SÍNDROME DE BLOOM


Belén Segovia y Gabriela Zapata
El Síndrome de Bloom (BS) es una enfermedad hereditaria autosómica recesiva, rara causada por una mutación en el gen BLM, que deriva en dos características principales:
- Predisposición a desarrollar todo tipo de cánceres a una edad temprana.
- Inestabilidad genética caracterizada por anomalías citogenéticas que incluyen numerosas roturas cromosómicas y un alto número de intercambios de cromátidas hermanas.
Se caracteriza por que los individuos que lo padecen tienen una estatura más baja de lo normal acompañada de facciones características de la enfermedad como son una cara larga y estrecha, nariz y orejas prominentes, con manifestaciones dermatológicas como es una erupción facial desencadenada por la exposición al sol (fotosensibilidad). Todo esto acompañado por una voz aguda, mayor susceptibilidad de padecer infecciones (inmunodeficiencia), y en ellos aumenta el riesgo de padecer cáncer. También padecen de problemas de fertilidad debido al hipogadismo, dificultades de aprendizaje, y retraso en el crecimiento y desarrollo.
Este es un síndrome de inestabilidad cromosómica, y las mutaciones somáticas que se producen como consecuencia de esa inestabilidad, son responsables del mayor riesgo de padecer cáncer. Entre todas las complicaciones, quizá las neoplasias malignas sean las más prominentes y potencialmente mortales. De hecho, los pacientes con BS tienen entre 150 y 300 veces más probabilidades de desarrollar una neoplasia maligna que la población general. Muchos de estos pacientes desarrollan algún tipo de cáncer a lo largo de su vida, y entre ellos destacan la leucemia, linfoma o cáncer colorrectal, de laringe, de mama y de piel.
Además de los síntomas anteriores, los pacientes con BS presentan niveles de inmunoglobulinas reducidas, lo que conlleva a una mayor susceptibilidad a neumonía, bronquiectasias y enfermedad pulmonar crónica. Aquellos que lo padecen también pueden presentar anomalías endocrinas particularmente anomalías del metabolismo de los carbohidratos, resistencias a la insulina y susceptibilidad a diabetes tipo II, dislipidemia e hipotiroidismo.
Este síndrome prevalece en la población judía Ashkenazi del este de Europa e Israel, lo que explica aproximadamente un tercio de los casos del síndrome de Bloom. De los judíos ashkenazis, 1 de cada 48000 personas lo presentan, y un 1% son portadores de este síndrome. En Estados Unidos se han notificado 170 casos, y raramente se ha notificado en otros países.
- ENZIMA (GENÉTICA, FUNCIÓN NORMAL, MECANISMO DE ACCIÓN Y FORMAS INTERMEDIAS)
El Síndrome de Bloom surge por mutaciones en ambas copias del gen BLM, localizado en el brazo largo del cromosoma 15 (15q26.1), por lo tanto la mutación que causa el síndrome es autosómica y, en este caso, recesiva. En esta región del cromosoma se han encontrado diversas patologías normalmente relacionadas con retrasos en el crecimiento y en el desarrollo mental por deleciones o duplicaciones.

Imagen 1. Esquema del cromosoma 15 y localización de BLM. Rescatada de: (https://www.rarechromo.org/media/information/Chromosome%2015/15q26%20deletions%20FTNW.pdf)
El gen situado en ese loci corresponde a una proteína de la familia de las helicasas RecQ, la RecQL3. Esta familia está formada por un solo miembro de origen procariota representado por la proteína RecQ de Escherichia coli, dos miembros de origen eucariota inferior representados por las proteínas Sgs1 y Rqh1, y cinco miembros de origen humano, las proteínas WRN (RecQL2), BLM (RecQL3), RecQ4 (RecQL4), RecQL (RecQL1) y RecQ5 (RecQL5).
Las helicasas son un grupo diverso de enzimas que se unen al DNA y desenrollan las dos hebras en espiral de la molécula del DNA. Este desenrollado es necesario en procesos en el núcleo de la célula, incluyendo la copia del DNA en preparación para la división celular y reparación del DNA dañado.
Cuando una célula se prepara para dividirse, el DNA que compone los cromosomas se copia de forma que cada nueva célula tendrá dos copias de cada cromosoma, una de cada progenitor. El DNA está dispuesto en las cromátidas hermanas, que están unidas una a otra durante las primeras etapas de la división celular. Las cromátidas hermanas de vez en cuando intercambian pequeñas secciones de DNA durante este tiempo, un proceso denominado intercambio de cromátidas hermanas. Se cree que estos intercambios pueden ser una respuesta al daño de DNA durante el proceso de copia.
