Progeria: El curioso caso de Benjamin Button.


Por Marina Casado Grandía y Tomás Cantis López (3ºB de Biología Sanitaria, UAH).
Si has visto El curioso caso de Benjamin Button, seguramente te haya impresionado la condición que sufría este niño, que nació en el cuerpo de una persona de 80 años, y conforme iba «creciendo», rejuvencía. Pues bien, esta película está inspirada en una enfermedad real, llamada progeria o síndrome de Hutchinson-Gilford. Aquí te contamos sus causas, mecanismos moleculares, y sus posibles tratamientos.


Es sabido que los núcleos de las células eucariotas son muy importantes y cumplen muchísimas funciones diferentes. Esto se debe en parte a las laminas nucleares, redes filamentosas compuestas por proteínas situadas en la cara interna de la membrana nuclear. Esta estructura cumple funciones mecanoquímicas, de organización estructural para la cromatina, de regulación epigenética y señalización de muchas rutas, así como de regulación de la expresión génica.
La lamina nuclear está formada por una gran cantidad de proteínas filamentosas de tipo V (filamentos intermedios), llamadas laminas. Estas proteínas están constituidas por una región central de hélice α o coiled coil rodeada por 2 regiones globulares, los extremos C y N terminal. El extremo N terminal es algo más grande, por lo que recibe el nombre de cabeza, mientras que el extremo C terminal es la cola. En esta cola, compuesta por láminas β, presentan además una secuencia señal de direccionamiento hacia el núcleo (llamada NLS, de Nuclear Localization Sequence), con una estructura altamente conservada similar a la de una inmunoglobulina G. Generalmente, esta secuencia está formada por una caja CaaX (donde “C” es una cisteína, “a” es un aminoácido alifático, y “X” es un aminoácido cualquiera). (Vidak, S. et al., 2016).
Para su ensamblaje, las laminas primero se encajan individualmente entre sí para formar dímeros, en los que los motivos centrales (coiled coil) se enrollan mutuamente. Entonces interaccionan las cabezas de un dímero con las colas de otro dímero, de forma que se generan polímeros. Por último se forma el filamento o red, en el que varios polímeros se posicionan paralelamente. (2)

Imagen tomada de (Cooper G. et al, 2006).
Se han descrito 2 grandes grupos de estas proteínas:
- Laminas A y C: están codificadas por el mismo gen, LMNA, cuyo ARN mensajero sufre 2 vías de splicing para dar lugar a los dos tipos de laminas (A y C). Intervienen en la rigidez de los núcleos, y mutaciones en este tipo de laminas provocan las enfermedades progeroides.
- Laminas B: están codificadas por los genes LMNB1 (que produce las laminas B1) y LMNB2 (que produce las laminas B2 y B3). Todas ellas intervienen en la elasticidad nuclear. (Vidak, S. et al., 2016).
Procesamiento de las laminas A
Este proceso de maduración de la pre-lamina A (proteína precursora de la lamina A madura, en el núcleo) es muy importante, ya que en la mayoría de casos, la progeria está causada por mutaciones que afectan en estos pasos.
