del sentido del tacto, no se ven afectados. (11) (12)
El gen NTRK1 codifica para el receptor de tirosina quinasa (TRKA) para el factor de crecimiento nervioso (NGF), el cual induce el desarrollo de axones y dendritas, además de promover la supervivencia de las neuronas sensoriales y simpáticas embrionarias. En estudios realizados con ratones que carecen del gen NTRK1 presentan características clínicas muy similares a las de CIPA (aunque la anhidrosis no es evidente). (13)
El sistema NGF-TRKA tiene un papel esencial en el desarrollo y la función de los nociceptores, así como en la termorregulación mediante la sudoración en humanos. (13)
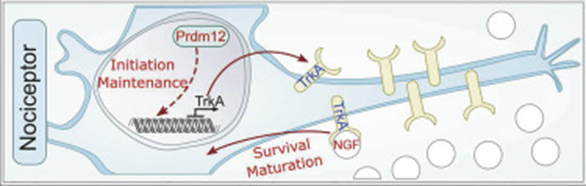
Ilustración gráfica de la interacción y función de TRKA-NGF. Imagen tomada de Desiderio, S., Vermeiren, S., Van Campenhout, C., Kricha, S., Malki, E., Richts, S., … & Bellefroid, E. J. (2019). Prdm12 directs nociceptive sensory neuron development by regulating the expression of the NGF receptor TrkA. Cell Reports, 26(13), 3522-3536.
Bibliografía:
- NIH-GARD. (2018, March 9). Insensibilidad congénita al dolor. Retrieved from https://rarediseases.info.nih.gov/espanol/12147/insensibilidad-congenita-al-dolor
- Schon, K. R., Parker, A. P. J., & Woods, C. G. (2020). Congenital insensitivity to pain overview.
- Drissi, I., Woods, W. A., & Woods, C. G. (2020). Understanding the genetic basis of congenital insensitivity to pain. British Medical Bulletin.
- Nahorski, M. S., Chen, Y. C., & Woods, C. G. (2015). New Mendelian disorders of painlessness. Trends in neurosciences, 38(11), 712-724.
- Minett, M. S., Pereira, V., Sikandar, S., Matsuyama, A., Lolignier, S., Kanellopoulos, A. H., … & Wood, J. N. (2015). Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1. 7. Nature communications, 6(1), 1-8.
- MacDonald, D. I., Sikandar, S., Weiss, J., Pyrski, M., Luiz, A. P., Millet, Q., … & Wood, J. N. (2021). A central mechanism of analgesia in mice and humans lacking the sodium channel NaV1. 7. Neuron, 109(9), 1497-1512.
- Elsana, B., Imtirat, A., Yagev, R., Gradstein, L., Majdalani, P., Iny, O., … & Tsumi, E. (2022). Ocular manifestations among patients with congenital insensitivity to pain due to variants in PRDM12 and SCN9A genes. American Journal of Medical Genetics Part A, 188(12), 3463-3468.
- Rienzo, M., Di Zazzo, E., Casamassimi, A., Gazzerro, P., Perini, G., Bifulco, M., & Abbondanza, C. (2021). PRDM12 in Health and Diseases. International journal of molecular sciences, 22(21), 12030.
- Desiderio, S., Vermeiren, S., Van Campenhout, C., Kricha, S., Malki, E., Richts, S., … & Bellefroid, E. J. (2019). Prdm12 directs nociceptive sensory neuron development by regulating the expression of the NGF receptor TrkA. Cell Reports, 26(13), 3522-3536.
- Imhof, S., Kokotović, T., & Nagy, V. (2020). PRDM12: new opportunity in pain research. Trends in Molecular Medicine, 26(10), 895-897.
- Li, S., Hu, H. Y., Xu, J. J., Feng, Z. K., Sun, Y. Q., Chen, X., … & Zhang, D. L. (2021). Identification of novel variations in the NTRK1 gene causing congenital insensitivity to pain with anhidrosis. Molecular Genetics & Genomic Medicine, 9(11), e1839.
- Indo, Y. (2020). NTRK1 congenital insensitivity to pain with anhidrosis.
- Indo, Y., Tsuruta, M., Hayashida, Y., Karim, M. A., Ohta, K., Kawano, T., … & Matsuda, I. (1996). Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nature genetics, 13(4), 485-488.
 Entrada anterior
Entrada anterior