BLM es una helicasa de ADN RecQ dependiente de ATP 3′-5 ′, uno de los estabilizadores del genoma más esenciales involucrados en la regulación de la replicación del ADN, recombinación y vías homólogas y no homólogas de reparación de rotura de doble cadena. Ayuda a evitar el exceso de intercambios de las cromátidas hermanas y también participa en otros procesos que ayudan a mantener la estabilidad del DNA durante el proceso de copia.
Se han identificado 151 mutaciones diferentes del gen BLM en las personas con síndrome de Bloom con diferentes orígenes étnicos. Principalmente las mutaciones dan lugar a un codón de paro prematuro que da lugar a una proteína truncada (80% de los casos), o se crea una proteína sin su actividad helicasa (20% de los casos).
Existe una mutación genética particular que elimina seis nucleótidos y los reemplaza con otros siete en la posición 2281 del DNA. Es la mutación blmAsh, en población judía Ashkenazi, que da lugar a la síntesis de una versión anormalmente corta, no funcional de la proteína BLM. Aparte de esta mutación, hay otras mutaciones del gen BLM que cambian aminoácidos en la secuencia de la proteína o crean una señal de parada precoz en su codificación. Como consecuencia de la ausencia o deficiencia de proteína BLM funcional, la frecuencia de intercambio de las cromátidas de las cromátidas hermanas es de alrededor de 10 veces más alta que el promedio.
Debido a la mutación de esta proteína, puede ocurrir la rotura de los cromosomas por la zona de telómeros, que se produce con mayor frecuencia en los individuos afectados. Estas roturas, que ocurrirían por una mala interacción de la BLM con los G-cuadruplexos, alteran las actividades normales de las células y provocan los problemas de salud asociados con este síndrome. Sin la proteína BLM, la célula es menos capaz de reparar el daño del ADN provocado por la luz UV, lo que deriva en una mayor sensibilidad a la luz solar.
Estos cambios genéticos permiten a las células dividirse de forma incontrolada dando lugar a una predisposición en el desarrollo de cánceres.Por otro lado, además de las mutaciones el gen de la BLM sufre otros tipos de alteraciones incluyendo un aumento en el número de copias transcripcionales, aumentando el número de proteínas en muchos cánceres.
De esta manera, se ha visto que las mutaciones en la BLM inducen al desarrollo de tumores y déficits en el desarrollo debido a la ausencia de su acción, lo que provoca intercambios entre las cromátidas hermanas y cromosomas homólogos que acaba provocando graves mutaciones en los individuos. Estas mutaciones se han visto en la línea hematopoyética, pero esto abriría nuevas preguntas para investigar cómo: ¿en qué células pueden darse estas modificaciones?, ¿en qué estado del desarrollo ocurre la recombinación? o ¿están estas mutaciones restringidas al precursor hematopoyético o podrían ocurrir en cualquier estado de desarrollo?

2. ANOMALÍA EN EL SÍNDROME A NIVEL BIOMOLECULAR
La BLM es una proteína que se compone de un conjunto de dominios que median sus funciones. Es una helicasa que utiliza la energía de la hidrólisis del ATP para moverse por una cadena simple de DNA y para poder romper los puentes de hidrógeno. Las helicasas están implicadas en la apertura del dúplex, lo cual se incluye los procesos de replicación, transcripción, recombinación homóloga, y otras formas de reparación del DNA, y todas ellas participan en el mantenimiento de la integridad del genoma.
Como se predice por su homología con las helicasas RecQ, la función de la BLM es hidrolizar el ATP para desenrollar el ADN moviéndose en una sola hebra en la dirección 3´-5´, y prefiere los sustratos que incluyen moléculas que simulan la replicación del DNA e intermediarios de recombinación (incluyendo horquillas de replicación, uniones de Holiday, bucles de desplazamiento y G-cuadruplexos). Es importante para la estabilidad de la horquilla de reparación, incluso en procesos de daño del DNA. Para ello está implicada en las siguientes procesos:

- Participa en la apertura de las dos cadenas para crean cadenas simples que son un sustrato de la RAD51, una enzima muy importante en la reparación del DNA.
- Implicada en reclutamiento de proteínas para estabilizar la apertura de la horquilla como las ssDNA-binding protein (RPA y RAD51). Estas son recombinasas que previenen que se formen apareamientos incorrectos por recombinación homóloga.