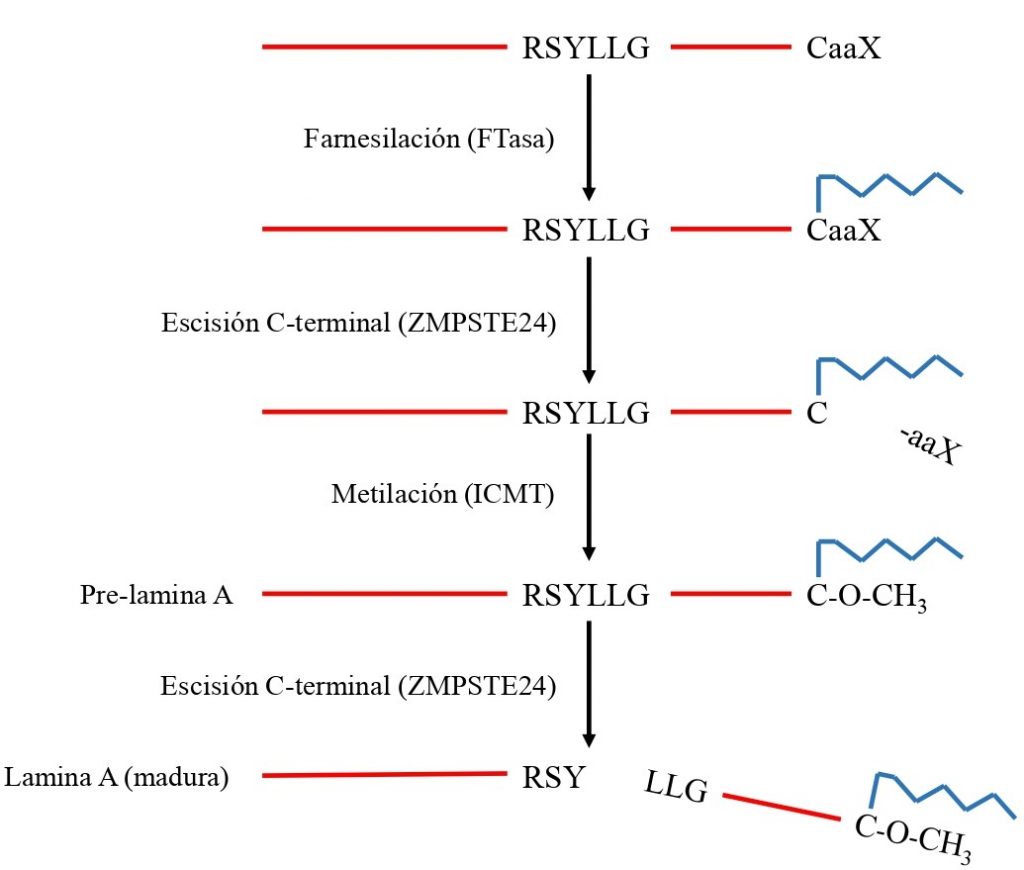
Al transcribirse el gen LMNA, se obtiene un transcrito de mRNA que es traducido para producir la pre-lamina A. Esta proteína sufre una serie de modificaciones:
- Primero es farnesilada (se añade un grupo farnesilo, una estructura que facilita su inserción en la membrana) por una enzima FTasa (farnesiltransferasa) en el residuo de cisteína de la caja CaaX.
- Una proteasa (FACE1/ZMPSTE24) elimina los 3 aminoácidos del final (aaX), quedando en el extremo C-terminal la cisteína unida al farnesilo. Este farnesilo que queda en la cisteína puede interaccionar fuertemente con la membrana nuclear, por su naturaleza lipídica.
- Después, la cisteína es carboxilada (se añade un grupo metilo) por una enzima llamada ICMT (isoprenil-cisteína.carboxil-metiltransferasa).
- Por último, la misma proteasa de antes elimina 15 aminoácidos del extremo C terminal, incluida la cisteína (carboximetilada y farnesilada).

Las laminopatías son aquellas enfermedades que afectan a la lamina nuclear, causadas por mutaciones en los genes que codifican las laminas. Estos trastornos afectan a muchos tejidos, pero suelen estar asociados al envejecimiento prematuro, y algunos ejemplos de estas enfermedades son el síndrome de Hutchinson-Gilford (progeria) o el síndrome de Charcot-Mary-Tooth 2B1, que afecta a los nervios periféricos. (Méndez-López, I., 2012).
El envejecimiento se puede definir como la degeneración de las funciones celulares y tisulares, que fisiológicamente, está asociada al tiempo. Los principales eventos que caracterizan el envejecimiento son 9:
- Inestabilidad genómica y defectos en la arquitectura nuclear (fallos en el ADN).
- Acortamiento de los telómeros, los extremos de los cromosomas.
- Alteraciones epigenéticas y remodelación de la cromatina.
- Pérdida de la proteostasis (regulación de las proteínas).