- Se encarga de desplazar las uniones de Holliday y de disolverlas sin sobrecruzamiento.
- Tiene funciones relacionadas con la interacción con estructuras como los G-cuadruplexos, encargados de mantener la estabilidad del genoma en regiones como los telómeros o los rRNA.
- La BLM en conjunto con la topoisomerasa IIIα desenrolla las moléculas de DNA con dos uniones de Holliday y las disuelve sin crear productos de recombinación. La topoisomerasa I es una enzima que se une a la doble hebra de DNA y permite el relajamiento del superenrollamiento que se crea en la hebra de DNA. La topoisomerasa IIIα es un tipo de topoisomerasa I, por lo que tiene actividad de corte y empalme que se da en una sola hebra en ssDNA.
La BLM forma un complejo con la topoisomerasa IIIα y otros componentes. La región de la BLM que interactúa con la topoisomerasa se encuentra localizada en el extremo N-terminal de los 200 últimos aminoácidos de la proteína. De esta manera las proteínas BLM que presentan deleciones en esta región no son capaces de interactuar con la topoisomerasa IIIα.
Una vez analizados los procesos en los que media la proteína, podemos analizar sus funciones celulares más detalladamente:
A. REPLICACIÓN
La helicasa BLM juega un papel esencial en varios eventos que tienen lugar durante la replicación del ADN. Cuando se produce un daño en la replicación, el ADN se vuelve más propenso a romperse y cualquier estrés replicativo podría ser una fuente de una transformación tumoral temprana
- Progresión y reparación de la horquilla de replicación: el daño inducido durante la replicación puede ser inducido por estructuras de ADN atípicas que pueden bloquear la progresión de la horquilla, como G-quadruplex, uniones de Holliday, complejos específicos de proteína-ADN, híbridos de ARN, etc. La enzima BLM es necesaria para la progresión y la estabilidad de la horquilla de replicación en condiciones fisiológicas normales y también es fundamental, después del daño en la replicación, para la estabilización y progreso de las horquillas bloqueadas al desenrollar estructuras de ADN inusuales, evitar la recombinación inapropiada y la supresión de la activación de un nuevo origen.
- DNA telomérico: las secuencias teloméricas repetidas forman estructuras muy ordenadas, como T-Loops, horquillas y G-quadruplexos, los sustratos preferidos para BLM, que deben disolverse para permitir la replicación adecuada del ADN telomérico.La BLM también participa en el mantenimiento de la integridad de los telómeros por la vía de alargamiento alternativo de los telómeros (ALT), la cual es independiente de la vía de la telomerasa.
- DNA ribosomal: las secuencias repetidas del DNA ribosomal se replican de manera única. La alta actividad de transcripción de estas regiones del genoma induce la formación de un R-Loop que si no se resuelve provoca rupturas del DNAr. BLM podría estar implicada en el mantenimiento de la integridad de este DNA, seguramente desenrollando las estructuras complejas que se formen.
B. RECOMBINACIÓN HOMÓLOGA
BLM interacciona con varios componentes de la maquinaria de recombinación homóloga (HR). La vía de la recombinación homóloga utiliza una secuencia homóloga como plantilla para reparar el ADN dañado o reiniciar las horquillas de replicación bloqueadas. BLM es una de esas proteínas necesarias que actúan en la replicación y recombinación desempeñando funciones tanto pro como anti-recombinogénicas.
- Reanuda el bloqueo de las horquillas de replicación cuando disuelve las uniones de Holliday.
- Reparación de la ruptura de la doble hebra: la ruptura de doble hebra es una de las formas más letales de daño en el DNA. En el primer paso, BLM actúa estimulando la actividad nucleasa de la exonucleasa 1 (EXO1) y la helicasa / endoexonucleasa DNA2 para asegurar la resección extensa 5′ de los extremos del ADN generados por la ruptura de doble hebra. Los overhangs creados se recubren rápidamente con la proteína RPA para estabilizarlas y protegerlas de la degradación (BLM tiene actividad recombinasa en este paso).
El overhang invade la hebra opuesta para buscar un fragmento homólogo a partir del cual pueda darse la reparación. Esta invasión de la hebra es seguida por la síntesis de ADN en el extremo invasor y conduce al desplazamiento de la hebra paralela, formando un D-Loop.
Si la homología encontrada es suficiente, BLM permitirá que continúe la reparación por la vía de recombinación homóloga. Las etapas finales de esta reparación se llevarán a cabo mediante la resolución de las uniones de Holliday (HJs), ya sea por el complejo disolvasoma BLM sin recombinación (actividad anti recombinasa de BLM), o por resolución, lo cual generará productos de recombinación.
El complejo BLM disolvasoma está compuesto por diversas enzimas: la BLM, topoisomerasa IIIα y y RMI1 y RMI 2, para procesar las uniones dobles de Holliday (dHJ) generadas durante el paso de invasión de la cadena de la vía de reparación por disolución.
- Vía alternativa de alargamiento de telómeros (ALT): en algunos cánceres se utiliza esta vía, en la que BLM interacciona con los telómeros y otras proteínas de esta vía.

Imagen 4. BLM en la recombinación homóloga. Extraída de https://pubmed.ncbi.nlm.nih.gov/33736941/
C. TRANSCRIPCIÓN
BLM desenrolla los dúplex híbridos de ARN-ADN, conocidos como R-Loop, y G4, en sitios transcripcionalmente activos, específicamente después del daño del ADN. La resolución de estas estructuras es esencial para permitir el acceso de la maquinaria de reparación a las lesiones inducidas, asegurando así una reanudación normal de la transcripción.

3. ESTRUCTURA DE LA BLM
El gen que codifica para la proteína BLM es un gen de 1417 aminoácidos ubicado en el cromosoma 15q26.1, que posee una actividad helicasa dependiente de ATP 3′ → 5 ‘. Su expresión está muy regulada en el ciclo celular, expresándose niveles más altos en las fases S y G2.
La proteína BLM, como ya hemos mencionado, es una helicasa de tipo RecQ, cuya función se basa en desenrollar el DNA, una actividad que utiliza ATP para moverse a lo largo de una hebra simple y para romper los enlaces de hidrógeno que mantienen unidas las dos cadenas de DNA. Su actividad dependiente de ATP es muy relevante en procesos tales como la replicación, transcripción, recombinación y reparación del ADN, por lo que estas proteínas presentan un sitio de unión a ATP. Las RecQ pertenecen a la superfamilia de las helicasas SF2 y juegan un papel crucial en el metabolismo del ADN, asegurando así el mantenimiento de la integridad y la estructura del genoma.
Sin embargo, el dominio helicasa no es suficiente para la actividad de la BLM. Asociado a este dominio helicasa está el dominio RQC, y ambos forman el núcleo catalítico de la BLM. El dominio helicasa es el más conservado en la familia de las RecQ, ya que es común a las proteínas de la familia SF2.
Este RQC se compone de un motivo de unión Zn 2+ y un dominio de “alas hélice de ADN” (winged helix domain) que conjuntamente actúan como el lugar de anclaje a la doble hebra del DNA. En este motivo se encuentra también una pinza-β que actúa insertando el complejo en el DNA y causando la separación del dúplex. El extremo C-terminal del RQC es el dominio “helicasa y ribonucleasa D C-terminal (HRDC)” que no se une directamente al DNA pero puede ayudar en las actividades helicasa y ATPasa. Es necesario para las disoluciones de las uniones de Holiday.
El extremo N- terminal de la proteína presenta la región de reconocimiento (la BLM forma un complejo con numerosas enzimas como son la topoisomerasa IIIα, RM1 y RM2, RPA…). La combinación de la actividad de desenrollamiento y reconocimiento cataliza la función del intercambio entre cadenas, que requiere promover previamente la disolución de las uniones holiday o el retroceso de las horquillas de replicación estancadas. No se han encontrado mutaciones que causen el síndrome de Bloom fuera de los dominios de RQC y helicasa.
Los miembros de la familia RecQ tienen siete motivos helicasa conservados, insertados entre dominios N-terminales y C-terminales únicos de tamaño variable. Existen cinco RecQ en humanos, las cuales tienen un dominio de helicasa estructuralmente conservado que contiene cajas Walker A y B y una caja DEAH que funciona en el desenrollamiento, siendo este dependiente de ATP y Mg2 +. Otros dominios como el RQC (RecQ C-terminal) y HDRC también son comunes a las proteínas de esta familia.
Estudios recientes basados en diversas bases de datos hacen ver que existe un pequeño dominio de unión a ácido nucleico en la región C-terminal de todos los miembros de la familia RecQ, lo que sugiere características comunes de reconocimiento de ADN en las helicasas.