- Disminución en la sensibilidad a los nutrientes.
- Disfunción mitocondrial (déficit energético).
- Senescencia (envejecimiento) celular.
- Agotamiento de las células madre.
- Alteración en la comunicación entre células.
(Vidak, S. et al., 2016).

1. SIGNOS Y SÍNTOMAS
En el momento del nacimiento, los pacientes con progeria no muestran ninguna característica diferencial que haga sospechar de la enfermedad, pero cumplido el año de edad, aparecen los primeros síntomas. Estos comienzan con retardo en el crecimiento, pérdida de pelo (alopecia) y de grasa subcutánea (que puede conducir a una resistencia a la insulina y sensibilidad al frío), ojos y venas craneales prominentes, piel envejecida, rigidez articular, baja densidad ósea, esclerodermatitis (piel gruesa y endurecida), displasia (fallos en el desarrollo) mandibular, macrocefalia, etc.
A medida que van creciendo, otros síntomas son osteoporosis, aterosclerosis (endurecimiento de las arterias) y enfermedades cardiovasculares. Entre los 14 y los 20 años de edad, se produce la muerte de estas personas, por infarto de miocardio, fallos cardiacos o aterosclerosis progresiva (que conduce a hemorragias letales).
(Ullrich, N. J. et al., 2015), (Goldman, R. et al., 2004).

Imagen tomada de (Kreienkamp, R. et al., 2020).
2. GENÉTICA Y PATOGÉNESIS MOLECULAR DE LA PROGERIA
La progeria es causada por una mutación autosómica (en cromosomas no sexuales) y heterocigótica (es suficiente una copia del gen mutada) en el gen LMNA. Con frecuencia esta mutación es el cambio de citosina a timina en la posición 1824 del gen, la cual activa una proteína que produce un corte de 150 pares de bases en el mRNA de la pre-lamina A.
Esta deleción provoca que en la proteína resultante (pre-lamina A) presente una deleción de 50 aminoácidos, incluido el sitio de reconocimiento para el segundo corte de la enzima. Con ello, no se pueden eliminar los últimos 18 aminoácidos, con la la cola y el farnesilo en la cisteína. (Ullrich, N. J. et al., 2015).
De esta manera, la pre-lamina A en su forma inmadura, acumulándose en estas células como progerina.
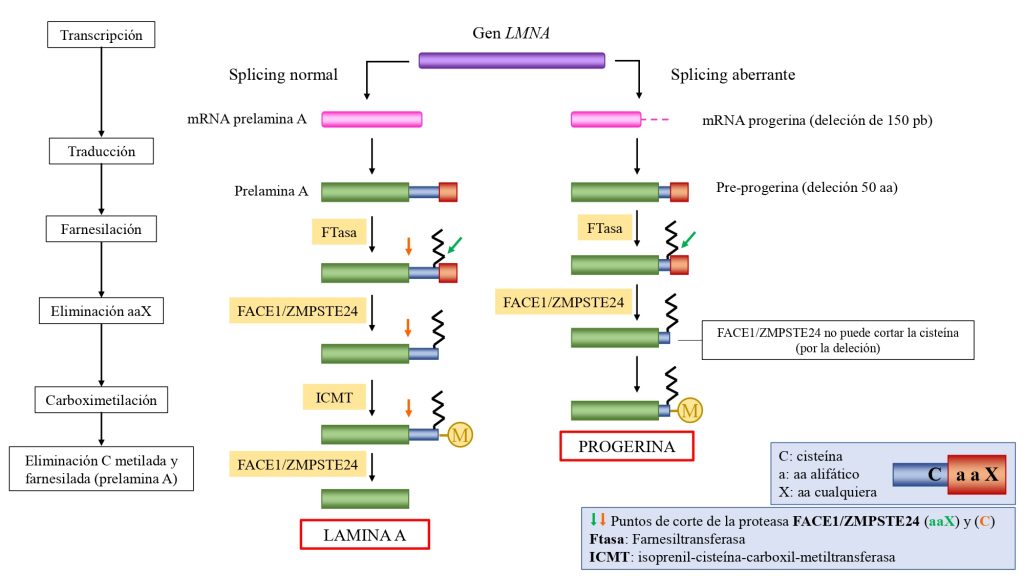
Esta progerina siempre se encuentra en la membrana nuclear (y no en el nucleoplasma), de forma que la lamina se ve engrosada y se producen cambios en la morfología del núcleo. La progerina, además, produce una disminución en los niveles de laminas B1, y de las proteínas HP1α y LAP2α, de gran importancia en numerosos procesos.