Las helicasas RecQ se pueden clasificar en 2 grupos: el grupo que contiene las proteínas WRN, BLM, RecQ4, Sgs1 y Rqh1 cuyos tamaños van desde 1208 a 1447 aminoácidos ácidos, y el grupo que contiene las proteínas RecQL y RecQ5 con 410 a 649 aminoácidos y que se componen prácticamente del dominio helicasa central. Cabe destacar que, como podemos visualizar en la imagen, las proteínas BLM, WRN, RecQL y RecQ5, presentan una señal de localización nuclear (NLS) en la región C-terminal.

Imagen 6. Proteínas de la familia de la RecQ. Imagen rescatada de: https://pubmed.ncbi.nlm.nih.gov/33736941/
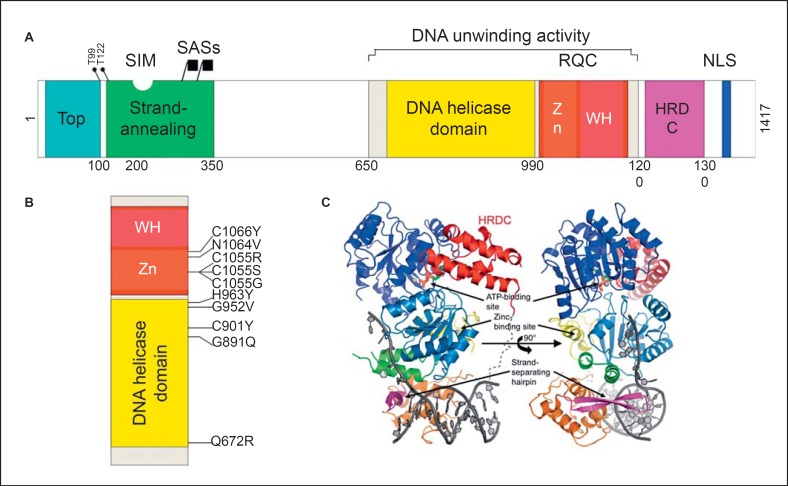
Imagen 7. Estructura de la BLM. Imagen rescatada de https://pubmed.ncbi.nlm.nih.gov/28232778/
Además se sabe que hasta hoy solo RecQ, RecQL, BLM, WRN y Sgs1 tienen una actividad ADN helicasa dependiente de ATP 3′-5 ’. La helicasa WRN tiene una actividad enzimática exonucleasa 3′-5 ′ adicional dentro de su región N-terminal.
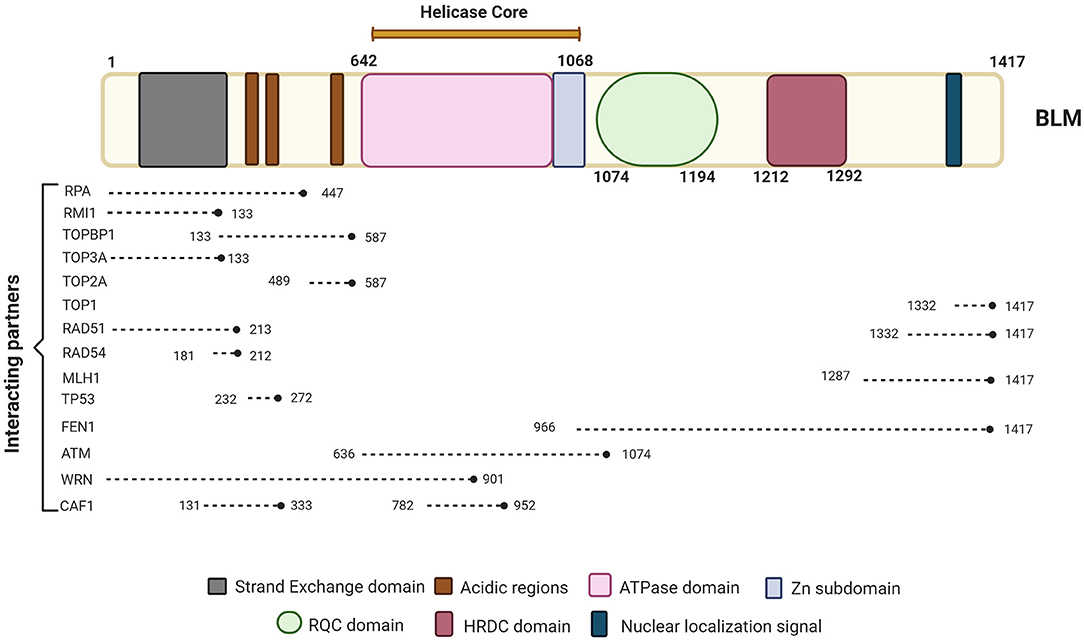
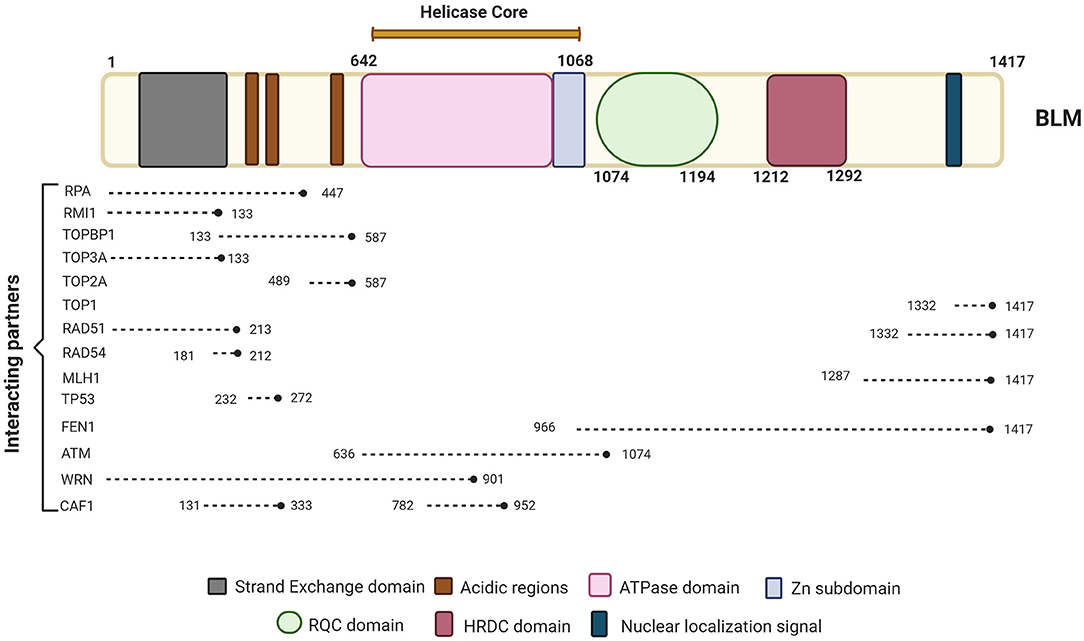
Imagen 8. Se representan las proteínas con las que interactúa la proteína BLM, la cual se compone de los dominios de núcleo de ATPasa / helicasa, RQC y HRDC. Podemos ver las proteínas clave involucradas en diferentes vías de reparación e interactuando con BLM junto con sus posiciones de unión a BLM. Extraída de: (https://www.frontiersin.org/files/Articles/634789/fgene-12-634789-HTML/image_m/fgene-12-634789-g001.jpg).
4. PATOLOGÍA Y CÁNCER
El cáncer es la complicación médica más frecuente y grave que aparece en pacientes con Síndrome de Bloom, y suele llevar a la muerte. En este síndrome se dan una amplia variedad de tipos de cáncer y ubicaciones anatómicas en personas con SB. Las células con mutaciones en BLM conducen a la acumulación de DNA con daños con alta sensibilidad hacia varios fármacos quimioterapéuticos.
La distribución de los cánceres es parecida a la que sucede en la población general, pero ocurren a edades más tempranas y una sola persona puede experimentar múltiples cánceres. En esta enfermedad los más frecuentes son la leucemia y el linfoma, y dentro de los tumores sólidos los más comunes son los cánceres del tracto digestivo, en particular el adenocarcinoma del tracto intestinal superior e inferior. Los carcinomas de células de cabeza y cuello también se han diagnosticado con frecuencia, especialmente en la base de la lengua, la epiglotis y el esófago.
Como dato curioso cabe destacar que la población judía Ashkenazi tiene alta prevalencia de cáncer colorrectal precisamente por la mutación en este gen. De acuerdo con su función de cuidar el genoma, se han visto mutaciones en el gen BLM asociadas con el riesgo de cáncer colorrectal. Aproximadamente el 0,11% de pacientes presentan la mutación heterocigótica BLM que confiere un riesgo de penetrancia de bajo a moderado para desarrollar cáncer colorrectal., y entre estos pacientes se encuentran personas con ascendencia judía asquenazí, portadores de alta frecuencia de la mutación heterocigótica BLMAsh.