La progerina se expresa únicamente en tejidos de origen mesenquimal (piel, hueso, músculo esquelético, tejido adiposo, corazón y arterias), y como consecuencia de ello, aparecen los siguientes efectos:
- Núcleos lobulados y con laminas engrosadas.
- Disminución y pérdida de heterocromatina periférica.
- Acumulación de daños en el ADN.
- Aberraciones teloméricas.
- Disfunción mitocondrial.
Es por ello que el organismo de estas personas sufre procesos senescentes prematuros, que conducen a la muerte del individuo antes de los 20 años de edad. (Vidak, S. et al., 2016).
3. DIAGNÓSTICO DE LA PROGERIA
La enfermedad se puede diagnosticar por el reconocimiento del fenotipo (exploración clínica). El análisis genético del gen LMNA puede servir para detectar mutaciones y confirmar el diagnóstico. (Ullrich, N. J. et al., 2015).
4. DIANAS TERAPÉUTICAS
Aunque la progeria no tiene cura, hay ciertos tratamientos que permiten ralentizar la evolución tan rápida de la enfermedad. Entre ellos, destacan:
- Inhibidores de la farnesiltransferasa (FTIs): Los FTIs son moléculas que se unen al farnesilo del centro activo de la farnesiltransferasa de manera reversible, inhibiendo así la farnesilación de la progerina y su posicionamiento en la membrana nuclear. Se ha probado in vivo en ratones y se han obtenido buenos resultados en los fenotipos como una mejor formación de los huesos, aumento de peso y con todo, alargamiento de la esperanza de vida. Aún así, la farnesilación no es la única forma de prenilación (adición de moléculas hidrofóbicas), sino que las proteínas también se pueden geranil-geranilar (lo que explica la eficacia moderada de los FTIs). Con ello, se remodeló la terapia a inhibir farnesil-pirofosfatasa sintasa (que genera el farnesil pirofosfato) con aminobifosfonato, o la HMG-CoA reductasa (que interviene en rutas sintéticas de isoprenoides) con estatinas. (Ullrich, N. J. et al., 2015), (Vidak, S. et al., 2016). Un ejemplo de FTI es el L-778, 123. (Reid, T. S. et al., 2004).
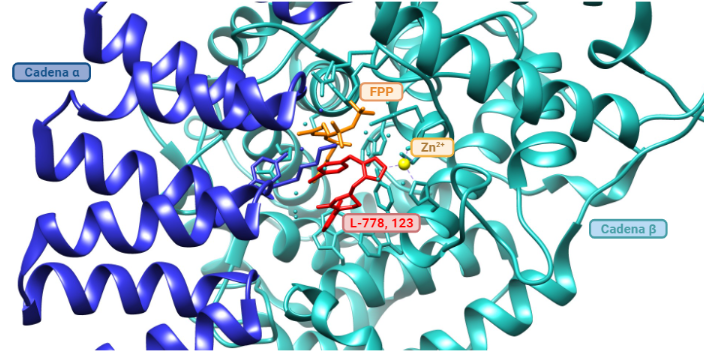
Imagen obtenida mediante Chimera y BioRender, a partir de la entrada de PDB 1SA4 (Reid, T. S. et al., 2004).
- Activación de la macroautofagia: la autofagia lleva proteínas defectuosas y organelos dañados a lisosomas para su degradación, y a veces está hiper-estimulada (por ejemplo, en condiciones de estrés o ayuno), con el fin de obtener aminoácidos. La rapamicina es un inhibidor de la vía de mTOR, que aumenta la autofagia, y con ello la vida media de las células HGPS (al eliminar la progerina de estas células). También tiene un efecto de reducción de los fenotipos más comunes de la progeria (núcleo lobulado, niveles de LAP-2α y daño en el ADN). Los FTIs también pueden actuar en esta vía, ya que al inhibir la farnesilación de la proteína Rheb GTPasa, activadora de la vía mTOR, también se reducen los niveles de progerina. (Vidak, S. et al., 2016).
- Sulforafano: es un antioxidante obtenido de crucíferas (plantas como el brócoli, la col, etc.) que estimula la actividad proteosomal y la autofagia en cultivos de fibroblastos. Con ello provoca la eliminación de la progerina, reduciéndose ciertos efectos de la enfermedad.
- Oligonucleótidos morfolinos: son polímeros de hasta 75 nucleótidos que bloquean esteáricamente el sitio de splicing en el exón 11 del pre-mRNA de la lamina A, revirtiendo algunos síntomas o fenotipos, y aumentando la vida de células HGPS.
- Resveratrol: activa la deacetilasa SIRT1, y ha mostrado efectos beneficiosos en algunos modelos de ratón, aunque no se conoce con exactitud su mecanismo de acción. Se cree que en presencia de progerina o laminas A, SIRT1 se asocia en menor medida a la matriz nuclear y disminuye su actividad deacetilasa. Con el resveratrol se ha visto que ocurre lo contrario, lo que en última instancia produce una disminución en la pérdida de masa corporal (aumentando la vida de los ratones).

La progeria es una enfermedad que se encuadra en aquellas consideradas «raras», ya que afecta a un número muy reducido de personas en todo el mundo. Sin embargo, a lo largo de los últimos años ha llamado mucho la atención de muchos investigadores e investigadoras, que tratan de comprender mejor los procesos moleculares que la producen, así como idear nuevos tratamientos que permitan aumentar la esperanza de vida de estas personas. Entendiendo este tipo de enfermedades, se podrá en un futuro mejorar o revertir otros problemas relacionados con el envejecimiento fisiológico, lo que también recibe mucha atención últimamente.

(1) Vidak, S., & Foisner, R. (2016). Molecular insights into the premature aging disease progeria. Histochemistry and Cell Biology, 145(4), 401–417. https://doi.org/10.1007/s00418-016-1411-1
(2) Cooper G., Hausman R. El núcleo. En: La Célula. 4ª edición. Marbán. 323-340. (2006).
(3) Méndez-López, I. (2012). Laminopatías. Enfermedades de la lámina nuclear. Medicina Clínica, 138(5), 208–214. https://doi.org/10.1016/j.medcli.2011.03.032
(4) Kreienkamp, R., & Gonzalo, S. (2020). Metabolic Dysfunction in Hutchinson–Gilford Progeria Syndrome. Cells, 9(2), 395. https://doi.org/10.3390/cells9020395
(5) Goldman, R. D., Shumaker, D. K., Erdos, M. R., Eriksson, M., Goldman, A. E., Gordon, L. B., Gruenbaum, Y., Khuon, S., Mendez, M., Varga, R., & Collins, F. S. (2004). Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome. Proceedings of the National Academy of Sciences, 101(24), 8963–8968. https://doi.org/10.1073/pnas.0402943101
(6) Ullrich, N. J., & Gordon, L. B. (2015). Hutchinson–Gilford progeria syndrome (pp. 249–264). https://doi.org/10.1016/B978-0-444-62702-5.00018-4
(7) Reid, T. S., Long, S. B., & Beese, L. S. (2004). Crystallographic Analysis Reveals that Anticancer Clinical Candidate L-778,123 Inhibits Protein Farnesyltransferase and Geranylgeranyltransferase-I by Different Binding Modes. Biochemistry, 43(28), 9000–9008. https://doi.org/10.1021/bi049280b
 Entrada anterior
Entrada anterior
Muy interesante!