Múltiples estudios muestran que la proteína BLM tiene una implicación directa en la regulación de varios oncogenes y genes supresores de tumores . BLM potencia la degradación y alteración de la función del factor de transcripción oncogénico c-Jun, moderando así el efecto de este último en la transformación neoplásica.
Es más, el gen c-Myc, que regula la transcripción de muchos genes fundamentales y se sobreexpresa en varios tipos de cánceres, también es un objetivo de BLM. De hecho, BLM disminuye los niveles de c-Myc y retrasa la iniciación del tumor en múltiples líneas de células de ratón. Es por esto que muchos autores denominan a la proteína como una “cuidadora de la supresión de tumores”
El efecto directo y específico de la función de BLM en la prevención del cáncer ya que BLM trabaja en conjunto con p53 para efectuar la apoptosis; por lo tanto, previene la transformación tumoral. Se han realizado múltiples estudios en células con síndrome de Bloom en los que se identifican anomalías tanto en la acumulación como en la activación de p53 inducida por daños en el ADN.
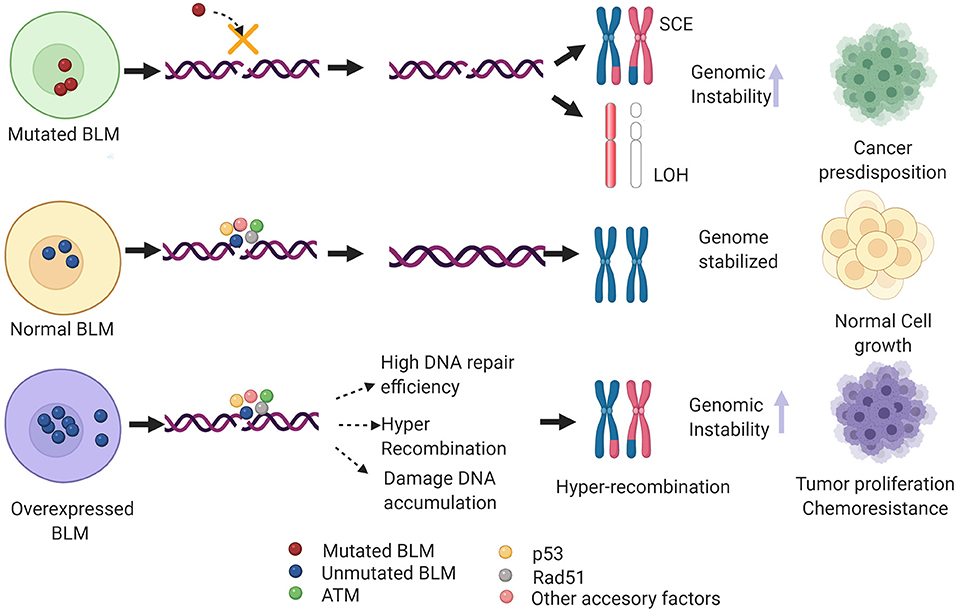
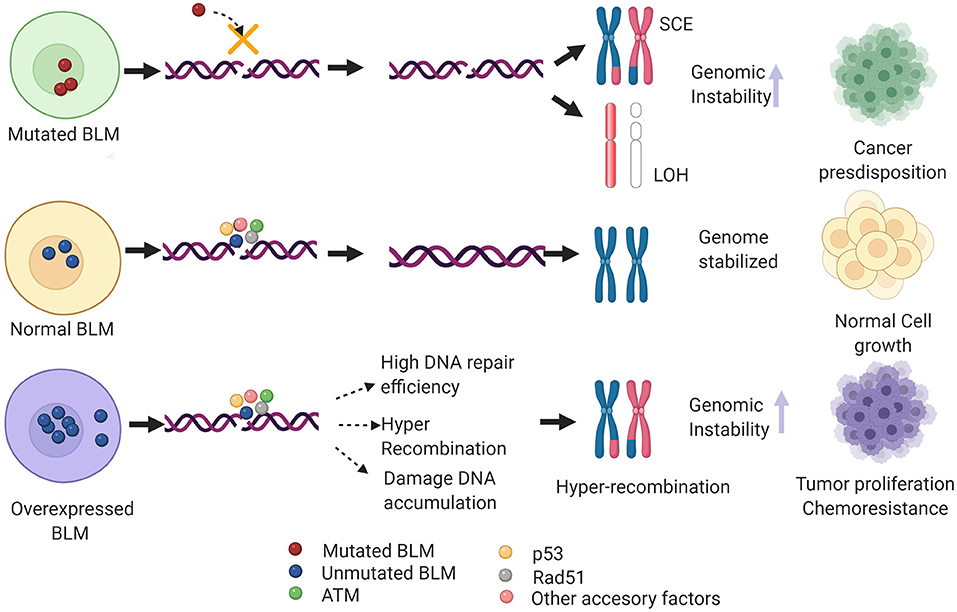
Imagen 9. Mecanismos implicados en neoplasia de BLM. Imagen rescatada de https://www.frontiersin.org/files/Articles/634789/fgene-12-634789-HTML/image_m/fgene-12-634789-g004.jpg
Además de la predisposición al cáncer que tienen los individuos que presentan mutaciones en la BLM, también existe esta predisposición en pacientes con sobreexpresión de la BLM. En estos últimos casos, hay una eficiencia muy alta de reparación así como de recombinación por la vía homóloga, lo que se traduce en una acumulación del daño en el DNA e hiper recombinación.
5. SISTEMA INMUNE
Se afirma también que la proteína BLM está involucrada en el desarrollo y mantenimiento del sistema inmune. En pacientes con el síndrome de Bloom se observan niveles de inmunoglobulinas IgM e IgA anormales y de IgG reducidos. Con los niveles disminuidos de BLM, las células precursoras linfoides de la médula ósea y las células B maduras del bazo y la cavidad peritoneal disminuyen significativamente, e incluso se ven fallos en el desarrollo de las células T.
Debido a la acumulación de ADN dañado en células deficientes en BLM, también se ha observado una mayor expresión del gen inflamatorio estimulado por interferón y niveles aumentados en sangre periférica. BLM juega un papel indispensable en el desarrollo, proliferación, mantenimiento, estabilidad y función de las células inmunes y contribuye a la inmunodeficiencia en pacientes afectados por BS.
6. MÉTODOS DIAGNÓSTICOS Y TRATAMIENTO
Debido a que este síndrome es raro, no existen pautas de tratamiento basadas en evidencias científicas. Entre las principales preocupaciones destacan las anomalías de la piel, los problemas de crecimiento y nutrición, las anomalías endocrinológicas y el riesgo de neoplasias malignas, que son la mayor causa de muerte.
Como remedio para los problemas de crecimiento se emplean preparados y alimentos hipercalóricos. Además el tratamiento con hormona de crecimiento podría mejorar el crecimiento lineal, aunque su uso es controvertido por algunos estudios que identifican un mayor riesgo para el desarrollo de cáncer en niños.
En algunos casos en los que hay una disminución de los niveles de inmunoglobulinas y se sufren infecciones, se pueden aplicar antibióticos y tratamiento con inmunoglobulinas intravenosas o subcutáneas.
Resulta fundamental el cuidado de la piel con cremas y protectores y su vigilancia en cuanto a la aparición de posibles cánceres.
La enfermedad sigue un patrón de herencia autosómico recesivo y es por eso que se ofrece consejo genético a las parejas en riesgo cuando ambos son portadores de la mutación, informándoles de un riesgo del 25% de tener un hijo afectado en cada embarazo.
El diagnóstico se intuye clínicamente en base a la identificación de síntomas propios del síndrome de Bloom y se confirma mediante la identificación de variantes patogénicas bialélicas del gen BLM en las pruebas moleculares. También puede confirmarse mediante un análisis citogenético que identifica un mayor número de intercambios entre cromátidas hermanas (esta prueba se puede realizar en el embarazo) o mediante técnicas de secuenciación y deleción/duplicación de BLM.
BIBLIOGRAFÍA
– Ababou M. (2021) Bloom syndrome and the underlying causes of genetic instability. Mol Genet Metab. 2021 May;133(1):35-48. doi: 10.1016/j.ymgme.2021.03.003. Epub 2021 Mar 10. PMID: 33736941.
– Cunniff C, Bassetti JA, Ellis NA. (2017) Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol Syndromol. 2017 Jan;8(1):4-23. doi: 10.1159/000452082. Epub 2016 Nov 5. PMID: 28232778; PMCID: PMC5260600.
– Kaur E, Agrawal R and Sengupta S (2021) Functions of BLM Helicase in Cells: Is It Acting Like a Double-Edged Sword? Front. Genet. 12:634789. doi: 10.3389/fgene.2021.634789

{kind=link}
{kind=link}